
p53/p73 Protein Network in Colorectal Cancer and Other Human Malignancies





Simple Summary
Abstract
1. Introduction


2. Gene Architecture and Generation of the p53/p73 Isoforms
3. Structure of the p53 Protein Isoforms
4. Structure of the p73 Protein Isoforms
5. Regulation of the p53 Isoforms’ Expression and Activity
6. Regulation of the p73 Isoforms’ Expression and Activity
7. Biological Activity and Functions of the p53 Isoforms
8. Biological Activity and Functions of the p73 Isoforms
9. Crosstalk between p53/p73 Isoforms
10. Alterations of the p53 and p73 Isoforms in Cancer
11. Alterations of the p53/p73 Isoforms Expression in Colorectal Cancer
12. Involvement of the p53/p73 Isoforms in CRC Development and Progression
13. Prognostic Relevance of the p53/p73 Isoforms Expression in CRC
14. Targeting the p53/p73 Family Isoforms in CRC
15. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lane, D. P53, Guardian of the Genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Kaghad, M.; Bonnet, H.; Yang, A.; Creancier, L.; Biscan, J.C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.M.; Dumont, X.; et al. Monoallelically Expressed Gene Related to P53 at 1p36, a Region Frequently Deleted in Neuroblastoma and Other Human Cancers. Cell 1997, 90, 809–819. [Google Scholar] [CrossRef]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dötsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. P63, a P53 Homolog at 3q27-29, Encodes Multiple Products with Transactivating, Death-Inducing, and Dominant-Negative Activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef]
- Moll, U.M.; Slade, N. P63 and P73: Roles in Development and Tumor Formation. Mol. Cancer Res. 2004, 2, 371–386. [Google Scholar]
- Murray-Zmijewski, F.; Lane, D.P.; Bourdon, J.C. P53/P63/P73 Isoforms: An Orchestra of Isoforms to Harmonise Cell Differentiation and Response to Stress. Cell Death Differ. 2006, 13, 962–972. [Google Scholar] [CrossRef]
- Campbell, J.D.; Yau, C.; Bowlby, R.; Liu, Y.; Brennan, K.; Fan, H.; Taylor, A.M.; Wang, C.; Walter, V.; Akbani, R.; et al. Genomic, Pathway Network, and Immunologic Features Distinguishing Squamous Carcinomas. Cell Rep. 2018, 23, 194–212.e6. [Google Scholar] [CrossRef]
- Candi, E.; Agostini, M.; Melino, G.; Bernassola, F. How the TP53 Family Proteins TP63 and TP73 Contribute to Tumorigenesis: Regulators and Effectors. Hum. Mutat. 2014, 35, 702–714. [Google Scholar] [CrossRef]
- Joruiz, S.M.; Bourdon, J.-C. P53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 2016, 6, a026039. [Google Scholar] [CrossRef]
- Ozretić, P.; Hanžić, N.; Proust, B.; Sabol, M.; Trnski, D.; Radić, M.; Musani, V.; Ciribilli, Y.; Milas, I.; Puljiz, Z.; et al. Expression Profiles of P53/P73, NME and GLI Families in Metastatic Melanoma Tissue and Cell Lines. Sci. Rep. 2019, 9, 12470. [Google Scholar] [CrossRef]
- Marcel, V.; Dichtel-Danjoy, M.L.; Sagne, C.; Hafsi, H.; Ma, D.; Ortiz-Cuaran, S.; Olivier, M.; Hall, J.; Mollereau, B.; Hainaut, P.; et al. Biological Functions of P53 Isoforms through Evolution: Lessons from Animal and Cellular Models. Cell Death Differ. 2011, 18, 1815–1824. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.-C.; Fernandes, K.; Murray-zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. P53 Isoforms Can Regulate P53 Transcriptional Activity. Genes Dev. 2005, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.S.; Grover, R.; Das, S. Two Internal Ribosome Entry Sites Mediate the Translation of P53 Isoforms. EMBO Rep. 2006, 7, 404–410. [Google Scholar] [CrossRef]
- Sharathchandra, A.; Katoch, A.; Das, S. IRES Mediated Translational Regulation of P53 Isoforms. Wiley Interdiscip. Rev. RNA 2014, 5, 131–139. [Google Scholar] [CrossRef]
- Anbarasan, T.; Bourdon, J.C. The Emerging Landscape of P53 Isoforms in Physiology, Cancer and Degenerative Diseases. Int. J. Mol. Sci. 2019, 20, 6257. [Google Scholar] [CrossRef]
- Yin, Y.; Stephen, C.W.; Luciani, M.G.; Fåhraeus, R. P53 Stability and Activity Is Regulated By Mdm2-Mediated Induction of Alternative P53 Translation Products. Nat. Cell Biol. 2002, 4, 462–467. [Google Scholar] [CrossRef]
- Ghosh, A.; Stewart, D.; Matlashewski, G. Regulation of Human P53 Activity and Cell Localization by Alternative Splicing. Mol. Cell. Biol. 2004, 24, 7987–7997. [Google Scholar] [CrossRef] [PubMed]
- Candeias, M.M.; Powell, D.J.; Roubalova, E.; Apcher, S.; Bourougaa, K.; Vojtesek, B.; Bruzzoni-Giovanelli, H.; Fåhraeus, R. Expression of P53 and P53/47 Are Controlled by Alternative Mechanisms of Messenger RNA Translation Initiation. Oncogene 2006, 25, 6936–6947. [Google Scholar] [CrossRef]
- Marcel, V.; Perrier, S.; Aoubala, M.; Ageorges, S.; Groves, M.J.; Diot, A.; Fernandes, K.; Tauro, S.; Bourdon, J.C. Δ160p53 Is a Novel N-Terminal P53 Isoform Encoded by Δ133p53 Transcript. FEBS Lett. 2010, 584, 4463–4468. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Mondal, A.M.; Horikawa, I.; Nguyen, G.H.; Kumamoto, K.; Sohn, J.J.; Bowman, E.D.; Mathe, E.A.; Schetter, A.J.; Pine, S.R.; et al. P53 Isoforms Δ133p53 and P53β Are Endogenous Regulators of Replicative Cellular Senescence. Nat. Cell Biol. 2009, 11, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Fragou, A.; Tzimagiorgis, G.; Karageorgopoulos, C.; Barbetakis, N.; Lazopoulos, A.; Papaioannou, M.; Haitoglou, C.; Kouidou, S. Increased Δ133p53 MRNA in Lung Carcinoma Corresponds with Reduction of P21 Expression. Mol. Med. Rep. 2017, 15, 1455–1460. [Google Scholar] [CrossRef]
- Hofstetter, G.; Berger, A.; Schuster, E.; Wolf, A.; Hager, G.; Vergote, I.; Cadron, I.; Sehouli, J.; Braicu, E.I.; Mahner, S.; et al. Δ133p53 Is an Independent Prognostic Marker in P53 Mutant Advanced Serous Ovarian Cancer. Br. J. Cancer 2011, 105, 1593–1599. [Google Scholar] [CrossRef]
- Nutthasirikul, N.; Limpaiboon, T.; Leelayuwat, C.; Patrakitkomjorn, S.; Jearanaikoon, P. Ratio Disruption of the Δ133p53 and TAp53 Isoform Equilibrium Correlates with Poor Clinical Outcome in Intrahepatic Cholangiocarcinoma. Int. J. Oncol. 2013, 42, 1181–1188. [Google Scholar] [CrossRef]
- Avery-Kiejda, K.A.; Morten, B.; Wong-Brown, M.W.; Mathe, A.; Scott, R.J. The Relative MRNA Expression of P53 Isoforms in Breast Cancer Is Associated with Clinical Features and Outcome. Carcinogenesis 2014, 35, 586–596. [Google Scholar] [CrossRef]
- Takahashi, R.; Giannini, C.; Sarkaria, J.N.; Schroeder, M.; Rogers, J.; Mastroeni, D.; Scrable, H. P53 Isoform Profiling in Glioblastoma and Injured Brain. Oncogene 2013, 32, 3165–3174. [Google Scholar] [CrossRef] [PubMed]
- Boldrup, L.; Bourdon, J.C.; Coates, P.J.; Sjöström, B.; Nylander, K. Expression of P53 Isoforms in Squamous Cell Carcinoma of the Head and Neck. Eur. J. Cancer 2007, 43, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Huo, S.W.; Lü, J.J.; Liu, Z.; Fang, X.L.; Jin, X.B.; Yuan, M.Z. Expression of P53 Isoforms in Renal Cell Carcinoma. Chin. Med. J. 2009, 122, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Mondal, A.M.; Horikawa, I.; Nguyen, G.H.; Sohn, J.J.; Bowman, E.D.; Mathe, E.A.; Schetter, A.J.; Pine, R.; Ji, H.; et al. P53 Isoforms in Cellular Senescence-and Ageing-Associated Biological and Physiological Functions. Int. J. Mol. Sci. 2019, 20, 6023. [Google Scholar] [CrossRef]
- Zaika, A.I.; Slade, N.; Erster, S.H.; Sansome, C.; Joseph, T.W.; Pearl, M.; Chalas, E.; Moll, U.M. ΔNp73, a Dominant-Negative Inhibitor of Wild-Type P53 and TAp73, Is up-Regulated in Human Tumors. J. Exp. Med. 2002, 196, 765–780. [Google Scholar] [CrossRef]
- Ishimoto, O.; Kawahara, C.; Enjo, K.; Obinata, M.; Nukiwa, T.; Ikawa, S. Possible Oncogenic Potential of ΔNp73: A Newly Identified Isoform of Human P73. Cancer Res. 2002, 62, 636–641. [Google Scholar]
- Stiewe, T.; Zimmermann, S.; Frilling, A.; Esche, H.; Pützer, B.M. Transactivation-Deficient ΔTA-P73 Acts as an Oncogene. Cancer Res. 2002, 62, 3598–3602. [Google Scholar] [PubMed]
- Stiewe, T.; Theseling, C.C.; Pützer, B.M. Transactivation-Deficient ΔTA-P73 Inhibits P53 by Direct Competition for DNA Binding. Implications for Tumorigenesis. J. Biol. Chem. 2002, 277, 14177–14185. [Google Scholar] [CrossRef] [PubMed]
- Pützer, B.M.; Tuve, S.; Tannapfel, A.; Stiewe, T. Increased ΔN-P73 Expression in Tumors by Upregulation of the E2F1-Regulated, TA-Promoter-Derived ΔN′-P73 Transcript. Cell Death Differ. 2003, 10, 612–614. [Google Scholar] [CrossRef][Green Version]
- Sayan, A.E.; Roperch, J.P.; Sayan, B.S.; Rossi, M.; Pinkoski, M.J.; Knight, R.A.; Willis, A.E.; Melino, G. Generation of ΔTAp73 Proteins by Translation from a Putative Internal Ribosome Entry Site. Ann. N. Y. Acad. Sci. 2007, 1095, 315–324. [Google Scholar] [CrossRef] [PubMed]
- De Laurenzi, V.; Costanzo, A.; Barcaroli, D.; Terrinoni, A.; Falco, M.; Annicchiarico-Petruzzelli, M.; Levrero, M.; Melino, G. Two New P73 Splice Variants, γ and δ, with Different Transcriptional Activity. J. Exp. Med. 1998, 188, 1763–1768. [Google Scholar] [CrossRef]
- De Laurenzi, V.; Catani, M.V.; Terrinoni, A.; Corazzari, M.; Melino, G.; Constanzo, A.; Levrero, M.; Knight, R.A. Additional Complexity in P73: Induction by Mitogens in Lymphoid Cells and Identification of Two New Splicing Variants ε and ζ. Cell Death Differ. 1999, 6, 389–390. [Google Scholar] [CrossRef]
- Zaika, A.I.; Kovalev, S.; Marchenko, N.D.; Moll, U.M. Overexpression of the Wild Type P73 Gene in Breast Cancer Tissues and Cell Lines. Cancer Res. 1999, 59, 3257–3263. [Google Scholar] [PubMed]
- Vilgelm, A.E.; Washington, M.K.; Wei, J.; Chen, H.; Prassolov, V.S.; Zaika, A.I. Interactions of the P53 Protein Family in Cellular Stress Response in Gastrointestinal Tumors. Mol. Cancer Ther. 2010, 9, 693–705. [Google Scholar] [CrossRef]
- Tschan, M.P.; Grob, T.J.; Peters, U.R.; de Laurenzi, V.; Huegli, B.; Kreuzer, K.A.; Schmidt, C.A.; Melino, G.; Fey, M.F.; Tobler, A.; et al. Enhanced P73 Expression during Differentiation and Complex P73 Isoforms in Myeloid Leukemia. Biochem. Biophys. Res. Commun. 2000, 277, 62–65. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically Unstructured Proteins and Their Functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Chillemi, G.; Kehrloesser, S.; Bernassola, F.; Desideri, A.; Dötsch, V.; Levine, A.J.; Melino, G. Structural Evolution and Dynamics of the P53 Proteins. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef]
- Raj, N.; Attardi, L.D. The Transactivation Domains of the P53 Protein. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef]
- Brady, C.A.; Jiang, D.; Mello, S.S.; Johnson, T.M.; Jarvis, L.A.; Kozak, M.M.; Broz, D.K.; Basak, S.; Park, E.J.; McLaughlin, M.E.; et al. Distinct P53 Transcriptional Programs Dictate Acute DNA-Damage Responses and Tumor Suppression. Cell 2011, 145, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Tsutsumi, S.; Chen, Y.; Ozeki, C.; Okabe, A.; Kawase, T.; Aburatani, H.; Ohki, R. Identification and Characterization of the Binding Sequences and Target Genes of P53 Lacking the 1st Transactivation Domain. Cancer Sci. 2020, 111, 451–466. [Google Scholar] [CrossRef]
- Yu, H.; Chen, J.K.; Feng, S.; Dalgarno, D.C.; Brauer, A.W.; Schreiber, S.L. Structural Basis for the Binding of Proline-Rich Peptides to SH3 Domains. Cell 1994, 76, 933–945. [Google Scholar] [CrossRef]
- Walker, K.K.; Levine, A.J. Identification of a Novel P53 Functional Domain That Is Necessary for Efficient Growth Suppression. Proc. Natl. Acad. Sci. USA 1996, 93. [Google Scholar] [CrossRef] [PubMed]
- Dornan, D.; Shimizu, H.; Burch, L.; Smith, A.J.; Hupp, T.R. The Proline Repeat Domain of P53 Binds Directly to the Transcriptional Coactivator P300 and Allosterically Controls DNA-Dependent Acetylation of P53. Mol. Cell. Biol. 2003, 23, 8846–8861. [Google Scholar] [CrossRef] [PubMed]
- Sakamuro, D.; Sabbatini, P.; White, E.; Prendergast, G.C. The Polyproline Region of P53 Is Required to Activate Apoptosis but Not Growth Arrest. Oncogene 1997, 15, 887–898. [Google Scholar] [CrossRef]
- Baptiste, N.; Friedlander, P.; Chen, X.; Prives, C. The Proline-Rich Domain of P53 Is Required for Cooperation with Anti-Neoplastic Agents to Promote Apoptosis of Tumor Cells. Oncogene 2002, 21, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Zorić, A.; Horvat, A.; Balija, M.; Slade, N. The Arg72Pro Polymorphism of TP53 in Croatian Population. Croat. Chem. Acta 2012, 85, 239–243. [Google Scholar] [CrossRef]
- Elshazli, R.M.; Toraih, E.A.; Elgaml, A.; Kandil, E.; Fawzy, M.S. Genetic Polymorphisms of TP53 (Rs1042522) and MDM2 (Rs2279744) and Colorectal Cancer Risk: An Updated Meta-Analysis Based on 59 Case-Control Studies. Gene 2020, 734, 144391. [Google Scholar] [CrossRef] [PubMed]
- Pavletich, N.P.; Chambers, K.A.; Pabo, C.O. The DNA-Binding Domain of 53 Contains the Four Conserved Regions the Major Mutation Hot Spots. Genes Dev. 1993, 7, 2556–2564. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal Structure of a P53 Tumor Suppressor-DNA Complex: Understanding Tumorigenic Mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef]
- Lei, J.; Qi, R.; Tang, Y.; Wang, W.; Wei, G.; Nussinov, R.; Ma, B. Conformational Stability and Dynamics of the Cancer-Associated Isoform Δ133p53β Are Modulated by P53 Peptides and P53-Specific DNA. FASEB J. 2019, 33, 4225–4235. [Google Scholar] [CrossRef]
- Hainaut, P.; Pfeifer, G.P. Somatic TP53 Mutations in the Era of Genome Sequencing. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef]
- Schlereth, K.; Beinoraviciute-Kellner, R.; Zeitlinger, M.K.; Bretz, A.C.; Sauer, M.; Charles, J.P.; Vogiatzi, F.; Leich, E.; Samans, B.; Eilers, M.; et al. DNA Binding Cooperativity of P53 Modulates the Decision between Cell-Cycle Arrest and Apoptosis. Mol. Cell 2010, 38, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Timofeev, O.; Schlereth, K.; Wanzel, M.; Braun, A.; Nieswandt, B.; Pagenstecher, A.; Rosenwald, A.; Elsässer, H.P.; Stiewe, T. P53 DNA Binding Cooperativity Is Essential for Apoptosis and Tumor Suppression InVivo. Cell Rep. 2013, 3, 1512–1525. [Google Scholar] [CrossRef]
- Marine, J.C. P53 Stabilization: The Importance of Nuclear Import. Cell Death Differ. 2010, 17, 191–192. [Google Scholar] [CrossRef]
- Marchenko, N.; Hanel, W.; Li, D.; Becker, K.; Reich, N.; Moll, U. Stress-Mediated Nuclear Stabilization of P53 Is Regulated by Ubiquitination and Importin-A3 Binding. Cell Death Differ. 2010, 17, 255–267. [Google Scholar] [CrossRef]
- Aurelio, O.N.; Cajot, J.F.; Hua, M.L.H.; Khwaja, Z.; Stanbridge, E.J. Germ-Line-Derived Hinge Domain P53 Mutants Have Lost Apoptotic but Not Cell Cycle Arrest Functions. Cancer Res. 1998, 58, 2190–2195. [Google Scholar]
- Kong, X.T.; Gao, H.; Stanbridge, E.J. Mechanisms of Differential Activation of Target Gene Promoters by P53 Hinge Domain Mutants with Impaired Apoptotic Function. J. Biol. Chem. 2001, 276, 32990–33000. [Google Scholar] [CrossRef] [PubMed]
- Scoumanne, A.; Harms, K.L.; Chen, X. Structural Basis for Gene Activation by P53 Family Members. Cancer Biol. Ther. 2005, 4, 1178–1185. [Google Scholar] [CrossRef]
- Jeffrey, P.D.; Gorina, S.; Pavletich, N.P. Crystal Structure of the Tetramerization Domain of the P53 Tumor Suppressor at 1.7 Angstroms. Science 1995, 267, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Stommel, J.M.; Marchenko, N.D.; Jimenez, G.S.; Moll, U.M.; Hope, T.J.; Wahl, G.M. A Leucine-Rich Nuclear Export Signal in the P53 Tetramerization Domain: Regulation of Subcellular Localization and P53 Activity by NES Masking. EMBO J. 1999, 18, 1660–1672. [Google Scholar] [CrossRef] [PubMed]
- Beckerman, R.; Yoh, K.; Mattia-Sansobrino, M.; Zupnick, A.; Laptenko, O.; Karni-Schmidt, O.; Ahn, J.; Byeon, I.J.; Keezer, S.; Prives, C. Lysines in the Tetramerization Domain of P53 Selectively Modulate G1 Arrest. Cell Cycle 2016, 15, 1425–1438. [Google Scholar] [CrossRef] [PubMed]
- Gencel-Augusto, J.; Lozano, G. P53 Tetramerization: At the Center of the Dominant-Negative Effect of Mutant P53. Genes Dev. 2020, 34, 1128–1146. [Google Scholar] [CrossRef]
- Lang, V.; Pallara, C.; Zabala, A.; Lobato-Gil, S.; Lopitz-Otsoa, F.; Farrás, R.; Hjerpe, R.; Torres-Ramos, M.; Zabaleta, L.; Blattner, C.; et al. Tetramerization-Defects of P53 Result in Aberrant Ubiquitylation and Transcriptional Activity. Mol. Oncol. 2014, 8, 1026–1042. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W.; Anderson, C.W. Posttranslational Modification of P53: Cooperative Integrators of Function. Cold Spring Harb. Perspect. Biol. 2009, 1, 1–16. [Google Scholar] [CrossRef]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The Multiple Mechanisms That Regulate P53 Activity and Cell Fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Laptenko, O.; Shiff, I.; Freed-Pastor, W.; Zupnick, A.; Mattia, M.; Freulich, E.; Shamir, I.; Kadouri, N.; Kahan, T.; Manfredi, J.; et al. The P53 C Terminus Controls Site-Specific DNA Binding and Promotes Structural Changes within the Central DNA Binding Domain. Mol. Cell 2015, 57. [Google Scholar] [CrossRef]
- Harms, K.L.; Chen, X. The Functional Domains in P53 Family Proteins Exhibit Both Common and Distinct Properties. Cell Death Differ. 2006, 13, 890–897. [Google Scholar] [CrossRef]
- Burge, S.; Teufel, D.P.; Townsley, F.M.; Freund, S.M.V.; Bycroft, M.; Fersht, A.R. Molecular Basis of the Interactions between the P73 N Terminus and P300: Effects on Transactivation and Modulation by Phosphorylation. Proc. Natl. Acad. Sci. USA 2009, 106, 3142–3147. [Google Scholar] [CrossRef]
- Krauskopf, K.; Gebel, J.; Kazemi, S.; Tuppi, M.; Löhr, F.; Schäfer, B.; Koch, J.; Güntert, P.; Dötsch, V.; Kehrloesser, S. Regulation of the Activity in the P53 Family Depends on the Organization of the Transactivation Domain. Structure 2018, 26, 1091–1100.e4. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Nozell, S.; Xiao, H.; Chen, X. ΔNp73β Is Active in Transactivation and Growth Suppression. Mol. Cell. Biol. 2004, 24, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Toh, W.H.; Logette, E.; Corcos, L.; Sabapathy, K. TAp73β and DNp73β Activate the Expression of the Pro-Survival Caspase-2S. Nucleic Acids Res. 2008, 36, 4498–4509. [Google Scholar] [CrossRef]
- Kartasheva, N.N.; Lenz-Bauer, C.; Hartmann, O.; Schäfer, H.; Eilers, M.; Dobbelstein, M. ΔNp73 Can Modulate the Expression of Various Genes in a P53-Independent Fashion. Oncogene 2003, 22, 8246–8254. [Google Scholar] [CrossRef] [PubMed]
- Niemantsverdriet, M.; Nagle, P.; Chiu, R.K.; Langendijk, J.A.; Kampinga, H.H.; Coppes, R.P. ΔNp73 Enhances Promoter Activity of TGF-β Induced Genes. PLoS ONE 2012, 7, e50815. [Google Scholar] [CrossRef]
- Scott Beeler, J.; Marshall, C.B.; Gonzalez-Ericsson, P.I.; Shaver, T.M.; Santos Guasch, G.L.; Lea, S.T.; Johnson, K.N.; Jin, H.; Venters, B.J.; Sanders, M.E.; et al. P73 Regulates Epidermal Wound Healing and Induced Keratinocyte Programming. PLoS ONE 2019, 14. [Google Scholar] [CrossRef]
- Nozell, S.; Wu, Y.; McNaughton, K.; Liu, G.; Willis, A.; Paik, J.C.; Chen, X. Characterization of P73 Functional Domains Necessary for Transactivation and Growth Suppression. Oncogene 2003, 22, 4333–4347. [Google Scholar] [CrossRef]
- Yuan, Z.M.; Shioya, H.; Ishiko, T.; Sun, X.; Gu, J.; Huang, Y.; Lu, H.; Kharbanda, S.; Weichselbaum, R.; Kufe, D. P73 Is Regulated by Tyrosine Kinase C-Abl in the Apoptotic Response to DNA Damage. Nature 1999, 399, 814–817. [Google Scholar] [CrossRef]
- Agami, R.; Blandino, G.; Oren, M.; Shaul, Y. Interaction of C-Abl and P73α and Their Collaboration to Induce Apoptosis. Nature 1999, 399, 809–812. [Google Scholar] [CrossRef]
- Canning, P.; Von Delft, F.; Bullock, A.N. Structural Basis for ASPP2 Recognition by the Tumor Suppressor P73. J. Mol. Biol. 2012, 423, 515–527. [Google Scholar] [CrossRef][Green Version]
- Ramos, A.; Tse, P.W.; Wang, J.; Ethayathulla, A.S.; Viadiu, H. Sequence Variation in the Response Element Determines Binding by the Transcription Factor P73. Biochemistry 2015, 54, 6961–6972. [Google Scholar] [CrossRef]
- Coutandin, D.; Löhr, F.; Niesen, F.H.; Ikeya, T.; Weber, T.A.; Schäfer, B.; Zielonka, E.M.; Bullock, A.N.; Yang, A.; Güntert, P.; et al. Conformational Stability and Activity of P73 Require a Second Helix in the Tetramerization Domain. Cell Death Differ. 2009, 16, 1582–1589. [Google Scholar] [CrossRef]
- Chi, S.W.; Ayed, A.; Arrowsmith, C.H. Solution Structure of a Conserved C-Terminal Domain of P73 with Structural Homology to the SAM Domain. EMBO J. 1999, 18, 4438–4445. [Google Scholar] [CrossRef] [PubMed]
- Vikhreva, P.; Melino, G.; Amelio, I. P73 Alternative Splicing: Exploring a Biological Role for the C-Terminal Isoforms. J. Mol. Biol. 2018, 430, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Chen, X. The C-Terminal Sterile α Motif and the Extreme C Terminus Regulate the Transcriptional Activity of the α Isoform of P73. J. Biol. Chem. 2005, 280, 20111–20119. [Google Scholar] [CrossRef]
- Bellini, I.; Pitto, L.; Marini, M.G.; Porcu, L.; Moi, P.; Garritano, S.; Boldrini, L.; Rainaldi, G.; Fontanini, G.; Chiarugi, M.; et al. ΔN133p53 Expression Levels in Relation to Haplotypes of the TP53 Internal Promoter Region. Hum. Mutat. 2010, 31, 456–465. [Google Scholar] [CrossRef]
- Eiholzer, R.A.; Mehta, S.; Kazantseva, M.; Drummond, C.J.; McKinney, C.; Young, K.; Slater, D.; Morten, B.C.; Avery-Kiejda, K.A.; Lasham, A.; et al. Intronic TP53 Polymorphisms Are Associated with Increased ∆133TP53 Transcript, Immune Infiltration and Cancer Risk. Cancers 2020, 12, 2472. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Vijayakumar, V.; Fernández-Cuesta, L.; Hafsi, H.; Sagne, C.; Hautefeuille, A.; Olivier, M.; Hainaut, P. P53 Regulates the Transcription of Its Δ133p53 Isoform through Specific Response Elements Contained within the TP53 P2 Internal Promoter. Oncogene 2010, 29, 2691–2700. [Google Scholar] [CrossRef]
- Aoubala, M.; Murray-Zmijewski, F.; Khoury, M.P.; Fernandes, K.; Perrier, S.; Bernard, H.; Prats, A.C.; Lane, D.P.; Bourdon, J.C. P53 Directly Transactivates Δ133p53α, Regulating Cell Fate Outcome in Response to DNA Damage. Cell Death Differ. 2011, 18, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Fernandes, K.; Terrier, O.; Lane, D.P.; Bourdon, J.C. Modulation of P53β and P53γ Expression by Regulating the Alternative Splicing of TP53 Gene Modifies Cellular Response. Cell Death Differ. 2014, 21, 1377–1387. [Google Scholar] [CrossRef]
- Marcel, V.; Petit, I.; Murray-Zmijewski, F.; Goullet De Rugy, T.; Fernandes, K.; Meuray, V.; Diot, A.; Lane, D.P.; Aberdam, D.; Bourdon, J.C. Diverse P63 and P73 Isoforms Regulate Δ133p53 Expression through Modulation of the Internal TP53 Promoter Activity. Cell Death Differ. 2012, 19, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Noto, J.; Zaika, E.; Romero-Gallo, J.; Correa, P.; El-Rifai, W.; Peek, R.M.; Zaika, A. Pathogenic Bacterium Helicobacter Pylori Alters the Expression Profile of P53 Protein Isoforms and P53 Response to Cellular Stresses. Proc. Natl. Acad. Sci. USA 2012, 109, E2543–E2550. [Google Scholar] [CrossRef]
- Tang, Y.; Horikawa, I.; Ajiro, M.; Robles, A.I.; Fujita, K.; Mondal, A.M.; Stauffer, J.K.; Zheng, Z.M.; Harris, C.C. Downregulation of Splicing Factor SRSF3 Induces P53β, an Alternatively Spliced Isoform of P53 That Promotes Cellular Senescence. Oncogene 2013, 32, 2792–2798. [Google Scholar] [CrossRef] [PubMed]
- Mermoud, J.E.; Cohen, P.T.W.; Lamond, A.I. Regulation of Mammalian Spliceosome Assembly by a Protein Phosphorylation Mechanism. EMBO J. 1994, 13, 5679–5688. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, G.; Adams, J.A. Phosphorylation Mechanism and Structure of Serine-Arginine Protein Kinases. FEBS J. 2011, 278, 587–597. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, D.; Qin, X.; Owzar, K.; McCann, J.J.; Kastan, M.B. Dna-Damage-Induced Alternative Splicing of P53. Cancers 2021, 13, 251. [Google Scholar] [CrossRef]
- Chen, J.; Crutchley, J.; Zhang, D.; Owzar, K.; Kastan, M.B. Identification of a DNA Damage–Induced Alternative Splicing Pathway That Regulates P53 and Cellular Senescence Markers. Cancer Discov. 2017, 7, 766–781. [Google Scholar] [CrossRef]
- Xie, N.; Chen, M.; Dai, R.; Zhang, Y.; Zhao, H.; Song, Z.; Zhang, L.; Li, Z.; Feng, Y.; Gao, H.; et al. SRSF1 Promotes Vascular Smooth Muscle Cell Proliferation through a Δ133p53/EGR1/KLF5 Pathway. Nat. Commun. 2017, 8, 16016. [Google Scholar] [CrossRef]
- Marcel, V.; Tran, P.L.T.; Sagne, C.; Martel-Planche, G.; Vaslin, L.; Teulade-Fichou, M.P.; Hall, J.; Mergny, J.L.; Hainaut, P.; van Dyck, E. G-Quadruplex Structures in TP53 Intron 3: Role in Alternative Splicing and in Production of P53 MRNA Isoforms. Carcinogenesis 2011, 32, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Perriaud, L.; Marcel, V.; Sagne, C.; Favaudon, V.; Guédin, A.; De Rache, A.; Guetta, C.; Hamon, F.; Teulade-Fichou, M.P.; Hainaut, P.; et al. Impact of G-Quadruplex Structures and Intronic Polymorphisms Rs17878362 and Rs1642785 on Basal and Ionizing Radiation-Induced Expression of Alternative P53 Transcripts. Carcinogenesis 2014, 35, 2706–2715. [Google Scholar] [CrossRef]
- Sagne, C.; Marcel, V.; Amadou, A.; Hainaut, P.; Olivier, M.; Hall, J. A Meta-Analysis of Cancer Risk Associated with the TP53 Intron 3 Duplication Polymorphism (Rs17878362): Geographic and Tumor-Specific Effects. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef]
- Morten, B.C.; Wong-Brown, M.W.; Scott, R.J.; Avery-Kiejda, K.A. The Presence of the Intron 3 16 Bp Duplication Polymorphism of P53 (Rs17878362) in Breast Cancer Is Associated with a Low Δ40p53:P53 Ratio and Better Outcome. Carcinogenesis 2016, 37, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Porubiaková, O.; Bohálová, N.; Inga, A.; Vadovičová, N.; Coufal, J.; Fojta, M.; Brázda, V. The Influence of Quadruplex Structure in Proximity to P53 Target Sequences on the Transactivation Potential of P53 Alpha Isoforms. Int. J. Mol. Sci. 2020, 21, 127. [Google Scholar] [CrossRef] [PubMed]
- Monti, P.; Brazda, V.; Bohálová, N.; Porubiaková, O.; Menichini, P.; Speciale, A.; Bocciardi, R.; Inga, A.; Fronza, G. Evaluating the Influence of a G-Quadruplex Prone Sequence on the Transactivation Potential by Wild-Type and/or Mutant P53 Family Proteins through a Yeast-Based Functional Assay. Genes 2021, 12, 277. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.Q.; Halaby, M.J.; Zhang, Y. The Identification of an Internal Ribosomal Entry Site in the 5′-Untranslated Region of P53 MRNA Provides a Novel Mechanism for the Regulation of Its Translation Following DNA Damage. Oncogene 2006, 25, 4613–4619. [Google Scholar] [CrossRef]
- Takagi, M.; Absalon, M.J.; McLure, K.G.; Kastan, M.B. Regulation of P53 Translation and Induction after DNA Damage by Ribosomal Protein L26 and Nucleolin. Cell 2005, 123, 49–63. [Google Scholar] [CrossRef]
- Grover, R.; Ray, P.S.; Das, S. Polypyrimidine Tract Binding Protein Regulates IRES-Mediated Translation of P53 Isoforms. Cell Cycle 2008, 7, 2189–2198. [Google Scholar] [CrossRef]
- Khan, D.; Katoch, A.; Das, A.; Sharathchandra, A.; Lal, R.; Roy, P.; Das, S.; Chattopadhyay, S.; Das, S. Reversible Induction of Translational Isoforms of P53 in Glucose Deprivation. Cell Death Differ. 2015, 22, 1203–1218. [Google Scholar] [CrossRef]
- Bellodi, C.; Kopmar, N.; Ruggero, D. Deregulation of Oncogene-Induced Senescence and P53 Translational Control in X-Linked Dyskeratosis Congenita. EMBO J. 2010, 29, 1865–1876. [Google Scholar] [CrossRef]
- Halaby, M.-J.; Harris, B.R.E.; Miskimins, W.K.; Cleary, M.P.; Yang, D.-Q. Deregulation of Internal Ribosome Entry Site-Mediated P53 Translation in Cancer Cells with Defective P53 Response to DNA Damage. Mol. Cell. Biol. 2015, 35, 4006–4017. [Google Scholar] [CrossRef]
- Halaby, M.-J.; Li, Y.; Harris, B.R.; Jiang, S.; Miskimins, W.K.; Cleary, M.P.; Yang, D.Q. Translational Control Protein 80 Stimulates IRES-Mediated Translation of P53 MRNA in Response to DNA Damage. Biomed Res. Int. 2015, 2015. [Google Scholar] [CrossRef]
- Weingarten-Gabbay, S.; Khan, D.; Liberman, N.; Yoffe, Y.; Bialik, S.; Das, S.; Oren, M.; Kimchi, A. The Translation Initiation Factor DAP5 Promotes IRES-Driven Translation of P53 MRNA. Oncogene 2014, 33, 611–618. [Google Scholar] [CrossRef]
- Sharathchandra, A.; Lal, R.; Khan, D.; Das, S. Annexin A2 and PSF Proteins Interact with P53 IRES and Regulate Translation of P53 MRNA. RNA Biol. 2012, 9, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.; Sharathchandra, A.; Ponnuswamy, A.; Grover, R.; Das, S. Effect of a Natural Mutation in the 5′ Untranslated Region on the Translational Control of P53 MRNA. Oncogene 2013, 32, 4148–4159. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guo, K.; Kastan, M.B. Interactions of Nucleolin and Ribosomal Protein L26 (RPL26) in Translational Control of Human P53 MRNA. J. Biol. Chem. 2012, 287, 16467–16476. [Google Scholar] [CrossRef] [PubMed]
- Solomon, H.; Bräuning, B.; Fainer, I.; Ben-Nissan, G.; Rabani, S.; Goldfinger, N.; Moscovitz, O.; Shakked, Z.; Rotter, V.; Sharon, M. Post-Translational Regulation of P53 Function through 20S Proteasome-Mediated Cleavage. Cell Death Differ. 2017, 24, 2187–2198. [Google Scholar] [CrossRef]
- Olshina, M.A.; Ben-Nissan, G.; Sharon, M. Functional Regulation of Proteins by 20S Proteasome Proteolytic Processing. Cell Cycle 2018, 17, 393–394. [Google Scholar] [CrossRef]
- Zorić, A.; Horvat, A.A.; Slade, N. Differential Effects of Diverse P53 Isoforms on TAp73 Transcriptional Activity and Apoptosis. Carcinogenesis 2013, 34, 522–529. [Google Scholar] [CrossRef][Green Version]
- Reinhardt, L.S.; Zhang, X.; Wawruszak, A.; Groen, K.; De Iuliis, G.N.; Avery-Kiejda, K.A. Good Cop, Bad Cop: Defining the Roles of ∆40p53 in Cancer and Aging. Cancers 2020, 12, 1659. [Google Scholar] [CrossRef]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 Oncoprotein Bound to the P53 Tumor Suppressor Transactivation Domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 Is a Ubiquitin Ligase E3 for Tumor Suppressor P53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- Camus, S.; Menéndez, S.; Fernandes, K.; Kua, N.; Liu, G.; Xirodimas, D.P.; Lane, D.P.; Bourdon, J. The P53 Isoforms Are Differentially Modified by Mdm2. Cell Cycle 2012, 11, 1646–1655. [Google Scholar] [CrossRef][Green Version]
- Horikawa, I.; Fujita, K.; Jenkins, L.M.M.; Hiyoshi, Y.; Mondal, A.M.; Vojtesek, B.; Lane, D.P.; Appella, E.; Harris, C.C. Autophagic Degradation of the Inhibitory P53 Isoform δ 133p53α as a Regulatory Mechanism for P53-Mediated Senescence. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Candeias, M.M.; Hagiwara, M.; Matsuda, M. Cancer-specific Mutations in P53 Induce the Translation of Δ160p53 Promoting Tumorigenesis. EMBO Rep. 2016, 17, 1542–1551. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zheng, Y.; Zhu, J.; Jiang, J.; Wang, J. P73 Is Transcriptionally Regulated by DNA Damage, P53, and P73. Oncogene 2001, 20, 769–774. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pediconi, N.; Ianari, A.; Costanzo, A.; Belloni, L.; Gallo, R.; Cimino, L.; Porcellini, A.; Screpanti, I.; Balsano, C.; Alesse, E.; et al. Differential Regulation of E2F1 Apoptotic Target Genes in Response to DNA Damage. Nat. Cell Biol. 2003, 5, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Brooks, L.A.; Sullivan, A.; O’Nions, J.; Bell, A.; Dunne, B.; Tidy, J.A.; Evans, D.J.; Osin, P.; Vousden, K.H.; Gusterson, B.; et al. E7 Proteins from Oncogenic Human Papillomavirus Types Transactivate P73: Role in Cervical Intraepithelial Neoplasia. Br. J. Cancer 2002, 86, 263–268. [Google Scholar] [CrossRef]
- Das, S.; El-Deiry, W.S.; Somasundaram, K. Regulation of the P53 Homolog P73 by Adenoviral Oncogene E1A. J. Biol. Chem. 2003, 278, 18313–18320. [Google Scholar] [CrossRef] [PubMed]
- Fontemaggi, G.; Gurtner, A.; Strano, S.; Higashi, Y.; Sacchi, A.; Piaggio, G.; Blandino, G. The Transcriptional Repressor ZEB Regulates P73 Expression at the Crossroad between Proliferation and Differentiation. Mol. Cell. Biol. 2001, 21, 8461–8470. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wimmer, K.; Kuick, R.; Lamb, B.J.; Motyka, S.; Jasty, R.; Castle, V.P.; Hanash, S.M. N-Myc Modulates Expression of P73 in Neuroblastoma. Neoplasia 2002, 4, 432–439. [Google Scholar] [CrossRef][Green Version]
- Irwin, M.; Marin, M.C.; Phillips, A.C.; Seelan, R.S.; Smith, D.I.; Liu, W.; Flores, E.R.; Tsai, K.Y.; Jacks, T.; Vousden, K.H.; et al. Role for the P53 Homologue P73 in E2F-1-Induced Apoptosis. Nature 2000, 407, 645–648. [Google Scholar] [CrossRef]
- Stiewe, T.; Putzer, B.M. Role of the P53-Homologue P73 in E2F1-Induced Apoptosis. Nat. Genet. 2000, 26, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Pediconi, N.; Guerrieri, F.; Vossio, S.; Bruno, T.; Belloni, L.; Schinzari, V.; Scisciani, C.; Fanciulli, M.; Levrero, M. HSirT1-Dependent Regulation of the PCAF-E2F1-P73 Apoptotic Pathway in Response to DNA Damage. Mol. Cell. Biol. 2009, 29, 1989–1998. [Google Scholar] [CrossRef] [PubMed]
- Marabese, M.; Vikhanskaya, F.; Rainelli, C.; Sakai, T.; Broggini, M. DNA Damage Induces Transcriptional Activation of P73 by Removing C-EBPα Repression on E2F1. Nucleic Acids Res. 2003, 31, 6624–6632. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Peña, C.; Garcia, J.M.; Dominguez, G.; Silva, J.; Garcia, V.; Carcereny, E.; Vargas, J.; Provencio, M.; España, P.; Bonilla, F. Intronic Deletion Affecting a Negative Regulatory Region of TP73 Is Related to Breast and Colorectal Carcinomas. Genes Chromosomes Cancer 2004, 39, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, G.; Peña, C.; Silva, J.; García, J.; García, V.; Rodríguez, R.; Cantos, B.; Citores, M.; España, P.; Bonilla, F. The Presence of an Intronic Deletion Inp73 and High Levels OfZEB1 Alter TheTAp73/ΔTAp73 Ratio in Colorectal Carcinomas. J. Pathol. 2006, 210, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Kim, E.J.; Um, S.J. Transcriptional Regulation of the P73 Gene, a Member of the P53 Family, by Early Growth Response-1 (Egr-1). Biochem. Biophys. Res. Commun. 2007, 362, 1044–1050. [Google Scholar] [CrossRef]
- Lai, J.; Nie, W.; Zhang, W.; Wang, Y.; Xie, R.; Wang, Y.; Gu, J.; Xu, J.; Song, W.; Yang, F.; et al. Transcriptional Regulation of the P73 Gene by Nrf-2 and Promoter CpG Methylation in Human Breast Cancer. Oncotarget 2014, 5, 6909–6922. [Google Scholar] [CrossRef]
- Lalioti, M.E.; Arbi, M.; Loukas, I.; Kaplani, K.; Kalogeropoulou, A.; Lokka, G.; Kyrousi, C.; Mizi, A.; Georgomanolis, T.; Josipovic, N.; et al. GemC1 Governs Multiciliogenesis through Direct Interaction with and Transcriptional Regulation of P73. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Castillo, J.; Goñi, S.; Latasa, M.U.; Perugorría, M.J.; Calvo, A.; Muntané, J.; Bioulac-Sage, P.; Balabaud, C.; Prieto, J.; Avila, M.A.; et al. Amphiregulin Induces the Alternative Splicing of P73 Into Its Oncogenic Isoform ΔEx2p73 in Human Hepatocellular Tumors. Gastroenterology 2009, 137, 1805–1815.e4. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zhang, J.; Zhang, Y.; Jung, Y.-S.; Chen, X. P73 Expression Is Regulated by RNPC1, a Target of the P53 Family, via MRNA Stability. Mol. Cell. Biol. 2012, 32, 2336–2348. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, J.; Yan, W.; Chen, X. P73 Expression Is Regulated by Ribosomal Protein RPL26 through MRNA Translation and Protein Stability. Oncotarget 2016, 7, 78255–78268. [Google Scholar] [CrossRef] [PubMed]
- Conforti, F.; Sayan, A.E.; Sreekumar, R.; Sayan, B.S. Regulation of P73 Activity by Post-Translational Modifications. Cell Death Dis. 2012, 3, e285. [Google Scholar] [CrossRef]
- Ramos, H.; Raimundo, L.; Saraiva, L. P73: From the P53 Shadow to a Major Pharmacological Target in Anticancer Therapy. Pharmacol. Res. 2020, 162. [Google Scholar] [CrossRef] [PubMed]
- Slade, N.; Horvat, A. Targeting P73—A Potential Approach in Cancer Treatment. Curr. Pharm. Des. 2011, 17, 591–602. [Google Scholar] [CrossRef]
- Bang, S.; Kaur, S.; Kurokawa, M. Regulation of the P53 Family Proteins by the Ubiquitin Proteasomal Pathway. Int. J. Mol. Sci. 2020, 21, 261. [Google Scholar] [CrossRef]
- Bálint, E.; Bates, S.; Vousden, K.H. Mdm2 Binds P73α without Targeting Degradation. Oncogene 1999, 18, 3923–3929. [Google Scholar] [CrossRef]
- Dobbelstein, M.; Wienzek, S.; König, C.; Roth, J. Inactivation of the P53-Homologue P73 by the Mdm2-Oncoprotein. Oncogene 1999, 18, 2101–2106. [Google Scholar] [CrossRef]
- Ongkeko, W.M.; Wang, X.Q.; Siu, W.Y.; Lau, A.W.S.; Yamashita, K.; Harris, A.L.; Cox, L.S.; Poon, R.Y.C. MDM2 and MDMX Bind and Stabilize the P53-Related Protein P73. Curr. Biol. 1999, 9, 829–832. [Google Scholar] [CrossRef]
- Zeng, X.; Chen, L.; Jost, C.A.; Maya, R.; Keller, D.; Wang, X.; Kaelin, W.G.; Oren, M.; Chen, J.; Lu, H. MDM2 Suppresses P73 Function without Promoting P73 Degradation. Mol. Cell. Biol. 1999, 19, 3257–3266. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; De Laurenzi, V.; Munarriz, E.; Green, D.R.; Liu, Y.C.; Vousden, K.H.; Cesareni, G.; Melino, G. The Ubiquitin-Protein Ligase Itch Regulates P73 Stability. EMBO J. 2005, 24, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A.; Malatesta, M.; Aqeilan, R.I.; Rossi, M.; Salomoni, P.; Murillas, R.; Sharma, P.; Kuehn, M.R.; Oren, M.; Croce, C.M.; et al. The Nedd4-Binding Partner 1 (N4BP1) Protein Is an Inhibitor of the E3 Ligase Itch. Proc. Natl. Acad. Sci. USA 2007, 104, 11280–11285. [Google Scholar] [CrossRef]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. The Yes-Associated Protein 1 Stabilizes P73 by Preventing Itch-Mediated Ubiquitination of P73. Cell Death Differ. 2007, 14, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Sayan, B.S.; Yang, A.L.; Conforti, F.; Tucci, P.; Piro, M.C.; Browne, G.J.; Agostini, M.; Bernardini, S.; Knight, R.A.; Mak, T.W.; et al. Differential Control of TAp73 and ΔNp73 Protein Stability by the Ring Finger Ubiquitin Ligase PIR2. Proc. Natl. Acad. Sci. USA 2010, 107, 12877–12882. [Google Scholar] [CrossRef]
- Dulloo, I.; Gopalan, G.; Melino, G.; Sabapathya, K. The Antiapoptotic DeltaNp73 Is Degraded in a C-Jun-Dependent Manner upon Genotoxic Stress through the Antizyme-Mediated Pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 4902–4907. [Google Scholar] [CrossRef]
- Abou Zeinab, R.; Wu, H.H.; Abuetabh, Y.; Leng, S.; Sergi, C.; Eisenstat, D.D.; Leng, R.P. Pirh2, an E3 Ligase, Regulates the AIP4-P73 Regulatory Pathway by Modulating AIP4 Expression and Ubiquitination. Carcinogenesis 2021, 42, 650–662. [Google Scholar] [CrossRef]
- Asher, G.; Tsvetkov, P.; Kahana, C.; Shaul, Y. A Mechanism of Ubiquitin-Independent Proteasomal Degradation of the Tumor Suppressors P53 and P73. Genes Dev. 2005, 19, 316–321. [Google Scholar] [CrossRef]
- Munarriz, E.; Bano, D.; Sayan, A.E.; Rossi, M.; Melino, G.; Nicotera, P. Calpain Cleavage Regulates the Protein Stability of P73. Biochem. Biophys. Res. Commun. 2005, 333, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Al-Bahlani, S.; Fraser, M.; Wong, A.Y.C.; Sayan, B.S.; Bergeron, R.; Melino, G.; Tsang, B.K. P73 Regulates Cisplatin-Induced Apoptosis in Ovarian Cancer Cells via a Calcium/Calpain-Dependent Mechanism. Oncogene 2011, 30, 4219–4230. [Google Scholar] [CrossRef]
- Nguyen, D.; Yang, K.; Chiao, L.; Deng, Y.; Zhou, X.; Zhang, Z.; Zeng, S.X.; Lu, H. Inhibition of Tumor Suppressor P73 by Nerve Growth Factor Receptor via Chaperone-Mediated Autophagy. J. Mol. Cell Biol. 2020, 12, 700–712. [Google Scholar] [CrossRef]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of Transcriptional Regulation by P53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef]
- Sammons, M.A.; Nguyen, T.A.T.; McDade, S.S.; Fischer, M. Tumor Suppressor P53: From Engaging DNA to Target Gene Regulation. Nucleic Acids Res. 2020, 48, 8848–8869. [Google Scholar] [CrossRef]
- Khoury, M.P.; Bourdon, J.C. The Isoforms of the P53 Protein. Cold Spring Harb. Perspect. Biol. 2010, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Joruiz, S.M.; Beck, J.A.; Horikawa, I.; Harris, C.C. The ∆133p53 Isoforms, Tuners of the P53 Pathway. Cancers 2020, 12, 3422. [Google Scholar] [CrossRef]
- Mondal, A.M.; Horikawa, I.; Pine, S.R.; Fujita, K.; Morgan, K.M.; Vera, E.; Mazur, S.J.; Appella, E.; Vojtesek, B.; Blasco, M.A.; et al. P53 Isoforms Regulate Aging- and Tumor-Associated Replicative Senescence in T Lymphocytes. J. Clin. Investig. 2013, 123, 5247–5257. [Google Scholar] [CrossRef]
- Von Muhlinen, N.; Horikawa, I.; Alam, F.; Isogaya, K.; Lissa, D.; Vojtesek, B.; Lane, D.P.; Harris, C.C. P53 Isoforms Regulate Premature Aging in Human Cells. Oncogene 2018, 37, 2379–2393. [Google Scholar] [CrossRef]
- Maier, B.; Gluba, W.; Bernier, B.; Turner, T.; Mohammad, K.; Guise, T.; Sutherland, A.; Thorner, M.; Scrable, H. Modulation of Mammalian Life Span by the Short Isoform of P53. Genes Dev. 2004, 18, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Gambino, V.; De Michele, G.; Venezia, O.; Migliaccio, P.; Dall’Olio, V.; Bernard, L.; Minardi, S.P.; Della Fazia, M.A.; Bartoli, D.; Servillo, G.; et al. Oxidative Stress Activates a Specific P53 Transcriptional Response That Regulates Cellular Senescence and Aging. Aging Cell 2013, 12, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.M.; Zhou, H.; Horikawa, I.; Suprynowicz, F.A.; Li, G.; Dakic, A.; Rosenthal, B.; Ye, L.; Harris, C.C.; Schlegel, R.; et al. Δ133P53A, a Natural P53 Isoform, Contributes To Conditional Reprogramming and Long-Term Proliferation of Primary Epithelial Cells. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef]
- Ungewitter, E.; Scrable, H. Δ40p53 Controls the Switch from Pluripotency to Differentiation by Regulating IGF Signaling in ESCs. Genes Dev. 2010, 24, 2408–2419. [Google Scholar] [CrossRef]
- Takahashi, R.; Markovic, S.N.; Scrable, H.J. Dominant Effects of Δ40p53 on P53 Function and Melanoma Cell Fate. J. Investig. Dermatol. 2014, 134, 791–800. [Google Scholar] [CrossRef]
- Ou, Z.; Yin, L.; Chang, C.; Peng, J.; Chen, J. Protein Interaction between P53 and Δ113p53 Is Required for the Anti-Apoptotic Function of Δ113p53. J. Genet. Genom. 2014, 41, 53–62. [Google Scholar] [CrossRef]
- Chen, J.; Ng, S.M.; Chang, C.; Zhang, Z.; Bourdon, J.-C.; Lane, D.P.; Peng, J. p53 isoform 113p53 is a p53 target gene that antagonizes p53 apoptotic activity via BclxL activation in zebrafish. Genes Dev. 2009, 23, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Arsic, N.; Ho-Pun-Cheung, A.; Evelyne, C.; Assenat, E.; Jarlier, M.; Anguille, C.; Colard, M.; Pezet, M.I.; Roux, P.; Gadea, G. The P53 Isoform Delta133p53β Regulates Cancer Cell Apoptosis in a RhoB-Dependent Manner. PLoS ONE 2017, 12, 1–15. [Google Scholar] [CrossRef]
- Gong, L.; Pan, X.; Chen, H.; Rao, L.; Zeng, Y.; Hang, H.; Peng, J.; Xiao, L.; Chen, J. P53 Isoform Δ133p53 Promotes Efficiency of Induced Pluripotent Stem Cells and Ensures Genomic Integrity during Reprogramming. Sci. Rep. 2016, 6, 37281. [Google Scholar] [CrossRef] [PubMed]
- Turnquist, C.; Horikawa, I.; Foran, E.; Major, E.O.; Vojtesek, B.; Lane, D.P.; Lu, X.; Harris, B.T.; Harris, C.C. P53 Isoforms Regulate Astrocyte-Mediated Neuroprotection and Neurodegeneration. Cell Death Differ. 2016, 23, 1515–1528. [Google Scholar] [CrossRef]
- Turnquist, C.; Beck, J.A.; Horikawa, I.; Obiorah, I.E.; Von Muhlinen, N.; Vojtesek, B.; Lane, D.P.; Grunseich, C.; Chahine, J.J.; Ames, H.M.; et al. Radiation-Induced Astrocyte Senescence Is Rescued by Δ133p53. Neuro-Oncol. 2019, 21, 474–485. [Google Scholar] [CrossRef]
- Gong, L.; Gong, H.; Pan, X.; Chang, C.; Ou, Z.; Ye, S.; Yin, L.; Yang, L.; Tao, T.; Zhang, Z.; et al. P53 Isoform Δ113p53/Δ133p53 Promotes DNA Double-Strand Break Repair to Protect Cell from Death and Senescence in Response to DNA Damage. Cell Res. 2015, 25, 351–369. [Google Scholar] [CrossRef]
- Gong, H.; Zhang, Y.; Jiang, K.; Ye, S.; Chen, S.; Zhang, Q.; Peng, J.; Chen, J. P73 Coordinates with Δ133p53 to Promote DNA Double-Strand Break Repair. Cell Death Differ. 2018, 25, 1063–1079. [Google Scholar] [CrossRef]
- Horikawa, I.; Park, K.Y.; Isogaya, K.; Hiyoshi, Y.; Li, H.; Anami, K.; Robles, A.I.; Mondal, A.M.; Fujita, K.; Serrano, M.; et al. Δ133P53 Represses P53-Inducible Senescence Genes and Enhances the Generation of Human Induced Pluripotent Stem Cells. Cell Death Differ. 2017, 24, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Arsic, N.; Gadea, G.; Lagerqvist, E.L.; Busson, M.; Cahuzac, N.; Brock, C.; Hollande, F.; Gire, V.; Pannequin, J.; Roux, P. The P53 Isoform Δ133p53β Promotes Cancer Stem Cell Potential. Stem Cell Rep. 2015, 4, 531–540. [Google Scholar] [CrossRef]
- Medrano, S.; Burns-Cusato, M.; Atienza, M.B.; Rahimi, D.; Scrable, H. Regenerative Capacity of Neural Precursors in the Adult Mammalian Brain Is under the Control of P53. Neurobiol. Aging 2009, 30, 483–497. [Google Scholar] [CrossRef]
- Gong, L.; Pan, X.; Lim, C.B.; De Polo, A.; Little, J.B.; Yuan, Z.M. A Functional Interplay between Δ133p53 and ΔNp63 in Promoting Glycolytic Metabolism to Fuel Cancer Cell Proliferation. Oncogene 2018, 37, 2150–2164. [Google Scholar] [CrossRef]
- Hinault, C.; Kawamori, D.; Liew, C.W.; Maier, B.; Hu, J.; Keller, S.R.; Mirmira, R.G.; Scrable, H.; Kulkarni, R.N. Δ40 Isoform of P53 Controls Β-Cell Proliferation and Glucose Homeostasis in Mice. Diabetes 2011, 60, 1210–1222. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.; Shi, Y.; Liu, K.; Qiao, L.; Guo, X.; Chen, D. Δ40p53 Is Involved in the Inactivation of Autophagy and Contributes to Inhibition of Cell Death in HCT116-Δ40p53 Cells. Oncotarget 2017, 8, 12754–12763. [Google Scholar] [CrossRef]
- Katoch, A.; Tripathi, S.K.; Pal, A.; Das, S. Regulation of MiR-186-YY1 Axis by the P53 Translational Isoform ∆40p53: Implications in Cell Proliferation. Cell Cycle 2021, 20, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Campbell, H.; Fleming, N.; Roth, I.; Mehta, S.; Wiles, A.; Williams, G.; Vennin, C.; Arsic, N.; Parkin, A.; Pajic, M.; et al. Δ133p53 Isoform Promotes Tumour Invasion and Metastasis via Interleukin-6 Activation of JAK-STAT and RhoA-ROCK Signaling. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Slatter, T.L.; Hung, N.; Campbell, H.; Rubio, C.; Mehta, R.; Renshaw, P.; Williams, G.; Wilson, M.; Engelmann, A.; Jeffs, A.; et al. Hyperproliferation, Cancer, and Inflammation in Mice Expressing a Δ133p53-like Isoform. Blood 2011, 117, 5166–5177. [Google Scholar] [CrossRef]
- Bernard, H.; Garmy-Susini, B.; Ainaoui, N.; Van Den Berghe, L.; Peurichard, A.; Javerzat, S.; Bikfalvi, A.; Lane, D.P.; Bourdon, J.C.; Prats, A.C. The P53 Isoform, Δ133p53, Stimulates Angiogenesis and Tumour Progression. Oncogene 2013, 32, 2150–2160. [Google Scholar] [CrossRef]
- Roth, I.; Campbell, H.; Rubio, C.; Vennin, C.; Wilson, M.; Wiles, A.; Williams, G.; Woolley, A.; Timpson, P.; Berridge, M.V.; et al. The Δ133p53 Isoform and Its Mouse Analogue Δ122p53 Promote Invasion and Metastasis Involving Pro-Inflammatory Molecules Interleukin-6 and CCL2. Oncogene 2016, 35, 4981–4989. [Google Scholar] [CrossRef]
- Gadea, G.; Arsic, N.; Fernandes, K.; Diot, A.; Joruiz, S.M.; Abdallah, S.; Meuray, V.; Vinot, S.; Anguille, C.; Remenyi, J.; et al. TP53 Drives Invasion through Expression of Its Δ133p53β Variant. Elife 2016, 5, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Kazantseva, M.; Eiholzer, R.A.; Mehta, S.; Taha, A.; Bowie, S.; Roth, I.; Zhou, J.; Joruiz, S.M.; Royds, J.A.; Hung, N.A.; et al. Elevation of the TP53 Isoform Δ133p53β in Glioblastomas: An Alternative to Mutant P53 in Promoting Tumor Development. J. Pathol. 2018, 246, 77–88. [Google Scholar] [CrossRef]
- Kazantseva, M.; Mehta, S.; Eiholzer, R.A.; Gimenez, G.; Bowie, S.; Campbell, H.; Reily-Bell, A.L.; Roth, I.; Ray, S.; Drummond, C.J.; et al. The Δ133p53β Isoform Promotes an Immunosuppressive Environment Leading to Aggressive Prostate Cancer. Cell Death Dis. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Courtois, S.; Verhaegh, G.; North, S.; Luciani, M.G.; Lassus, P.; Hibner, U.; Oren, M.; Hainaut, P. ΔN-P53, a Natural Isoform of P53 Lacking the First Transactivation Domain, Counteracts Growth Suppression by Wild-Type P53. Oncogene 2002, 21, 6722–6728. [Google Scholar] [CrossRef] [PubMed]
- Avery-Kiejda, K.A.; Xu, D.Z.; Adams, L.J.; Scott, R.J.; Vojtesek, B.; Lane, D.P.; Hersey, P. Small Molecular Weight Variants of P53 Are Expressed in Human Melanoma Cells and Are Induced by the DNA-Damaging Agent Cisplatin. Clin. Cancer Res. 2008, 14, 1659–1668. [Google Scholar] [CrossRef]
- Bourougaa, K.; Naski, N.; Boularan, C.; Mlynarczyk, C.; Candeias, M.M.; Marullo, S.; Fåhraeus, R. Endoplasmic Reticulum Stress Induces G2 Cell-Cycle Arrest via MRNA Translation of the P53 Isoform P53/47. Mol. Cell 2010, 38, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W. Tumour Suppression by P53: A Role for the DNA Damage Response? Nat. Rev. Cancer 2009, 9, 714–723. [Google Scholar] [CrossRef]
- Helton, E.S.; Chen, X. P53 Modulation of the DNA Damage Response. J. Cell. Biochem. 2007, 100, 883–896. [Google Scholar] [CrossRef]
- Gatz, S.A.; Wiesmüller, L. P53 in Recombination and Repair. Cell Death Differ. 2006, 13, 1003–1016. [Google Scholar] [CrossRef]
- Mekeel, K.L.; Tang, W.; Kachnic, L.A.; Luo, C.M.; DeFrank, J.S.; Powell, S.N. Inactivation of P53 Results in High Rates of Homologous Recombination. Oncogene 1997, 14, 1847–1857. [Google Scholar] [CrossRef]
- Akyüz, N.; Boehden, G.S.; Süsse, S.; Rimek, A.; Preuss, U.; Scheidtmann, K.-H.; Wiesmüller, L. DNA Substrate Dependence of P53-Mediated Regulation of Double-Strand Break Repair. Mol. Cell. Biol. 2002, 22, 6306–6317. [Google Scholar] [CrossRef] [PubMed]
- Keimling, M.; Wiesmüller, L. DNA Double-Strand Break Repair Activities in Mammary Epithelial Cells–Influence of Endogenous P53 Variants. Carcinogenesis 2009, 30, 1260–1268. [Google Scholar] [CrossRef]
- Arias-Lopez, C.; Lazaro-Trueba, I.; Kerr, P.; Lord, C.J.; Dexter, T.; Iravani, M.; Ashworth, A.; Silva, A. P53 Modulates Homologous Recombination by Transcriptional Regulation of the RAD51 Gene. EMBO Rep. 2006, 7, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.; Shalgi, R.; Liran, A.; Landan, G.; Korotayev, K.; Nguyen, G.H.; Enerly, E.; Johnsen, H.; Buganim, Y.; Solomon, H.; et al. P53-Repressed MiRNAs Are Involved with E2F in a Feed-Forward Loop Promoting Proliferation. Mol. Syst. Biol. 2008, 4, 1–15. [Google Scholar] [CrossRef]
- Wu, M.; Wang, X.; McGregor, N.; Pienta, K.J.; Zhang, J. Dynamic Regulation of Rad51 by E2F1 and P53 in Prostate Cancer Cells upon Drug-Induced DNA Damage under Hypoxia. Mol. Pharmacol. 2014, 85, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Itahana, Y.; Itahana, K. Emerging Roles of P53 Family Members in Glucose Metabolism. Int. J. Mol. Sci. 2018, 19, 776. [Google Scholar] [CrossRef]
- Gomes, A.S.; Ramos, H.; Soares, J.; Saraiva, L. P53 and Glucose Metabolism: An Orchestra To Be Directed in Cancer Therapy. Pharmacol. Res. 2018, 131, 75–86. [Google Scholar] [CrossRef]
- Lahalle, A.; Lacroix, M.; De Blasio, C.; Ciss, M.Y.; Linares, L.K.; Cam, L. Le The P53 Pathway and Metabolism: The Tree That Hides the Forest. Cancers 2021, 13, 133. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Brown, R.; Short, S.; Christopher, S. Colorectal Cancer and Metabolism. Curr. Colorectal Cancer Rep. 2018, 14, 226–241. [Google Scholar] [CrossRef]
- Neitzel, C.; Demuth, P.; Wittmann, S.; Fahrer, J. Targeting Altered Energy Metabolism in Colorectal Cancer: Oncogenic Reprogramming, the Central Role of the Tca Cycle and Therapeutic Opportunities. Cancers 2020, 12, 1731. [Google Scholar] [CrossRef]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C Selectively Kills KRAS and BRAF Mutant Colorectal Cancer Cells by Targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef]
- Ngo, B.; Van Riper, J.M.; Cantley, L.C.; Yun, J. Targeting Cancer Vulnerabilities with High-Dose Vitamin C. Nat. Rev. Cancer 2019, 19, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Nutthasirikul, N.V.; Hahnvajanawong, C.; Techasen, A.; Limpaiboon, T.; Wat, C.L.; Chau-In, S.; Jearanaikoon, P. Targeting the Δ133p53 Isoform Can Restore Chemosensitivity in 5-Fluorouracil-Resistant Cholangiocarcinoma Cells. Int. J. Oncol. 2015, 47, 2153–2164. [Google Scholar] [CrossRef][Green Version]
- Nemajerova, A.; Amelio, I.; Gebel, J.; Dötsch, V.; Melino, G.; Moll, U.M. Non-Oncogenic Roles of TAp73: From Multiciliogenesis to Metabolism. Cell Death Differ. 2018, 25, 144–153. [Google Scholar] [CrossRef]
- Yang, A.; Walker, N.; Bronson, R.; Kaghad, M.; Oosterwegel, M.; Bonnin, J.; Vagner, C.; Bonnet, H.; Dikkes, P.; Sharpe, A.; et al. P73-Deficient Mice Have Neurological, Pheromonal and Inflammatory Defects but Lack Spontaneous Tumours. Nature 2000, 404, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Sengupta, S.; Miller, J.B.; Newman, J.J.; Bronson, R.; Crowley, D.; Yang, A.; McKeon, F.; Jacks, T. Tumor Predisposition in Mice Mutant for P63 and P73: Evidence for Broader Tumor Suppressor Functions for the P53 Family. Cancer Cell 2005, 7, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Melino, G. Molecular Mechanisms and Function of the P53 Protein Family Member–P73. Biochemistry 2020, 85, 1202–1209. [Google Scholar] [CrossRef]
- Tomasini, R.; Tsuchihara, K.; Wilhelm, M.; Fujitani, M.; Rufini, A.; Cheung, C.C.; Khan, F.; Itie-Youten, A.; Wakeham, A.; Tsao, M.S.; et al. TAp73 Knockout Shows Genomic Instability with Infertility and Tumor Suppressor Functions. Genes Dev. 2008, 22, 2677–2691. [Google Scholar] [CrossRef]
- Talos, F.; Nemajerova, A.; Flores, E.R.; Petrenko, O.; Moll, U.M. P73 Suppresses Polyploidy and Aneuploidy in the Absence of Functional P53. Mol. Cell 2007, 27, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, E.M.; Moll, U.M. Role of P53 Family Members P73 and P63 in Human Hematological Malignancies. Leuk. Lymphoma 2012, 53, 2116–2129. [Google Scholar] [CrossRef] [PubMed]
- Nemajerova, A.; Petrenko, O.; Trümper, L.; Palacios, G.; Moll, U.M. Loss of P73 Promotes Dissemination of Myc-Induced B Cell Lymphomas in Mice. J. Clin. Investig. 2010, 120, 2070–2080. [Google Scholar] [CrossRef]
- Strano, S.; Monti, O.; Pediconi, N.; Baccarini, A.; Fontemaggi, G.; Lapi, E.; Mantovani, F.; Damalas, A.; Citro, G.; Sacchi, A.; et al. The Transcriptional Coactivator Yes-Associated Protein Drives P73 Gene-Target Specificity in Response to DNA Damage. Mol. Cell 2005, 18, 447–459. [Google Scholar] [CrossRef]
- Scian, M.J.; Carchman, E.H.; Mohanraj, L.; Stagliano, K.E.R.; Anderson, M.A.E.; Deb, D.; Crane, B.M.; Kiyono, T.; Windle, B.; Deb, S.P.; et al. Wild-Type P53 and P73 Negatively Regulate Expression of Proliferation Related Genes. Oncogene 2008, 27, 2583–2593. [Google Scholar] [CrossRef]
- Allocati, N.; Di Ilio, C.; De Laurenzi, V. P63/P73 in the Control of Cell Cycle and Cell Death. Exp. Cell Res. 2012, 318, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Lefkimmiatis, K.; Caratozzolo, M.F.; Merlo, P.; D’Erchia, A.M.; Navarro, B.; Levrero, M.; Sbisa’, E.; Tullo, A. P73 and P63 Sustain Cellular Growth by Transcriptional Activation of Cell Cycle Progression Genes. Cancer Res. 2009, 69, 8563–8571. [Google Scholar] [CrossRef]
- Lin, Y.L.; Sengupta, S.; Gurdziel, K.; Bell, G.W.; Jacks, T.; Flores, E.R. P63 and P73 Transcriptionally Regulate Genes Involved in DNA Repair. PLoS Genet. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Zaika, E.; Wei, J.; Yin, D.; Andl, C.; Moll, U.; El-Rifai, W.; Zaika, A.I. P73 Protein Regulates DNA Damage Repair. FASEB J. 2011, 25, 4406–4414. [Google Scholar] [CrossRef]
- Harms, K.; Nozell, S.; Chen, X. The Common and Distinct Target Genes of the P53 Family Transcription Factors. Cell. Mol. Life Sci. 2004, 61, 822–842. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.K.; Ha, J.H.; Lee, M.S.; Chi, S.W. Structure and Apoptotic Function of P73. BMB Rep. 2015, 48, 81–90. [Google Scholar] [CrossRef]
- Sayan, A.E.; Sayan, B.S.; Gogvadze, V.; Dinsdale, D.; Nyman, U.; Hansen, T.M.; Zhivotovsky, B.; Cohen, G.M.; Knight, R.A.; Melino, G. P73 and Caspase-Cleaved P73 Fragments Localize to Mitochondria and Augment TRAIL-Induced Apoptosis. Oncogene 2008, 27, 4363–4372. [Google Scholar] [CrossRef]
- Yoon, M.K.; Kim, B.Y.; Lee, J.Y.; Ha, J.H.; Kim, S.A.; Lee, D.H.; Lee, M.S.; Lee, M.K.; Choi, J.S.; Cho, J.H.; et al. Cytoplasmic Pro-Apoptotic Function of the Tumor Suppressor P73 Is Mediated through a Modified Mode of Recognition of the Anti-Apoptotic Regulator Bcl-XL. J. Biol. Chem. 2018, 293, 19546–19558. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.T.; Rufini, A.; Wetzel, M.K.; Tsuchihara, K.; Inoue, S.; Tomasini, R.; Itie-Youten, A.; Wakeham, A.; Arsenian-Henriksson, M.; Melino, G.; et al. Isoform-Specific P73 Knockout Mice Reveal a Novel Role for ΔNp73 in the DNA Damage Response Pathway. Genes Dev. 2010, 24, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Stiewe, T.; Stanelle, J.; Theseling, C.C.; Pollmeier, B.; Beitzinger, M.; Pützer, B.M. Inactivation of Retinoblastoma (RB) Tumor Suppressor by Oncogenic Isoforms of the P53 Family Member P73. J. Biol. Chem. 2003, 278, 14230–14236. [Google Scholar] [CrossRef] [PubMed]
- Petrenko, O.; Zaika, A.; Moll, U.M. ΔNp73 Facilitates Cell Immortalization and Cooperates with Oncogenic Ras in Cellular Transformation In Vivo. Mol. Cell. Biol. 2003, 23, 5540–5555. [Google Scholar] [CrossRef][Green Version]
- Vilgelm, A.; Wei, J.X.; Piazuelo, M.B.; Washington, M.K.; Prassolov, V.; El-Rifai, W.; Zaika, A. ΔNp73α Regulates MDR1 Expression by Inhibiting P53 Function. Oncogene 2008, 27, 2170–2176. [Google Scholar] [CrossRef]
- Sakil, H.A.M.; Stantic, M.; Wolfsberger, J.; Brage, S.E.; Hansson, J.; Wilhelm, M.T. ΔNp73 Regulates the Expression of the Multidrug-Resistance Genes ABCB1 and ABCB5 in Breast Cancer and Melanoma Cells—A Short Report. Cell. Oncol. 2017, 40, 631–638. [Google Scholar] [CrossRef]
- Sabapathy, K. P73: A Positive or Negative Regulator of Angiogenesis, or Both? Mol. Cell. Biol. 2016, 36, 848–854. [Google Scholar] [CrossRef]
- Dulloo, I.; Hooi, P.B.; Sabapathy, K. Hypoxia-Induced DNp73 Stabilization Regulates Vegf-A Expression and Tumor Angiogenesis Similar to TAp73. Cell Cycle 2015, 14, 3533–3539. [Google Scholar] [CrossRef] [PubMed]
- Dulloo, I.; Phang, B.H.; Othman, R.; Tan, S.Y.; Vijayaraghavan, A.; Goh, L.K.; Martin-Lopez, M.; Marques, M.M.; Li, C.W.; Wang, D.Y.; et al. Hypoxia-Inducible TAp73 Supports Tumorigenesis by Regulating the Angiogenic Transcriptome. Nat. Cell Biol. 2015, 17, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Stantic, M.; Sakil, H.A.M.; Zirath, H.; Fang, T.; Sanz, G.; Fernandez-Woodbridge, A.; Marin, A.; Susanto, E.; Mak, T.W.; Henriksson, M.A.; et al. TAp73 Suppresses Tumor Angiogenesis through Repression of Proangiogenic Cytokines and HIF-1α Activity. Proc. Natl. Acad. Sci. USA 2015, 112, 220–225. [Google Scholar] [CrossRef]
- Amelio, I.; Inoue, S.; Markert, E.K.; Levine, A.J.; Knight, R.A.; Mak, T.W.; Melino, G. TAp73 Opposes Tumor Angiogenesis by Promoting Hypoxia-Inducible Factor 1α Degradation. Proc. Natl. Acad. Sci. USA 2015, 112, 226–231. [Google Scholar] [CrossRef]
- Kovalev, S.; Marchenko, N.; Swendeman, S.; LaQuaglia, M.; Moll, U.M. Expression Level, Allelic Origin, and Mutation Analysis of the P73 Gene in Neuroblastoma Tumors and Cell Lines. Cell Growth Differ. 1998, 9, 897–903. [Google Scholar]
- Rufini, A.; Niklison-Chirou, M.V.; Inoue, S.; Tomasini, R.; Harris, I.S.; Marino, A.; Federici, M.; Dinsdale, D.; Knight, R.A.; Melino, G.; et al. TAp73 Depletion Accelerates Aging through Metabolic Dysregulation. Genes Dev. 2012, 26, 2009–2014. [Google Scholar] [CrossRef]
- Marini, A.; Rotblat, B.; Sbarrato, T.; Niklison-Chirou, M.V.; Knight, J.R.P.; Dudek, K.; Jones, C.; Bushell, M.; Knight, R.A.; Amelio, I.; et al. TAp73 Contributes to the Oxidative Stress Response by Regulating Protein Synthesis. Proc. Natl. Acad. Sci. USA 2018, 115, 6219–6224. [Google Scholar] [CrossRef]
- Agostini, M.; Niklison-Chirou, M.V.; Catani, M.V.; Knight, R.A.; Melino, G.; Rufini, A. TAp73 Promotes Anti-Senescence-Anabolism Not Proliferation. Aging 2014, 6, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Antonov, A.A.; Catani, M.V.; Massoud, R.; Bernassola, F.; Knight, R.A.; Melino, G.; Rufini, A. TAp73 Promotes Anabolism. Oncotarget 2014, 5, 12820–12834. [Google Scholar] [CrossRef] [PubMed]
- Iscan, E.; Ekin, U.; Yildiz, G.; Oz, O.; Keles, U.; Suner, A.; Cakan-Akdogan, G.; Ozhan, G.; Nekulova, M.; Vojtesek, B.; et al. TAp73β Can Promote Hepatocellular Carcinoma Dedifferentiation. Cancers 2021, 13, 783. [Google Scholar] [CrossRef]
- Amelio, I.; Markert, E.K.; Rufini, A.; Antonov, A.V.; Sayan, B.S.; Tucci, P.; Agostini, M.; Mineo, T.C.; Levine, A.J.; Melino, G. P73 Regulates Serine Biosynthesis in Cancer. Oncogene 2014, 33, 5039–5046. [Google Scholar] [CrossRef] [PubMed]
- Velletri, T.; Romeo, F.; Tucci, P.; Peschiaroli, A.; Annicchiarico-Petruzzelli, M.; Niklison-Chirou, M.V.; Amelio, I.; Knight, R.A.; Mak, T.W.; Melino, G.; et al. GLS2 Is Transcriptionally Regulated by P73 and Contributes to Neuronal Differentiation. Cell Cycle 2013, 12, 3564–3573. [Google Scholar] [CrossRef] [PubMed]
- Niklison-Chirou, M.V.; Erngren, I.; Engskog, M.; Haglöf, J.; Picard, D.; Remke, M.; McPolin, P.H.R.; Selby, M.; Williamson, D.; Clifford, S.C.; et al. TAp73 Is a Marker of Glutamine Addiction in Medulloblastoma. Genes Dev. 2017, 31, 1738–1753. [Google Scholar] [CrossRef] [PubMed]
- Venkatanarayan, A.; Raulji, P.; Norton, W.; Chakravarti, D.; Coarfa, C.; Su, X.; Sandur, S.K.; Ramirez, M.S.; Lee, J.; Kingsley, C.V.; et al. IAPP-Driven Metabolic Reprogramming Induces Regression of P53-Deficient Tumours in Vivo. Nature 2015, 517, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Niklison-Chirou, M.V.; Annicchiarico-Petruzzelli, M.M.; Grelli, S.; Di Daniele, N.; Pestlikis, I.; Knight, R.A.; Melino, G.; Rufini, A. P73 Regulates Primary Cortical Neuron Metabolism: A Global Metabolic Profile. Mol. Neurobiol. 2018, 55, 3237–3250. [Google Scholar] [CrossRef]
- Fedorova, N.E.; Chernoryzh, Y.Y.; Vinogradskaya, G.R.; Emelianova, S.S.; Zavalyshina, L.E.; Yurlov, K.I.; Zakirova, N.F.; Verbenko, V.N.; Kochetkov, S.N.; Kushch, A.A.; et al. Inhibitor of Polyamine Catabolism MDL72.527 Restores the Sensitivity to Doxorubicin of Monocytic Leukemia Thp-1 Cells Infected with Human Cytomegalovirus. Biochimie 2019, 158, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Nemajerova, A.; Moll, U.M. Tissue-Specific Roles of P73 in Development and Homeostasis. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Fujitani, M.; Cancino, G.I.; Dugani, C.B.; Weaver, I.C.G.; Gauthier-Fisher, A.; Paquin, A.; Mak, T.W.; Wojtowicz, M.J.; Miller, F.D.; Kaplan, D.R. TAp73 Acts via the BHLH Hey2 to Promote Long-Term Maintenance of Neural Precursors. Curr. Biol. 2010, 20, 2058–2065. [Google Scholar] [CrossRef]
- Agostini, M.; Tucci, P.; Killick, R.; Candi, E.; Sayan, B.S.; Di Val Cervo, P.R.; Nicoterad, P.; McKeon, F.; Knight, R.A.; Mak, T.W.; et al. Neuronal Differentiation by TAp73 Is Mediated by MicroRNA-34a Regulation of Synaptic Protein Targets. Proc. Natl. Acad. Sci. USA 2011, 108, 21093–21098. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Tucci, P.; Steinert, J.R.; Shalom-Feuerstein, R.; Rouleau, M.; Aberdam, D.; Forsythe, I.D.; Young, K.W.; Ventura, A.; Concepcion, C.P.; et al. MicroRNA-34a Regulates Neurite Outgrowth, Spinal Morphology, and Function. Proc. Natl. Acad. Sci. USA 2011, 108, 21099–21104. [Google Scholar] [CrossRef]
- Tissir, F.; Ravni, A.; Achouri, Y.; Riethmacher, D.; Meyer, G.; Goffinet, A.M. DeltaNp73 Regulates Neuronal Survival in Vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 16871–16876. [Google Scholar] [CrossRef]
- Pozniak, C.D.; Radinovic, S.; Yang, A.; McKeon, F.; Kaplan, D.R.; Miller, F.D. An Anti-Apoptotic Role for the P53 Family Member, P73, during Developmental Neuron Death. Science 2000, 289, 304–306. [Google Scholar] [CrossRef]
- Amelio, I.; Panatta, E.; Niklison-Chirou, M.V.; Steinert, J.R.; Agostini, M.; Morone, N.; Knight, R.A.; Melino, G. The c Terminus of P73 Is Essential for Hippocampal Development. Proc. Natl. Acad. Sci. USA 2020, 117, 15694–15701. [Google Scholar] [CrossRef]
- Fujitani, M.; Sato, R.; Yamashita, T. Loss of P73 in Ependymal Cells during the Perinatal Period Leads to Aqueductal Stenosis. Sci. Rep. 2017, 7, 12007. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Alonso, R.; Martin-Lopez, M.; Gonzalez-Cano, L.; Garcia, S.; Castrillo, F.; Diez-Prieto, I.; Fernandez-Corona, A.; Lorenzo-Marcos, M.E.; Li, X.; Claesson-Welsh, L.; et al. P73 Is Required for Endothelial Cell Differentiation, Migration and the Formation of Vascular Networks Regulating VEGF and TGFβ Signaling. Cell Death Differ. 2015, 22, 1287–1299. [Google Scholar] [CrossRef]
- Marshall, C.B.; Mays, D.J.; Beeler, J.S.; Rosenbluth, J.M.; Boyd, K.L.; Santos Guasch, G.L.; Shaver, T.M.; Tang, L.J.; Liu, Q.; Shyr, Y.; et al. P73 Is Required for Multiciliogenesis and Regulates the Foxj1-Associated Gene Network. Cell Rep. 2016, 14, 2289–2300. [Google Scholar] [CrossRef] [PubMed]
- Nemajerova, A.; Kramer, D.; Siller, S.S.; Herr, C.; Shomroni, O.; Pena, T.; Suazo, C.G.; Glaser, K.; Wildung, M.; Steffen, H.; et al. TAp73 Is a Central Transcriptional Regulator of Airway Multiciliogenesis. Genes Dev. 2016, 30, 1300–1312. [Google Scholar] [CrossRef]
- McLure, K.G.; Lee, P.W.K. How P53 Binds DNA as a Tetramer. EMBO J. 1998, 17. [Google Scholar] [CrossRef]
- Fillippovich, I.; Sorokina, N.; Gatei, M.; Haupt, Y.; Hobson, K.; Moallem, E.; Spring, K.; Mould, M.; Mcguckin, M.A.; Lavin, M.F.; et al. Transactivation-Deficient P73α (P73Δexon2) Inhibits Apoptosis and Competes with P53. Oncogene 2001, 20, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Munarriz, E.; Rossi, M.; Cristofanelli, B.; Shaul, Y.; Castagnoli, L.; Levine, A.J.; Sacchi, A.; Cesareni, G.; Oren, M.; et al. Physical and Functional Interaction between P53 Mutants and Different Isoforms of P73. J. Biol. Chem. 2000, 275, 29503–29512. [Google Scholar] [CrossRef] [PubMed]
- Di Como, C.J.; Gaiddon, C.; Prives, C. P73 Function Is Inhibited by Tumor-Derived P53 Mutants in Mammalian Cells. Mol. Cell. Biol. 1999, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Grob, T.J.; Novak, U.; Maisse, C.; Barcaroli, D.; Lüthi, A.U.; Pirnia, F.; Hügli, B.; Graber, H.U.; De Laurenzi, V.; Fey, M.F.; et al. Human ΔNp73 Regulates a Dominant Negative Feedback Loop for TAp73 and P53. Cell Death Differ. 2001, 8, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Rajagopalan, S.; Natan, E.; Veprintsev, D.B.; Robinson, C.V.; Fersht, A.R. Structural Evolution of P53, P63, and P73: Implication for Heterotetramer Formation. Proc. Natl. Acad. Sci. USA 2009, 106, 17705–17710. [Google Scholar] [CrossRef] [PubMed]
- Rufini, A.; Agostini, M.; Grespi, F.; Tomasini, R.; Sayan, B.S.; Niklison-Chirou, M.V.; Conforti, F.; Velletri, T.; Mastino, A.; Mak, T.W.; et al. P73 in Cancer. Genes Cancer 2011, 2, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Slade, N.; Zaika, A.I.; Erster, S.; Moll, U.M. ΔNp73 Stabilises TAp73 Proteins but Compromises Their Function Due to Inhibitory Hetero-Oligomer Formation. Cell Death Differ. 2004, 11, 357–360. [Google Scholar] [CrossRef]
- Nakagawa, T.; Takahashi, M.; Ozaki, T.; Watanabe, K.; Todo, S.; Mizuguchi, H.; Hayakawa, T.; Nakagawara, A. Autoinhibitory Regulation of P73 by ΔNp73 To Modulate Cell Survival and Death through a P73-Specific Target Element within the ΔNp73 Promoter. Mol. Cell. Biol. 2002, 22, 2575–2585. [Google Scholar] [CrossRef] [PubMed]
- Kartasheva, N.N.; Contente, A.; Lenz-Sto, C.; Roth, J.; Dobbelstein, M. P53 Induces the Expression of Its Antagonist P73DN, Establishing an Autoregulatory Feedback Loop. Oncogene 2002, 21, 4715–4727. [Google Scholar] [CrossRef] [PubMed]
- Melino, G.; De Laurenzi, V.; Vousden, K.H. p73: Friend or foe in tumorigenesis. Nat. Rev. Cancer 2002, 2, 605–615. [Google Scholar] [CrossRef]
- Hafsi, H.; Santos-Silva, D.; Courtois-Cox, S.; Hainaut, P. Effects of Δ40p53, an Isoform of P53 Lacking the N-Terminus, on Transactivation Capacity of the Tumor Suppressor Protein P53. BMC Cancer 2013, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Billant, O.; Léon, A.; Le Guellec, S.; Friocourt, G.; Blondel, M.; Voisset, C. The Dominant-Negative Interplay between P53, P63 and P73: A Family Affair. Oncotarget 2016, 7, 69549–69564. [Google Scholar] [CrossRef]
- Flores, E.R.; Tsai, K.Y.; Crowley, D.; Sengupta, S.; Yang, A.; McKeon, F.; Jacks, T. P63 and P73 Are Required for P53-Dependent Apoptosis in Response to DNA Damage. Nature 2002, 416, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.S.; Kondo, K.; Marin, M.C.; Cheng, L.S.; Hahn, W.C.; Kaelin, W.G. Chemosensitivity Linked to P73 Function. Cancer Cell 2003, 3, 403–410. [Google Scholar] [CrossRef]
- Silva, J.L.; Gallo, C.V.D.M.; Costa, D.C.F.; Rangel, L.P. Prion-like Aggregation of Mutant P53 in Cancer. Trends Biochem. Sci. 2014, 39, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.C.; et al. Gain of Function of Mutant P53 by Coaggregation with Multiple Tumor Suppressors. Nat. Chem. Biol. 2011, 7. [Google Scholar] [CrossRef]
- Maritschnegg, E.; Heinzl, N.; Wilson, S.; Deycmar, S.; Niebuhr, M.; Klameth, L.; Holzer, B.; Koziel, K.; Concin, N.; Zeillinger, R. Polymer-Ligand-Based ELISA for Robust, High-Throughput, Quantitative Detection of P53 Aggregates. Anal. Chem. 2018, 90, 13273–13279. [Google Scholar] [CrossRef]
- Rodríguez, N.; Peláez, A.; Barderas, R.; Domínguez, G. Clinical Implications of the Deregulated TP73 Isoforms Expression in Cancer. Clin. Transl. Oncol. 2018, 20, 827–836. [Google Scholar] [CrossRef]
- Hofstetter, G.; Berger, A.; Fiegl, H.; Slade, N.; Zori, A.; Holzer, B.; Schuster, E.; Mobus, V.J.; Reimer, D.; Daxenbichler, G.; et al. Alternative Splicing of P53 and P73: The Novel P53 Splice Variant P53 Is an Independent Prognostic Marker in Ovarian Cancer. Oncogene 2010, 29, 1997–2004. [Google Scholar] [CrossRef]
- Concin, N.; Becker, K.; Slade, N.; Erster, S.; Mueller-Holzner, E.; Ulmer, H.; Daxenbichler, G.; Zeimet, A.; Zeillinger, R.; Marth, C.; et al. Transdominant ΔTAp73 Isoforms Are Frequently Up-Regulated in Ovarian Cancer. Evidence for Their Role as Epigenetic P53 Inhibitors in Vivo. Cancer Res. 2004, 64, 2449–2460. [Google Scholar] [CrossRef]
- Knezović Florijan, M.; Ozretić, P.; Bujak, M.; Pezzè, L.; Ciribilli, Y.; Kaštelan, Ž.; Slade, N.; Hudolin, T. The Role of P53 Isoforms’ Expression and P53 Mutation Status in Renal Cell Cancer Prognosis. Urol. Oncol. Semin. Orig. Investig. 2019, 37, 578.e1–578.e10. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Deng, Y.; Wang, K.; Zhou, H.; Zheng, X.; Si, L.; Fu, Z. Profiles of Alternative Splicing in Colorectal Cancer and Their Clinical Significance: A Study Based on Large-Scale Sequencing Data. EBioMedicine 2018, 36, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Garranzo-Asensio, M.; Guzmán-Aránguez, A.; Povés, C.; Fernández-Aceñero, M.J.; Montero-Calle, A.; Ceron, M.Á.; Fernandez-Diez, S.; Rodríguez, N.; Gómez de Cedrón, M.; Ramírez de Molina, A.; et al. The Specific Seroreactivity to ∆Np73 Isoforms Shows Higher Diagnostic Ability in Colorectal Cancer Patients than the Canonical P73 Protein. Sci. Rep. 2019, 9, 13547. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.D.; Fan, C.; Zhang, H.; Sun, X. Artificial Intelligence–Based 5-year Survival Prediction and Prognosis of DNp73 Expression in Rectal Cancer Patients. Clin. Transl. Med. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Soldevilla, B.; Rodriguez, M.; Millán, C.S.; Garcia, V.; Fernández-Periañez, R.; Gil-Calderón, B.; Martin, P.; Garcia-Grande, A.; Silva, J.; Bonilla, F.; et al. Tumor-Derived Exosomes Are Enriched in ΔNp73, Which Promotes Oncogenic Potential in Acceptor Cells and Correlates with Patient Survival. Hum. Mol. Genet. 2014, 23, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Soldevilla, B.; Díaz, R.; Silva, J.; Campos-Martín, Y.; Muñoz, C.; García, V.; García, J.M.; Peña, C.; Herrera, M.; Rodriguez, M.; et al. Prognostic Impact of ΔTAp73 Isoform Levels and Their Target Genes in Colon Cancer Patients. Clin. Cancer Res. 2011, 17, 6029–6039. [Google Scholar] [CrossRef]
- Dominguez, G.; Garcia, J.M.; Peña, C.; Silva, J.; Garcia, V.; Martinez, L.; Maximiano, C.; Gómez, M.E.; Rivera, J.A.; Garcia-Andrade, C.; et al. ΔTAp73 Upregulation Correlates with Poor Prognosis in Human Tumors: Putative in Vivo Network Involving P73 Isoforms, P53, and E2F-1. J. Clin. Oncol. 2006, 24, 805–815. [Google Scholar] [CrossRef]
- Pfeifer, D.; Wallin, Å.; Holmlund, B.; Sun, X.F. Protein Expression Following γ-Irradiation Relevant to Growth Arrest and Apoptosis in Colon Cancer Cells. J. Cancer Res. Clin. Oncol. 2009, 135, 1583–1592. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting P53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Brady, C.A.; Attardi, L.D. P53 at a Glance. J. Cell Sci. 2010, 123, 2527–2532. [Google Scholar] [CrossRef]
- Güllülü, Ö.; Hehlgans, S.; Rödel, C.; Fokas, E.; Rödel, F. Tumor Suppressor Protein P53 and Inhibitor of Apoptosis Proteins in Colorectal Cancer-a Promising Signaling Network for Therapeutic Interventions. Cancers 2021, 13, 624. [Google Scholar] [CrossRef] [PubMed]
- Diaz, R.; González-Sancho, J.M.; Soldevilla, B.; Silva, J.; García, J.M.; García, V.; Peña, C.; Herrera, M.; Gómez, I.; Bonilla, F.; et al. Differential Regulation of TP73 Isoforms by 1α,25-Dihydroxyvitamin D3 and Survivin in Human Colon and Breast Carcinomas. Genes Chromosomes Cancer 2010, 49, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Dabiri, Y.; Kalman, S.; Gürth, C.M.; Kim, J.Y.; Mayer, V.; Cheng, X. The Essential Role of TAp73 in Bortezomib-Induced Apoptosis in P53-Deficient Colorectal Cancer Cells. Sci. Rep. 2017, 7, 5423. [Google Scholar] [CrossRef]
- Prabhu, V.V.; Hong, B.; Allen, J.E.; Zhang, S.; Lulla, A.R.; Dicker, D.T.; El-Deiry, W.S. Small-Molecule Prodigiosin Restores P53 Tumor Suppressor Activity in Chemoresistant Colorectal Cancer Stem Cells via c-Jun-Mediated Δnp73 Inhibition and P73 Activation. Cancer Res. 2016, 76, 1989–1999. [Google Scholar] [CrossRef]
- Marabese, M.; Marchini, S.; Sabatino, M.A.; Polato, F.; Vikhanskaya, F.; Marrazzo, E.; Riccardi, E.; Scanziani, E.; Broggini, M. Effects of Inducible Overexpression of DNp73α on Cancer Cell Growth and Response to Treatment in Vitro and in Vivo. Cell Death Differ. 2005, 12, 805–814. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sabatino, M.A.; Previdi, S.; Broggini, M. In Vivo Evaluation of the Role of DNp73α Protein in Regulating the P53-Dependent Apoptotic Pathway after Treatment with Cytotoxic Drugs. Int. J. Cancer 2007, 120, 506–513. [Google Scholar] [CrossRef]
- Marrazzo, E.; Marchini, S.; Previdi, S.; Broggini, M. Questioning the Oncogenic Role of ΔNp73α in Different Cell Lines Expressing P53 or Not. Cancer Biol. Ther. 2006, 5, 794–803. [Google Scholar] [CrossRef][Green Version]
- Lööf, J.; Pfeifer, D.; Ding, Z.; Sun, X.F.; Zhang, H. Effects of ΔNp73β on Cisplatin Treatment in Colon Cancer Cells. Mol. Carcinog. 2012, 51, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Díaz, R.; Peña, C.; Silva, J.; Lorenzo, Y.; García, V.; García, J.M.; Sánchez, A.; Espinosa, P.; Yuste, R.; Bonilla, F.; et al. P73 Isoforms Affect VEGF, VEGF165b and PEDF Expression in Human Colorectal Tumors: VEGF165b Downregulation as a Marker of Poor Prognosis. Int. J. Cancer 2008, 123, 1060–1067. [Google Scholar] [CrossRef]
- Du, W.; Jiang, P.; Mancuso, A.; Stonestrom, A.; Brewer, M.D.; Minn, A.J.; Mak, T.W.; Wu, M.; Yang, X. TAp73 Enhances the Pentose Phosphate Pathway and Supports Cell Proliferation. Nat. Cell Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Yang, X. A Critical Role of Glucose-6-Phosphate Dehydrogenase in TAp73-Mediated Cell Proliferation. Cell Cycle 2013. [Google Scholar] [CrossRef] [PubMed]
- D’Aguanno, S.; Barcaroli, D.; Rossi, C.; Zucchelli, M.; Ciavardelli, D.; Cortese, C.; De Cola, A.; Volpe, S.; D’Agostino, D.; Todaro, M.; et al. P63 Isoforms Regulate Metabolism of Cancer Stem Cells. J. Proteome Res. 2014, 13, 2120–2136. [Google Scholar] [CrossRef] [PubMed]
- Bahnassy, A.A.; Zekri, A.R.N.; Salem, S.E.; Abou-Bakr, A.A.; Sakr, M.A.; Abdel-Samiaa, A.G.; Al-Bradei, M. Differential Expression of P53 Family Proteins in Colorectal Adenomas and Carcinomas: Prognostic and Predictive Values. Histol. Histopathol. 2014, 29, 207–216. [Google Scholar] [CrossRef]
- Pham, T.; Fan, C.; Pfeifer, D.; Zhang, H.; Sun, X.F. Image-Based Network Analysis of DNp73 Expression by Immunohistochemistry in Rectal Cancer Patients. Front. Physiol. 2020, 10, 1551. [Google Scholar] [CrossRef] [PubMed]
- Butt, J.; Blot, W.J.; Visvanathan, K.; Le Marchand, L.; Wilkens, L.R.; Chen, Y.; Sesso, H.D.; Teras, L.; Ryser, M.D.; Hyslop, T.; et al. Auto-Antibodies to P53 and the Subsequent Development of Colorectal Cancer in a U.S. Prospective Cohort Consortium. Cancer Epidemiol. Prev. Biomark. 2020, 29, 2729–2734. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T. P53 Antibodies in the Sera of Patients with Various Types of Cancer: A Review. Cancer Res. 2000, 60, 1777–1788. [Google Scholar] [PubMed]
- Zhang, S.; Zhou, L.; Hong, B.; Van Den Heuvel, A.P.J.; Prabhu, V.V.; Warfel, N.A.; Kline, C.L.B.; Dicker, D.T.; Kopelovich, L.; El-Deiry, W.S. Small-Molecule NSC59984 Restores P53 Pathway Signaling and Antitumor Effects against Colorectal Cancer via P73 Activation and Degradation of Mutant P53. Cancer Res. 2015, 75, 3842–3852. [Google Scholar] [CrossRef] [PubMed]
- Millán, C.S.; Soldevilla, B.; Martín, P.; Gil-Calderón, B.; Compte, M.; Pérez-Sacristán, B.; Donoso, E.; Peña, C.; Romero, J.; Granado-Lorencio, F.; et al. β-Cryptoxanthin Synergistically Enhances the Antitumoral Activity of Oxaliplatin through ΔNP73 Negative Regulation in Colon Cancer. Clin. Cancer Res. 2015, 21, 4398–4409. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.M.S.; Nugent, J.K.; Zhao, X.; Irwin, M.S. HDM2 Antagonist Nutlin-3 Disrupts P73-HDM2 Binding and Enhances P73 Function. Oncogene 2008, 27, 997–1003. [Google Scholar] [CrossRef]





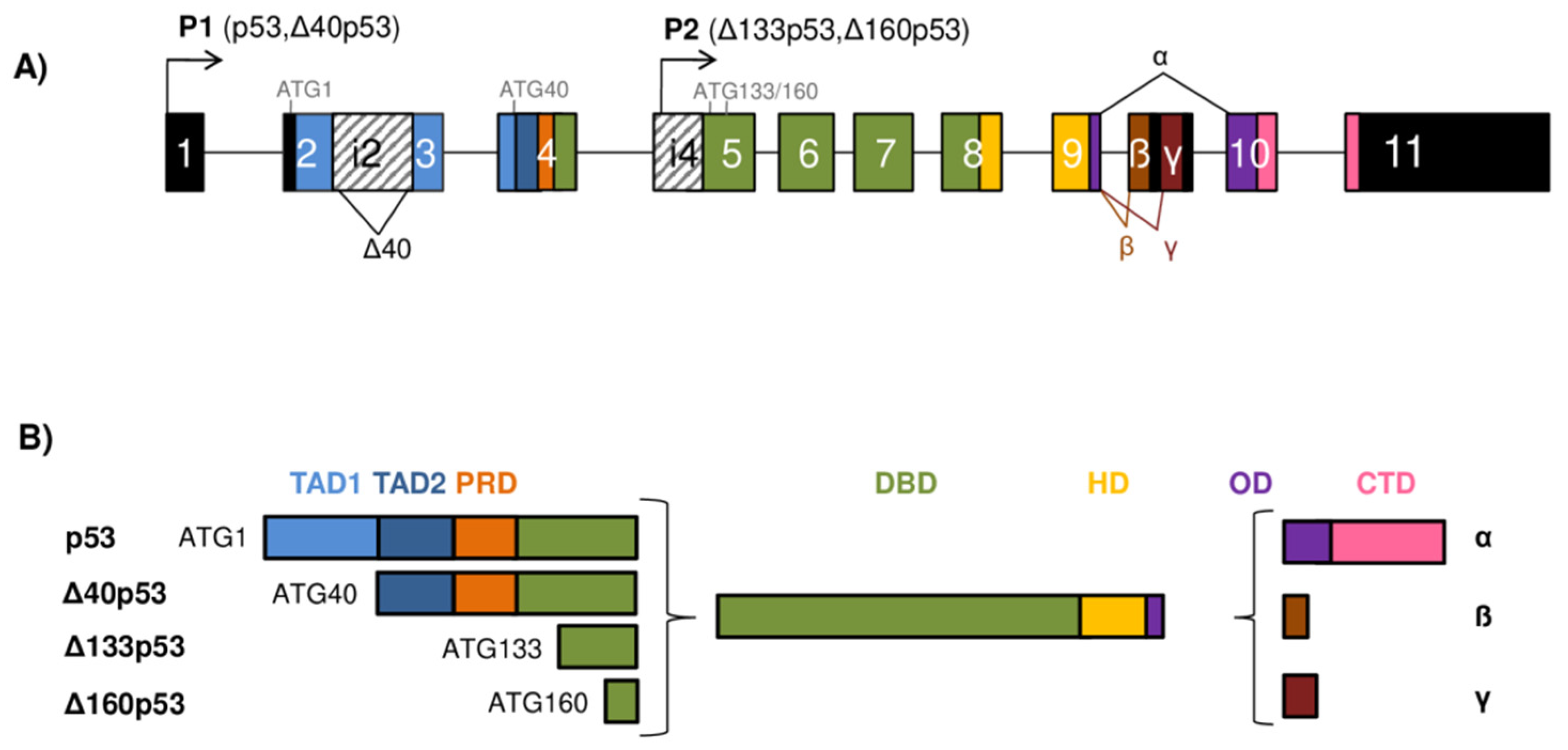{kind=link}
{kind=link}
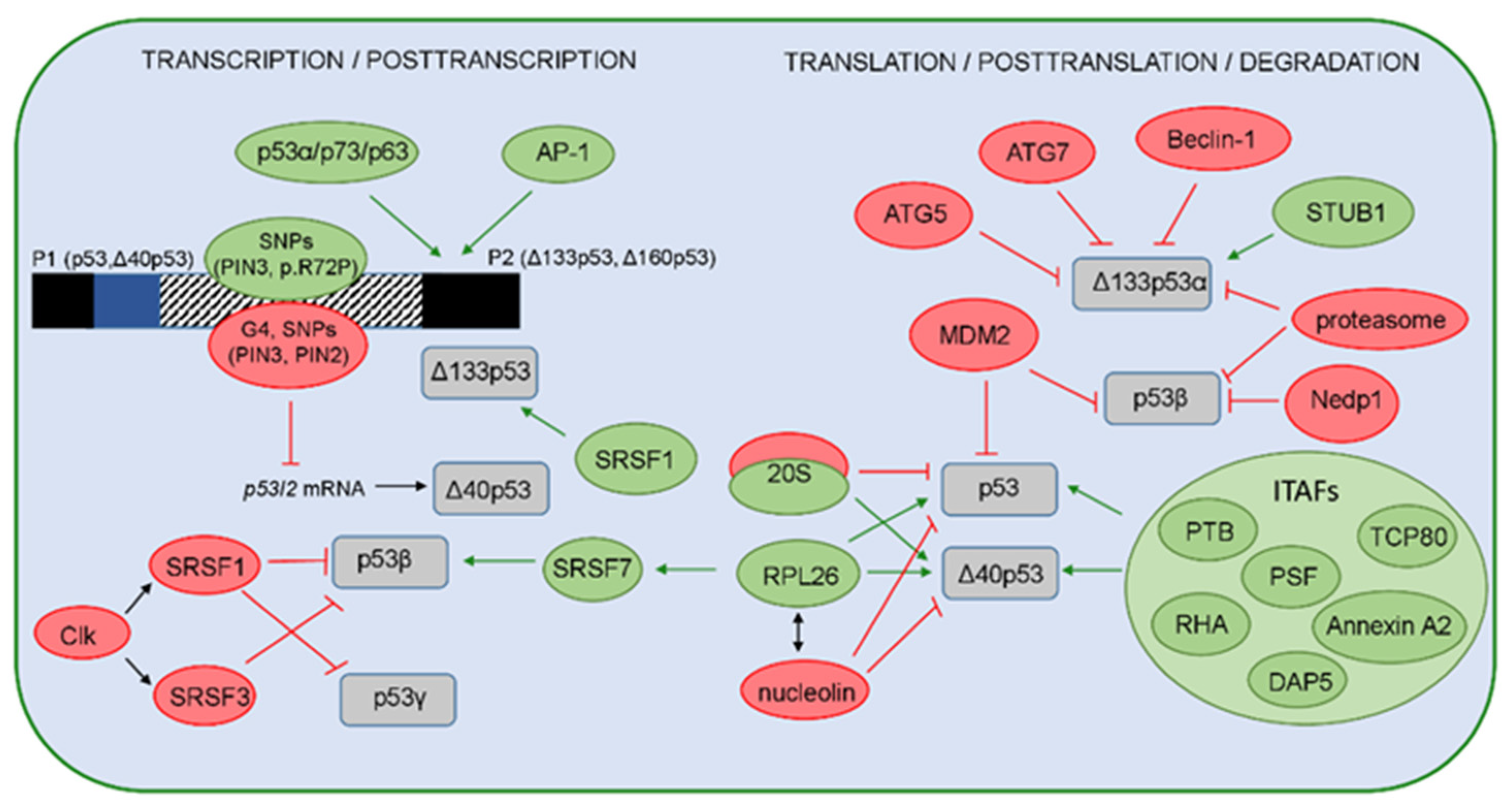{kind=link}
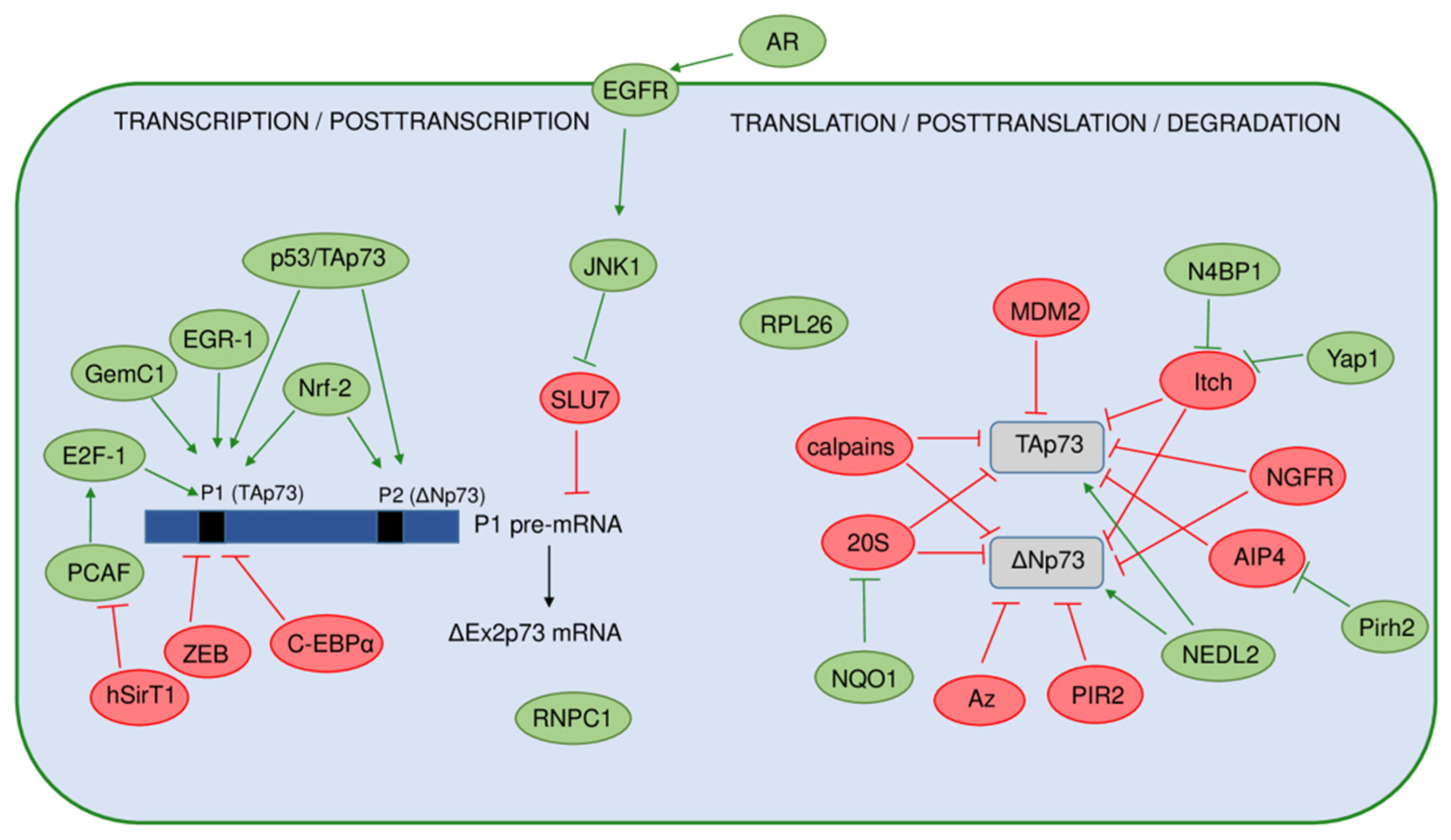{kind=link}
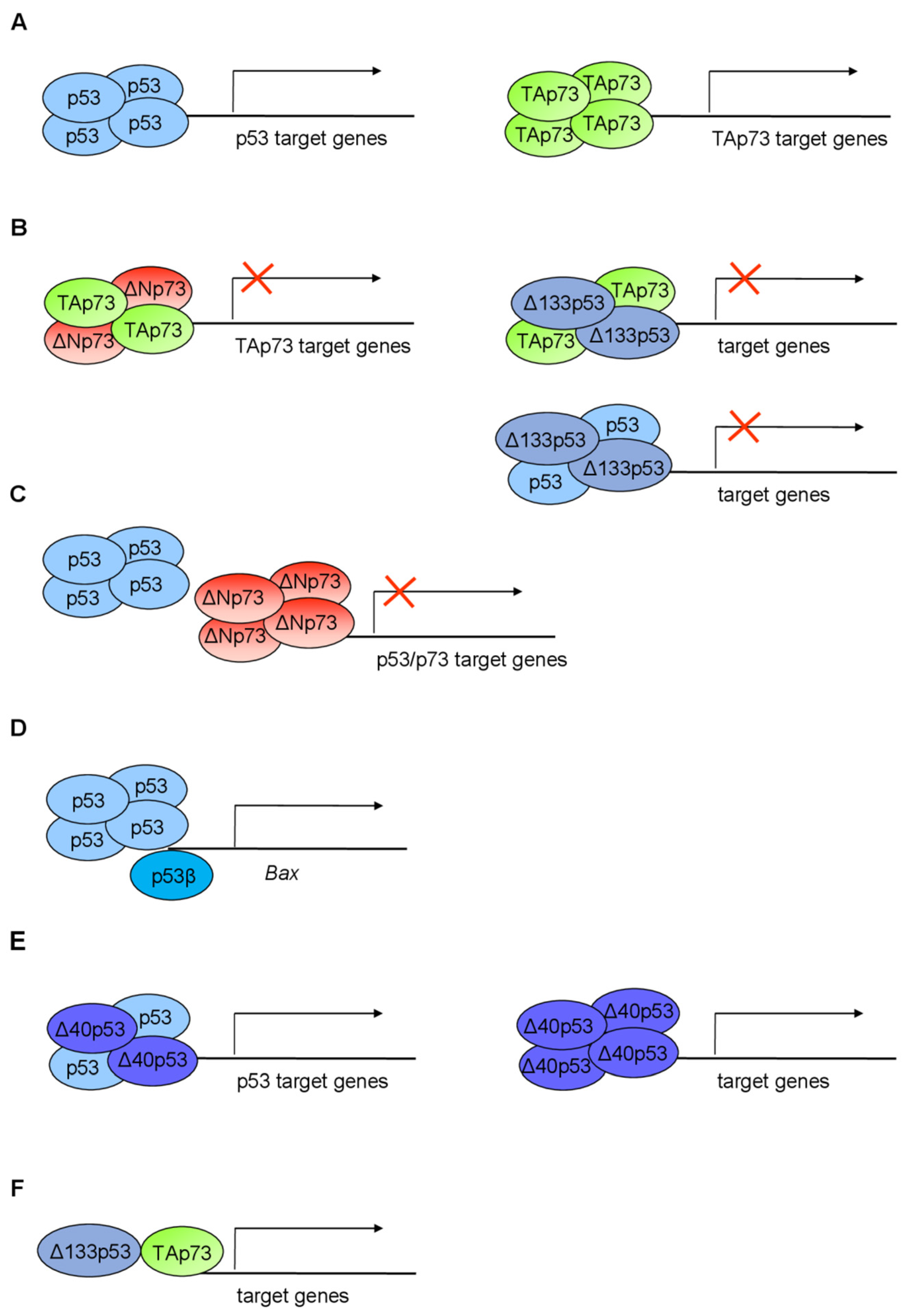{kind=link}
{kind=link}
| Cellular Function | Isoform | Model | Signalling | Effect | Reference |
|---|---|---|---|---|---|
| Apoptosis and sensitivity to antitumor drugs | ∆40p53 | HCT116−/− | ↑ miR-186 → ↓ YY1 | ↓ proliferation | [187] |
| ∆133p53ß | HCT116, SW480, LoVo, SW620 and Colo205 | ↓ RhoB | ↑ resistance to camptothecin-induced apoptosis | [175] | |
| TAp73, ∆Np73, ∆Ex2p73, ∆Ex2/3p73 | SW480-ADH and HCT116 | ↑ survivin → ↑ p73 isoforms | - | [299] | |
| TAp73 | HCT116 p53−/− and p53-mt HT-29 | Casp3 and PARP cleavage | ↑ bortezomib-induced apoptosis | [300] | |
| ΔNp73 | HCT116 | - | ↑ proliferation and resistance to oxaliplatin-induced apoptosis | [293] | |
| ΔNp73 | DLD1 and HCT116 | - | ↑ colonosphere formation and resistance to prodigiosin | [301] | |
| ΔNp73α | HCT116 and HCT116 xenograft | - | = cellular and tumor growth = sensitivity to cDDP or DX in vitro and in vivo | [302,303] | |
| ΔNp73α | HCT116 p53−/− | - | = cellular growth = sensitivity to cDDP, DX and UV light | [304] | |
| ΔNp73β | HCT116 and HCT116 p53−/− | - | ↑ cellular viability = sensitivity to cDDP | [305] | |
| Exosomal ΔNp73β | HCT116 and HCT116 xenografts | ↑ ΔNp73β mRNA in recipient cells | ↑ proliferation and resistance to oxaliplatin in vitro ↑ proliferation and tumor size | [292] | |
| Autophagy | Δ40p53 | HCT116 | ↓ PKR/ Eif2 and DRAM | ↓ starvation and methyl methane sulfonate-induced autophagy | [186] |
| DNA repair | Δ133p53 and TAp73α | HCT116 | ↑ RAD51, LIG4 and RAD52 | DNA double-strand break repair upon γ-irradiation | [180] |
| Inflammation | Δ133p53 | HCT116 | ↑ IL-6, IL-8, Bcl-2 | - | [93] |
| Invasion | ∆133p53 and Δ133p53β | HCT116, LoVo, SW480, SW620, Colo205 | ↓ E-cadherin and β1-integrin | ↑ invasion ↑ rounded phenotype ↓ adhesion ↑ epithelial–amoeboid transition | [192] |
| Δ133p53α, Δ133p53β and Δ133p53γ | HCT116 | ↑ RhoA and ROCKA | ↑ invasion ↑ rounded phenotype | [188] | |
| Angiogenesis | TAp73, ΔNp73 | CRC patient samples | Correlations with VEGF and VEGF165b | - | [306] |
| ∆Ex2/3p73 | Correlation with VEGF165b | ||||
| ∆Ex2p73 | Correlations with VEGF and VEGF165b Inverse correlation with PEDF | ||||
| Metabolism | p53 and Δ40p53 | HCT116 and A549 | Glucose starvation → ↑SMAR1 ↑ IRES ↑ TIGAR | - | [109] |
| TAp73 | HCT116 and HCT116 p53−/− | ↑ G6PD | ↑ proliferation | [307,308] | |
| TAp63 | Patient-derived CCSCs | - | ↑ glycolytic activity | [309] | |
| Senescence | ↑ p53β and ↓ Δ133p53 | CRA patient samples | - | ↑ senescence | [19] |
| ↓ p53β and ↑ Δ133p53 | CRC patient samples | Escape from senescence barrier |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horvat, A.; Tadijan, A.; Vlašić, I.; Slade, N. p53/p73 Protein Network in Colorectal Cancer and Other Human Malignancies. Cancers 2021, 13, 2885. https://doi.org/10.3390/cancers13122885
Horvat A, Tadijan A, Vlašić I, Slade N. p53/p73 Protein Network in Colorectal Cancer and Other Human Malignancies. Cancers. 2021; 13(12):2885. https://doi.org/10.3390/cancers13122885
Chicago/Turabian StyleHorvat, Anđela, Ana Tadijan, Ignacija Vlašić, and Neda Slade. 2021. "p53/p73 Protein Network in Colorectal Cancer and Other Human Malignancies" Cancers 13, no. 12: 2885. https://doi.org/10.3390/cancers13122885
APA StyleHorvat, A., Tadijan, A., Vlašić, I., & Slade, N. (2021). p53/p73 Protein Network in Colorectal Cancer and Other Human Malignancies. Cancers, 13(12), 2885. https://doi.org/10.3390/cancers13122885