
CBP/p300: Critical Co-Activators for Nuclear Steroid Hormone Receptors and Emerging Therapeutic Targets in Prostate and Breast Cancers


Simple Summary
Abstract
1. Introduction
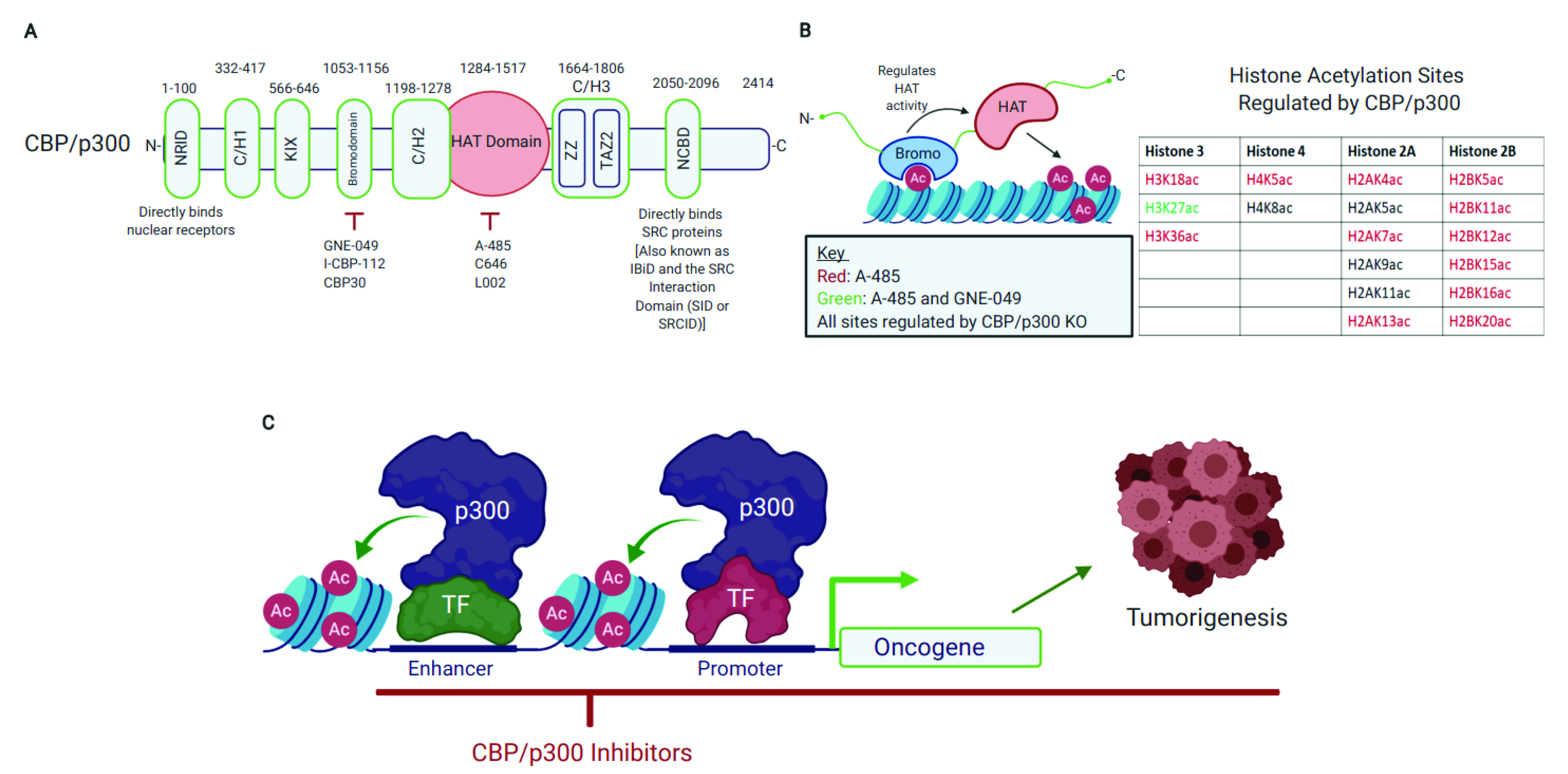
2. CBP/p300 Are Emerging Therapeutic Targets with Pharmacologically Tractable HAT Domain and Bromodomain
2.1. CBP/p300 Acetylate Diverse Proteins and Their HAT Domain Represents a Druggable Target
2.2. The CBP/p300 Bromodomain Binds Acetylated Lysine Residues, Regulates HAT Activity, and Is a Target for Pharmacological CBP/p300 Inhibition
3. The Role of CBP/p300 in AR Signaling in PCa
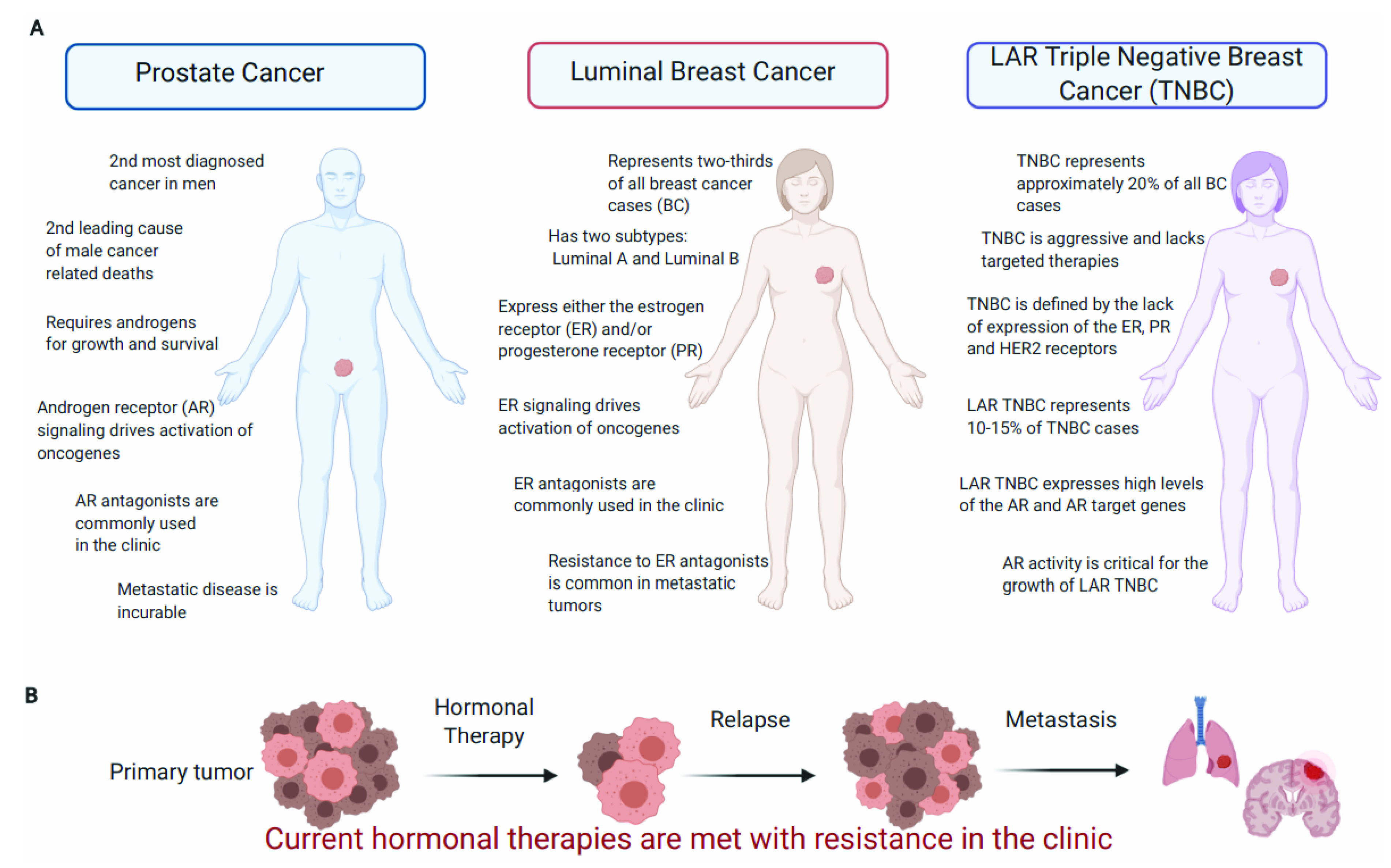
3.1. PCa Is the Most Common Noncutaneous Cancer in Men and Is Predominantly Driven by the AR
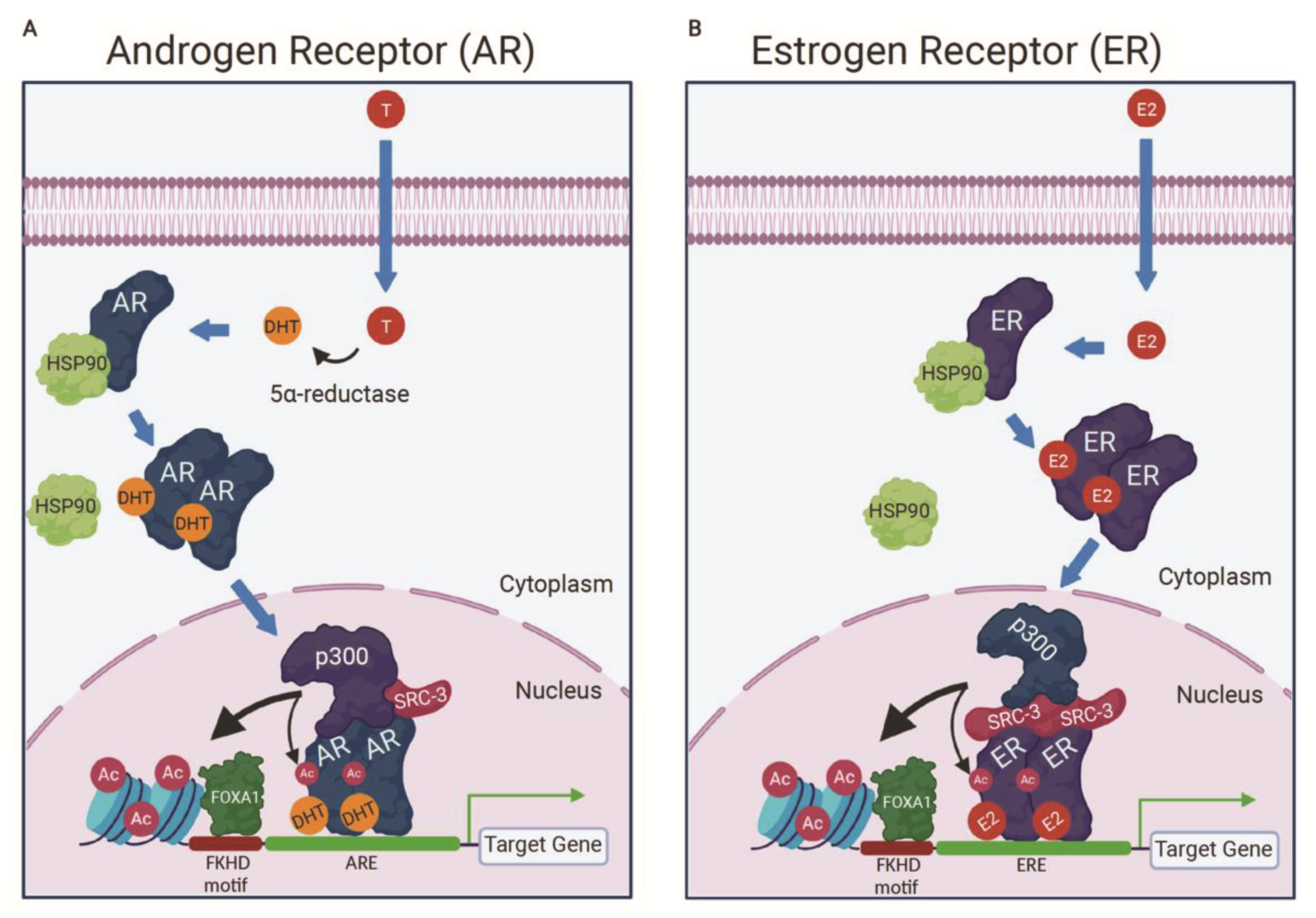
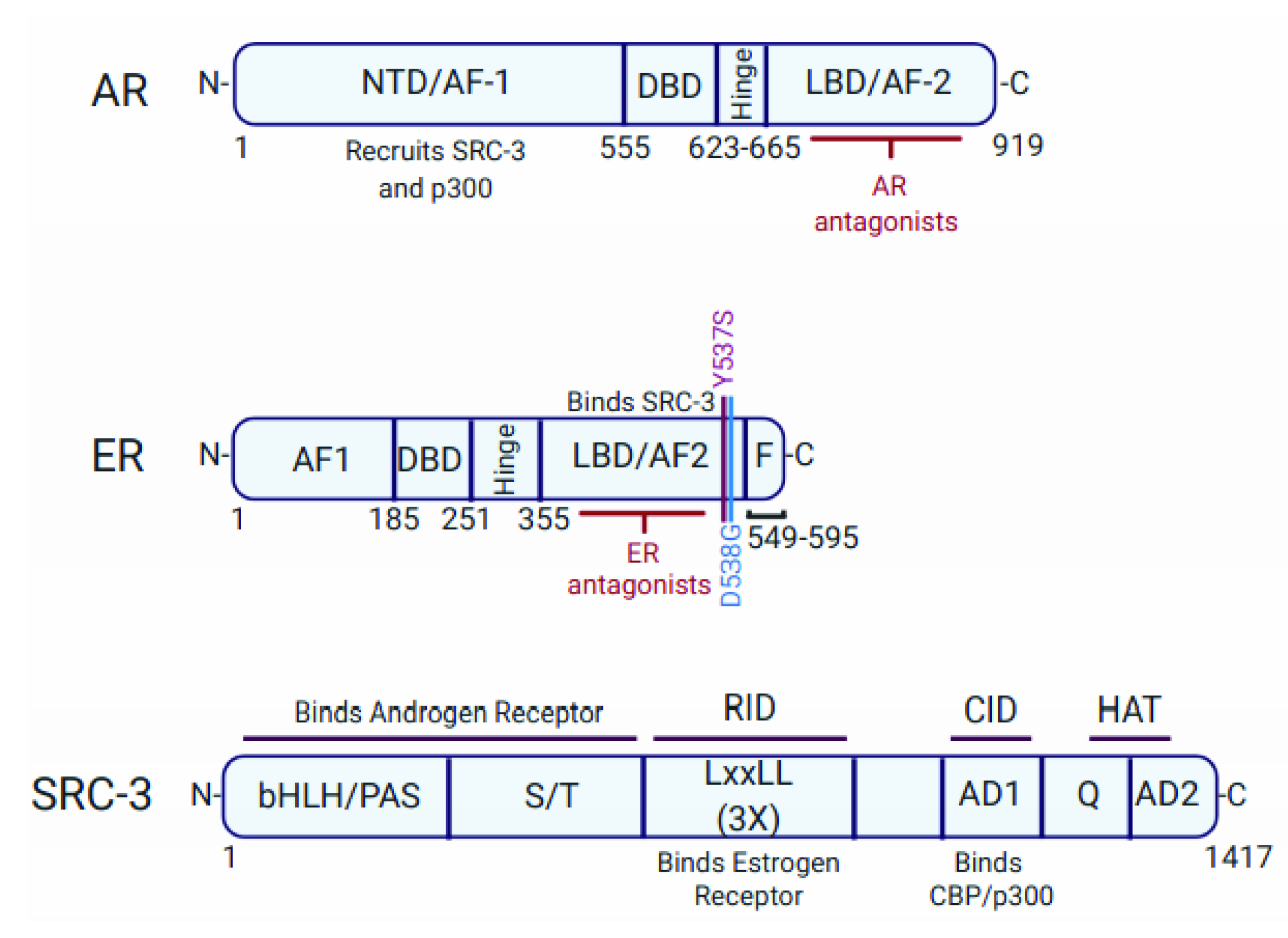
3.2. p300 Is a Component of the AR Activation Complex
3.3. p300 Co-Occupies Many Genomic Sites with the AR to Activate AR Target Gene Expression
3.4. p300 Stabilizes the AR While Androgen Deprivation Stabilizes CBP/p300
3.5. CBP/p300 Are AR Co-Activators in the Absence of Androgens
4. CBP/p300 Represent Rational Drug Targets in AR+ PCa
4.1. p300 Is Critical for PCa Tumor Growth
4.2. CBP/p300 BD Inhibitors Show Potent Anti-Proliferative Effects in Preclinical PCa Studies
4.3. The CBP/p300 HAT Inhibitor A-485 Is Effective against PCa In Vitro and In Vivo
5. The Role of CBP/p300 in ER Signaling in ER+ BC
5.1. BC Is a Diverse Disease with Hormone-Dependent and Independent Subtypes
5.2. ER Signaling Drives Tumor Growth of Luminal BC Subtypes and Is the Major Therapeutic Target
5.3. CBP/p300 Are Components of the ER Activation Complex
5.4. CBP/p300 Co-Bind with ER at Many Genomic Sites
5.5. The HAT Activity of CBP/p300 Is Critical to ER Signaling
5.6. p300 Contributes to ER-Mediated Transcriptional Repression
5.7. CBP/p300 Have Enhanced Interactions with Constitutively Active ER Mutants and p300 Is Essential for ER Mutant BC Cell Growth
6. CBP/p300 Represent Rational Drug Targets in BC
6.1. CBP/p300 Inhibitors Suppress ER Signaling and ER+ BC Growth
6.2. Targeting CBP/p300 to Inhibit AR Signaling as a Potential Therapeutic Strategy in AR+ BC
6.3. CBP/p300 Promotes Sex Hormone-Independent Oncogenic Signaling Pathways in BC
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yee, S.P.; Branton, P.E. Detection of cellular proteins associated with human adenovirus type 5 early region 1A polypeptides. Virology 1985, 147, 142–153. [Google Scholar] [CrossRef]
- Whyte, P.; Williamson, N.M.; Harlow, E. Cellular targets for transformation by the adenovirus E1A proteins. Cell 1989, 56, 67–75. [Google Scholar] [CrossRef]
- Eckner, R.; Ewen, M.E.; Newsome, D.; Gerdes, M.; DeCaprio, J.A.; Lawrence, J.B.; Livingston, D.M. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 1994, 8, 869–884. [Google Scholar] [CrossRef]
- Chrivia, J.C.; Kwok, R.P.; Lamb, N.; Hagiwara, M.; Montminy, M.R.; Goodman, R.H. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 1993, 365, 855–859. [Google Scholar] [CrossRef]
- Wang, L.; Tang, Y.; Cole, P.A.; Marmorstein, R. Structure and chemistry of the p300/CBP and Rtt109 histone acetyltransferases: Implications for histone acetyltransferase evolution and function. Curr. Opin. Struct. Biol. 2008, 18, 741–747. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, F.; Marshall, C.B.; Ikura, M. Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: Structural and functional versatility in target recognition. Cell Mol. Life Sci. 2013, 70, 3989–4008. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.P.; Oh, S.P.; Fuchs, M.; Zhou, N.D.; Ch’ng, L.E.; Newsome, D.; Bronson, R.T.; Li, E.; Livingston, D.M.; Eckner, R. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 1998, 93, 361–372. [Google Scholar] [CrossRef]
- Oike, Y.; Takakura, N.; Hata, A.; Kaname, T.; Akizuki, M.; Yamaguchi, Y.; Yasue, H.; Araki, K.; Yamamura, K.; Suda, T. Mice homozygous for a truncated form of CREB-binding protein exhibit defects in hematopoiesis and vasculo-angiogenesis. Blood 1999, 93, 2771–2779. [Google Scholar] [CrossRef]
- Kung, A.L.; Rebel, V.I.; Bronson, R.T.; Ch’ng, L.E.; Sieff, C.A.; Livingston, D.M.; Yao, T.P. Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev. 2000, 14, 272–277. [Google Scholar] [PubMed]
- Ding, L.; Chen, S.; Liu, P.; Pan, Y.; Zhong, J.; Regan, K.M.; Wang, L.; Yu, C.; Rizzardi, A.; Cheng, L.; et al. CBP loss cooperates with PTEN haploinsufficiency to drive prostate cancer: Implications for epigenetic therapy. Cancer Res. 2014, 74, 2050–2061. [Google Scholar] [CrossRef]
- Zhong, J.; Ding, L.; Bohrer, L.R.; Pan, Y.; Liu, P.; Zhang, J.; Sebo, T.J.; Karnes, R.J.; Tindall, D.J.; van Deursen, J.; et al. p300 acetyltransferase regulates androgen receptor degradation and PTEN-deficient prostate tumorigenesis. Cancer Res. 2014, 74, 1870–1880. [Google Scholar] [CrossRef] [PubMed]
- Dancy, B.M.; Cole, P.A. Protein lysine acetylation by p300/CBP. Chem. Rev. 2015, 115, 2419–2452. [Google Scholar] [CrossRef]
- Sterner, D.E.; Berger, S.L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Merika, M.; Williams, A.J.; Chen, G.; Collins, T.; Thanos, D. Recruitment of CBP/p300 by the IFN beta enhanceosome is required for synergistic activation of transcription. Mol. Cell 1998, 1, 277–287. [Google Scholar] [CrossRef]
- Phillips, D.M. The presence of acetyl groups of histones. Biochem. J. 1963, 87, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of Rna synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. The CBP co-activator is a histone acetyltransferase. Nature 1996, 384, 641–643. [Google Scholar] [CrossRef]
- Brownell, J.E.; Zhou, J.; Ranalli, T.; Kobayashi, R.; Edmondson, D.G.; Roth, S.Y.; Allis, C.D. Tetrahymena histone acetyltransferase A: A homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 1996, 84, 843–851. [Google Scholar] [CrossRef]
- Kuo, M.H.; Brownell, J.E.; Sobel, R.E.; Ranalli, T.A.; Cook, R.G.; Edmondson, D.G.; Roth, S.Y.; Allis, C.D. Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature 1996, 383, 269–272. [Google Scholar] [CrossRef]
- Ogryzko, V.V.; Schiltz, R.L.; Russanova, V.; Howard, B.H.; Nakatani, Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996, 87, 953–959. [Google Scholar] [CrossRef]
- Yang, X.J.; Ogryzko, V.V.; Nishikawa, J.; Howard, B.H.; Nakatani, Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature 1996, 382, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Weinert, B.T.; Narita, T.; Satpathy, S.; Srinivasan, B.; Hansen, B.K.; Scholz, C.; Hamilton, W.B.; Zucconi, B.E.; Wang, W.W.; Liu, W.R.; et al. Time-resolved analysis reveals rapid dynamics and broad scope of the CBP/p300 acetylome. Cell 2018, 174, 231–244 e212. [Google Scholar] [CrossRef] [PubMed]
- Raisner, R.; Kharbanda, S.; Jin, L.; Jeng, E.; Chan, E.; Merchant, M.; Haverty, P.M.; Bainer, R.; Cheung, T.; Arnott, D.; et al. Enhancer activity requires CBP/P300 bromodomain-dependent histone H3K27 acetylation. Cell Rep. 2018, 24, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chen, L.; Han, Y.; Wu, F.; Yang, W.S.; Zhang, Z.; Huo, T.; Zhu, Y.; Yu, C.; Kim, H.; et al. Acetylation of histone H3K27 signals the transcriptional elongation for estrogen receptor alpha. Commun. Biol. 2020, 3, 165. [Google Scholar] [CrossRef] [PubMed]
- Delvecchio, M.; Gaucher, J.; Aguilar-Gurrieri, C.; Ortega, E.; Panne, D. Structure of the p300 catalytic core and implications for chromatin targeting and HAT regulation. Nat. Struct. Mol. Biol. 2013, 20, 1040–1046. [Google Scholar] [CrossRef]
- Kaczmarska, Z.; Ortega, E.; Goudarzi, A.; Huang, H.; Kim, S.; Marquez, J.A.; Zhao, Y.; Khochbin, S.; Panne, D. Structure of p300 in complex with acyl-CoA variants. Nat. Chem. Biol. 2017, 13, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Stanfield, R.L.; Martinez-Yamout, M.A.; Dyson, H.J.; Wilson, I.A.; Wright, P.E. Role of the CBP catalytic core in intramolecular SUMOylation and control of histone H3 acetylation. Proc. Natl. Acad. Sci. USA 2017, 114, E5335–E5342. [Google Scholar] [CrossRef]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [Google Scholar] [CrossRef]
- Deng, Q.; Li, Y.; Tedesco, D.; Liao, R.; Fuhrmann, G.; Sun, P. The ability of E1A to rescue ras-induced premature senescence and confer transformation relies on inactivation of both p300/CBP and Rb family proteins. Cancer Res. 2005, 65, 8298–8307. [Google Scholar] [CrossRef]
- Somasundaram, K.; El-Deiry, W.S. Inhibition of p53-mediated transactivation and cell cycle arrest by E1A through its p300/CBP-interacting region. Oncogene 1997, 14, 1047–1057. [Google Scholar] [CrossRef]
- Gayther, S.A.; Batley, S.J.; Linger, L.; Bannister, A.; Thorpe, K.; Chin, S.F.; Daigo, Y.; Russell, P.; Wilson, A.; Sowter, H.M.; et al. Mutations truncating the EP300 acetylase in human cancers. Nat. Genet. 2000, 24, 300–303. [Google Scholar] [CrossRef]
- Attar, N.; Kurdistani, S.K. Exploitation of EP300 and CREBBP lysine acetyltransferases by cancer. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Fioretos, T.; Isaksson, M.; Samuelsson, U.; Billstrom, R.; Strombeck, B.; Mitelman, F.; Johansson, B. Fusion of the MORF and CBP genes in acute myeloid leukemia with the t(10;16)(q22;p13). Human Mol. Genet. 2001, 10, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Kitabayashi, I.; Aikawa, Y.; Yokoyama, A.; Hosoda, F.; Nagai, M.; Kakazu, N.; Abe, T.; Ohki, M. Fusion of MOZ and p300 histone acetyltransferases in acute monocytic leukemia with a t(8;22)(p11;q13) chromosome translocation. Leukemia 2001, 15, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gural, A.; Sun, X.J.; Zhao, X.; Perna, F.; Huang, G.; Hatlen, M.A.; Vu, L.; Liu, F.; Xu, H.; et al. The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science 2011, 333, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Ogiwara, H.; Sasaki, M.; Mitachi, T.; Oike, T.; Higuchi, S.; Tominaga, Y.; Kohno, T. Targeting p300 addiction in CBP-deficient cancers causes synthetic lethality by apoptotic cell death due to abrogation of MYC expression. Cancer Discov. 2016, 6, 430–445. [Google Scholar] [CrossRef]
- Koch, L.M.; Birkeland, E.S.; Battaglioni, S.; Helle, X.; Meerang, M.; Hiltbrunner, S.; Ibanez, A.J.; Peter, M.; Curioni-Fontecedro, A.; Opitz, I.; et al. Cytosolic pH regulates proliferation and tumour growth by promoting expression of cyclin D1. Nat. Metab. 2020, 2, 1212–1222. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019. [Google Scholar] [CrossRef]
- Krug, K.; Jaehnig, E.J.; Satpathy, S.; Blumenberg, L.; Karpova, A.; Anurag, M.; Miles, G.; Mertins, P.; Geffen, Y.; Tang, L.C.; et al. Proteogenomic landscape of breast cancer tumorigenesis and targeted therapy. Cell 2020, 183, 1436–1456.e1431. [Google Scholar] [CrossRef]
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, D.; Spring, D.J.; DePinho, R.A. Genetics and biology of prostate cancer. Genes Dev. 2018, 32, 1105–1140. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, A.; Duguay, S.R.; Ouellette, R.J.; Surette, M.E. 17beta-estradiol induces stearoyl-CoA desaturase-1 expression in estrogen receptor-positive breast cancer cells. BMC Cancer 2015, 15, 440. [Google Scholar] [CrossRef] [PubMed]
- Bernard, D.; Pourtier-Manzanedo, A.; Gil, J.; Beach, D.H. Myc confers androgen-independent prostate cancer cell growth. J. Clin. Investig. 2003, 112, 1724–1731. [Google Scholar] [CrossRef] [PubMed]
- Gonthier, K.; Poluri, R.T.K.; Audet-Walsh, E. Functional genomic studies reveal the androgen receptor as a master regulator of cellular energy metabolism in prostate cancer. J. Steroid Biochem. Mol. Biol. 2019, 191, 105367. [Google Scholar] [CrossRef] [PubMed]
- Prall, O.W.; Rogan, E.M.; Musgrove, E.A.; Watts, C.K.; Sutherland, R.L. c-Myc or cyclin D1 mimics estrogen effects on cyclin E-Cdk2 activation and cell cycle reentry. Mol. Cell. Biol. 1998, 18, 4499–4508. [Google Scholar] [CrossRef]
- Spratt, D.E.; Zumsteg, Z.S.; Feng, F.Y.; Tomlins, S.A. Translational and clinical implications of the genetic landscape of prostate cancer. Nat. Rev. Clin. Oncol. 2016, 13, 597–610. [Google Scholar] [CrossRef]
- Wang, C.; Mayer, J.A.; Mazumdar, A.; Fertuck, K.; Kim, H.; Brown, M.; Brown, P.H. Estrogen induces c-myc gene expression via an upstream enhancer activated by the estrogen receptor and the AP-1 transcription factor. Mol. Endocrinol. 2011, 25, 1527–1538. [Google Scholar] [CrossRef]
- Wang, Q.; Li, W.; Zhang, Y.; Yuan, X.; Xu, K.; Yu, J.; Chen, Z.; Beroukhim, R.; Wang, H.; Lupien, M.; et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 2009, 138, 245–256. [Google Scholar] [CrossRef]
- Wong, Y.N.; Ferraldeschi, R.; Attard, G.; de Bono, J. Evolution of androgen receptor targeted therapy for advanced prostate cancer. Nat. Rev. Clin. Oncol. 2014, 11, 365–376. [Google Scholar] [CrossRef]
- Yu, X.; Yi, P.; Hamilton, R.A.; Shen, H.; Chen, M.; Foulds, C.E.; Mancini, M.A.; Ludtke, S.J.; Wang, Z.; O’Malley, B.W. Structural insights of transcriptionally active, full-length androgen receptor coactivator complexes. Mol. Cell 2020, 79, 812–823.e814. [Google Scholar] [CrossRef]
- Yi, P.; Wang, Z.; Feng, Q.; Pintilie, G.D.; Foulds, C.E.; Lanz, R.B.; Ludtke, S.J.; Schmid, M.F.; Chiu, W.; O’Malley, B.W. Structure of a biologically active estrogen receptor-coactivator complex on DNA. Mol. Cell 2015, 57, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Garcia, J.; Chan, E.; de la Cruz, C.; Segal, E.; Merchant, M.; Kharbanda, S.; Raisner, R.; Haverty, P.M.; Modrusan, Z.; et al. Therapeutic targeting of the CBP/p300 bromodomain blocks the growth of castration-resistant prostate cancer. Cancer Res. 2017, 77, 5564–5575. [Google Scholar] [CrossRef] [PubMed]
- Zwart, W.; Theodorou, V.; Kok, M.; Canisius, S.; Linn, S.; Carroll, J.S. Oestrogen receptor-co-factor-chromatin specificity in the transcriptional regulation of breast cancer. EMBO J. 2011, 30, 4764–4776. [Google Scholar] [CrossRef]
- Kim, S.H.; Kang, H.J.; Na, H.; Lee, M.O. Trichostatin A enhances acetylation as well as protein stability of ERalpha through induction of p300 protein. Breast Cancer Res. 2010, 12, R22. [Google Scholar] [CrossRef] [PubMed]
- Waddell, A.R.; Mahmud, I.; Ding, H.; Huo, Z.; Liao, D. Pharmacological inhibition of CBP/p300 blocks estrogen recep-tor alpha (ERα) function through suppressing enhancer H3K27 acetylation in luminal breast cancer. Cancers 2021, 13, 2799. [Google Scholar] [CrossRef]
- Chang, M. Tamoxifen resistance in breast cancer. Biomol. Ther. 2012, 20, 256–267. [Google Scholar] [CrossRef]
- Vander Ark, A.; Cao, J.; Li, X. Mechanisms and approaches for overcoming enzalutamide resistance in prostate cancer. Front. Oncol. 2018, 8, 180. [Google Scholar] [CrossRef]
- Debes, J.D.; Schmidt, L.J.; Huang, H.; Tindall, D.J. p300 mediates androgen-independent transactivation of the androgen receptor by interleukin 6. Cancer Res. 2002, 62, 5632–5636. [Google Scholar] [PubMed]
- Jeselsohn, R.; Bergholz, J.S.; Pun, M.; Cornwell, M.; Liu, W.; Nardone, A.; Xiao, T.; Li, W.; Qiu, X.; Buchwalter, G.; et al. Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell 2018, 33, 173–186.e175. [Google Scholar] [CrossRef]
- Lee, S.O.; Chun, J.Y.; Nadiminty, N.; Lou, W.; Feng, S.; Gao, A.C. Interleukin-4 activates androgen receptor through CBP/p300. Prostate 2009, 69, 126–132. [Google Scholar] [CrossRef]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef]
- Zhang, Q.X.; Borg, A.; Wolf, D.M.; Oesterreich, S.; Fuqua, S.A. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res. 1997, 57, 1244–1249. [Google Scholar]
- Santer, F.R.; Hoschele, P.P.; Oh, S.J.; Erb, H.H.; Bouchal, J.; Cavarretta, I.T.; Parson, W.; Meyers, D.J.; Cole, P.A.; Culig, Z. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol. Cancer Ther. 2011, 10, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Teufel, D.P.; Freund, S.M.; Bycroft, M.; Fersht, A.R. Four domains of p300 each bind tightly to a sequence spanning both transactivation subdomains of p53. Proc. Natl. Acad. Sci. USA 2007, 104, 7009–7014. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kellis, M. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 2017, 12, 2478–2492. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Chng, W.J.; Zhou, J. Super-enhancers: Critical roles and therapeutic targets in hematologic malignancies. J. Hematol. Oncol. 2019, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Yu, L.R.; Wang, L.; Zhang, Z.; Kasper, L.H.; Lee, J.E.; Wang, C.; Brindle, P.K.; Dent, S.Y.; Ge, K. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011, 30, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.Y. Targeting super-enhancers for disease treatment and diagnosis. Mol. Cells 2018, 41, 506–514. [Google Scholar] [CrossRef]
- Pradeepa, M.M. Causal role of histone acetylations in enhancer function. Transcription 2017, 8, 40–47. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef]
- Ali, I.; Conrad, R.J.; Verdin, E.; Ott, M. Lysine acetylation goes global: From epigenetics to metabolism and therapeutics. Chem. Rev. 2018, 118, 1216–1252. [Google Scholar] [CrossRef]
- Reed, S.M.; Quelle, D.E. p53 acetylation: Regulation and consequences. Cancers 2014, 7, 30–69. [Google Scholar] [CrossRef]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef]
- Lill, N.L.; Grossman, S.R.; Ginsberg, D.; DeCaprio, J.; Livingston, D.M. Binding and modulation of p53 by p300/CBP coactivators. Nature 1997, 387, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Wang, C.; Reutens, A.T.; Wang, J.; Angeletti, R.H.; Siconolfi-Baez, L.; Ogryzko, V.; Avantaggiati, M.L.; Pestell, R.G. p300 and p300/cAMP-response element-binding protein-associated factor acetylate the androgen receptor at sites governing hormone-dependent transactivation. J. Biol. Chem. 2000, 275, 20853–20860. [Google Scholar] [CrossRef]
- Wang, C.; Fu, M.; Angeletti, R.H.; Siconolfi-Baez, L.; Reutens, A.T.; Albanese, C.; Lisanti, M.P.; Katzenellenbogen, B.S.; Kato, S.; Hopp, T.; et al. Direct acetylation of the estrogen receptor alpha hinge region by p300 regulates transactivation and hormone sensitivity. J. Biol. Chem. 2001, 276, 18375–18383. [Google Scholar] [CrossRef]
- Chen, L.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, M.; Tang, Y.; Laszkowska, M.; Roeder, R.G.; Gu, W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 2259–2264. [Google Scholar] [CrossRef]
- Kim, M.Y.; Woo, E.M.; Chong, Y.T.; Homenko, D.R.; Kraus, W.L. Acetylation of estrogen receptor alpha by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Mol. Endocrinol. 2006, 20, 1479–1493. [Google Scholar] [CrossRef]
- Wang, Z.A.; Cole, P.A. The chemical biology of reversible lysine post-translational modifications. Cell Chem. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- He, Z.X.; Wei, B.F.; Zhang, X.; Gong, Y.P.; Ma, L.Y.; Zhao, W. Current development of CBP/p300 inhibitors in the last decade. Eur. J. Med. Chem. 2021, 209, 112861. [Google Scholar] [CrossRef] [PubMed]
- Bowers, E.M.; Yan, G.; Mukherjee, C.; Orry, A.; Wang, L.; Holbert, M.A.; Crump, N.T.; Hazzalin, C.A.; Liszczak, G.; Yuan, H.; et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: Identification of a selective small molecule inhibitor. Chem. Biol. 2010, 17, 471–482. [Google Scholar] [CrossRef]
- Liu, X.; Wang, L.; Zhao, K.; Thompson, P.R.; Hwang, Y.; Marmorstein, R.; Cole, P.A. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature 2008, 451, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Hogg, S.J.; Motorna, O.; Cluse, L.A.; Johanson, T.M.; Coughlan, H.D.; Raviram, R.; Myers, R.M.; Costacurta, M.; Todorovski, I.; Pijpers, L.; et al. Targeting histone acetylation dynamics and oncogenic transcription by catalytic P300/CBP inhibition. Mol. Cell 2021, 81, 2183–2200.e2113. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; He, D.; Cheng, L.; Huang, C.; Zhang, Y.; Rao, X.; Kong, Y.; Li, C.; Zhang, Z.; Liu, J.; et al. p300/CBP inhibition enhances the efficacy of programmed death-ligand 1 blockade treatment in prostate cancer. Oncogene 2020, 39, 3939–3951. [Google Scholar] [CrossRef] [PubMed]
- Bosnakovski, D.; da Silva, M.T.; Sunny, S.T.; Ener, E.T.; Toso, E.A.; Yuan, C.; Cui, Z.; Walters, M.A.; Jadhav, A.; Kyba, M. A novel P300 inhibitor reverses DUX4-mediated global histone H3 hyperacetylation, target gene expression, and cell death. Sci. Adv. 2019, 5, eaaw7781. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, R.; Li, Z.; Mei, L.; Wan, S.; Ding, H.; Chen, Z.; Xing, J.; Feng, H.; Han, J.; et al. Discovery of highly potent, selective, and orally efficacious p300/CBP histone acetyltransferases inhibitors. J. Med. Chem. 2020, 63, 1337–1360. [Google Scholar] [CrossRef]
- Wu, F.; Hua, Y.; Kaochar, S.; Nie, S.; Lin, Y.L.; Yao, Y.; Wu, J.; Wu, X.; Fu, X.; Schiff, R.; et al. Discovery, structure-activity relationship, and biological activity of histone-competitive inhibitors of histone acetyltransferases P300/CBP. J. Med. Chem. 2020, 63, 4716–4731. [Google Scholar] [CrossRef]
- Ji, Z.; Clark, R.F.; Bhat, V.; Matthew Hansen, T.; Lasko, L.M.; Bromberg, K.D.; Manaves, V.; Algire, M.; Martin, R.; Qiu, W.; et al. Discovery of spirohydantoins as selective, orally bioavailable inhibitors of p300/CBP histone acetyltransferases. Bioorg. Med. Chem. Lett. 2021, 39, 127854. [Google Scholar] [CrossRef]
- Romero, F.A.; Murray, J.; Lai, K.W.; Tsui, V.; Albrecht, B.K.; An, L.; Beresini, M.H.; de Leon Boenig, G.; Bronner, S.M.; Chan, E.W.; et al. GNE-781, a highly advanced potent and selective bromodomain inhibitor of cyclic adenosine monophosphate response element binding protein, binding protein (CBP). J. Med. Chem. 2017, 60, 9162–9183. [Google Scholar] [CrossRef]
- Welti, J.; Sharp, A.; Brooks, N.; Yuan, W.; McNair, C.; Chand, S.N.; Pal, A.; Figueiredo, I.; Riisnaes, R.; Gurel, B.; et al. Targeting the p300/CBP axis in lethal prostate cancer. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Vannam, R.; Sayilgan, J.; Ojeda, S.; Karakyriakou, B.; Hu, E.; Kreuzer, J.; Morris, R.; Herrera Lopez, X.I.; Rai, S.; Haas, W.; et al. Targeted degradation of the enhancer lysine acetyltransferases CBP and p300. Cell Chem. Biol. 2021, 28, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Cochran, A.G.; Conery, A.R.; Sims, R.J., 3rd. Bromodomains: A new target class for drug development. Nat. Rev. Drug. Discov. 2019, 18, 609–628. [Google Scholar] [CrossRef] [PubMed]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Perez-Salvia, M.; Esteller, M. Bromodomain inhibitors and cancer therapy: From structures to applications. Epigenetics Off. J. DNA Methylation Soc. 2017, 12, 323–339. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Manickavinayaham, S.; Velez-Cruz, R.; Biswas, A.K.; Bedford, E.; Klein, B.J.; Kutateladze, T.G.; Liu, B.; Bedford, M.T.; Johnson, D.G. E2F1 acetylation directs p300/CBP-mediated histone acetylation at DNA double-strand breaks to facilitate repair. Nat. Commun. 2019, 10, 4951. [Google Scholar] [CrossRef] [PubMed]
- Mujtaba, S.; He, Y.; Zeng, L.; Yan, S.; Plotnikova, O.; Sachchidanand; Sanchez, R.; Zeleznik-Le, N.J.; Ronai, Z.; Zhou, M.M. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol. Cell 2004, 13, 251–263. [Google Scholar] [CrossRef]
- Olzscha, H.; Fedorov, O.; Kessler, B.M.; Knapp, S.; La Thangue, N.B. CBP/p300 bromodomains regulate amyloid-like protein aggregation upon aberrant lysine acetylation. Cell Chem. Biol. 2017, 24, 9–23. [Google Scholar] [CrossRef]
- Liao, D. CBP/p300 bromodomain mediates amyloid formation. Cell Chem. Biol. 2017, 24, 128–129. [Google Scholar] [CrossRef] [PubMed]
- Hammitzsch, A.; Tallant, C.; Fedorov, O.; O’Mahony, A.; Brennan, P.E.; Hay, D.A.; Martinez, F.O.; Al-Mossawi, M.H.; de Wit, J.; Vecellio, M.; et al. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc. Natl. Acad. Sci. USA 2015, 112, 10768–10773. [Google Scholar] [CrossRef] [PubMed]
- Hay, D.A.; Fedorov, O.; Martin, S.; Singleton, D.C.; Tallant, C.; Wells, C.; Picaud, S.; Philpott, M.; Monteiro, O.P.; Rogers, C.M.; et al. Discovery and optimization of small-molecule ligands for the CBP/p300 bromodomains. J. Am. Chem. Soc. 2014, 136, 9308–9319. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.; Fedorov, O.; Thanasopoulou, A.; Leonards, K.; Jones, K.; Meier, J.; Olzscha, H.; Monteiro, O.; Martin, S.; Philpott, M.; et al. Generation of a selective small molecule inhibitor of the CBP/p300 bromodomain for leukemia therapy. Cancer Res. 2015, 75, 5106–5119. [Google Scholar] [CrossRef]
- Rooney, T.P.; Filippakopoulos, P.; Fedorov, O.; Picaud, S.; Cortopassi, W.A.; Hay, D.A.; Martin, S.; Tumber, A.; Rogers, C.M.; Philpott, M.; et al. A series of potent CREBBP bromodomain ligands reveals an induced-fit pocket stabilized by a cation-pi interaction. Angew. Chem. Int. Ed. Engl. 2014, 252, 6126–6130. [Google Scholar] [CrossRef]
- Taylor, A.M.; Cote, A.; Hewitt, M.C.; Pastor, R.; Leblanc, Y.; Nasveschuk, C.G.; Romero, F.A.; Crawford, T.D.; Cantone, N.; Jayaram, H.; et al. Fragment-based discovery of a selective and cell-active benzodiazepinone CBP/EP300 bromodomain inhibitor (CPI-637). ACS Med. Chem. Lett. 2016, 7, 531–536. [Google Scholar] [CrossRef]
- Xiang, Q.; Wang, C.; Zhang, Y.; Xue, X.; Song, M.; Zhang, C.; Li, C.; Wu, C.; Li, K.; Hui, X.; et al. Discovery and optimization of 1-(1H-indol-1-yl)ethanone derivatives as CBP/EP300 bromodomain inhibitors for the treatment of castration-resistant prostate cancer. Eur. J. Med. Chem. 2018, 147, 238–252. [Google Scholar] [CrossRef]
- Yan, Y.; Ma, J.; Wang, D.; Lin, D.; Pang, X.; Wang, S.; Zhao, Y.; Shi, L.; Xue, H.; Pan, Y.; et al. The novel BET-CBP/p300 dual inhibitor NEO2734 is active in SPOP mutant and wild-type prostate cancer. EMBO Mol. Med. 2019, 11, e10659. [Google Scholar] [CrossRef]
- Zou, L.J.; Xiang, Q.P.; Xue, X.Q.; Zhang, C.; Li, C.C.; Wang, C.; Li, Q.; Wang, R.; Wu, S.; Zhou, Y.L.; et al. Y08197 is a novel and selective CBP/EP300 bromodomain inhibitor for the treatment of prostate cancer. Acta Pharmacol. Sin. 2019, 40, 1436–1447. [Google Scholar] [CrossRef]
- Popp, T.A.; Tallant, C.; Rogers, C.; Fedorov, O.; Brennan, P.E.; Muller, S.; Knapp, S.; Bracher, F. Development of selective CBP/P300 benzoxazepine bromodomain inhibitors. J. Med. Chem. 2016, 59, 8889–8912. [Google Scholar] [CrossRef] [PubMed]
- Zucconi, B.E.; Luef, B.; Xu, W.; Henry, R.A.; Nodelman, I.M.; Bowman, G.D.; Andrews, A.J.; Cole, P.A. Modulation of p300/CBP acetylation of nucleosomes by bromodomain ligand I-CBP112. Biochemistry 2016, 55, 3727–3734. [Google Scholar] [CrossRef]
- Das, C.; Roy, S.; Namjoshi, S.; Malarkey, C.S.; Jones, D.N.; Kutateladze, T.G.; Churchill, M.E.; Tyler, J.K. Binding of the histone chaperone ASF1 to the CBP bromodomain promotes histone acetylation. Proc. Natl. Acad. Sci. USA 2014, 111, E1072–E1081. [Google Scholar] [CrossRef] [PubMed]
- Kraus, W.L.; Manning, E.T.; Kadonaga, J.T. Biochemical analysis of distinct activation functions in p300 that enhance transcription initiation with chromatin templates. Mol. Cell. Biol. 1999, 19, 8123–8135. [Google Scholar] [CrossRef]
- Thompson, P.R.; Wang, D.; Wang, L.; Fulco, M.; Pediconi, N.; Zhang, D.; An, W.; Ge, Q.; Roeder, R.G.; Wong, J.; et al. Regulation of the p300 HAT domain via a novel activation loop. Nat. Struct. Mol. Biol. 2004, 11, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Ortega, E.; Rengachari, S.; Ibrahim, Z.; Hoghoughi, N.; Gaucher, J.; Holehouse, A.S.; Khochbin, S.; Panne, D. Transcription factor dimerization activates the p300 acetyltransferase. Nature 2018, 562, 538–544. [Google Scholar] [CrossRef]
- Murakami, S.; Nagari, A.; Kraus, W.L. Dynamic assembly and activation of estrogen receptor alpha enhancers through coregulator switching. Genes Dev. 2017, 31, 1535–1548. [Google Scholar] [CrossRef]
- Garcia-Carpizo, V.; Ruiz-Llorente, S.; Sarmentero, J.; Graña-Castro, O.; Pisano, D.G.; Barrero, M.J. CREBBP/EP300 bromodomains are critical to sustain the GATA1/MYC regulatory axis in proliferation. Epigenetics Chromatin 2018, 11, 30. [Google Scholar] [CrossRef]
- Zucconi, B.E.; Makofske, J.L.; Meyers, D.J.; Hwang, Y.; Wu, M.; Kuroda, M.I.; Cole, P.A. Combination targeting of the bromodomain and acetyltransferase active site of p300/CBP. Biochemistry 2019, 58, 2133–2143. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, J.; Zhao, L.Y.; Chen, X.; Zheng, G.; Zhang, X.; Liao, D. Discovery of histone deacetylase 3 (HDAC3)-specific PROTACs. Chem. Commun. 2020, 56, 9866–9869. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Crews, C.M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; He, B. Androgen receptor signaling in the development of castration-resistant prostate cancer. Front. Oncol. 2019, 9, 858. [Google Scholar] [CrossRef] [PubMed]
- Lonergan, P.E.; Tindall, D.J. Androgen receptor signaling in prostate cancer development and progression. J. Carcinog. 2011, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.L.; Carroll, J.S. FoxA1 is a key mediator of hormonal response in breast and prostate cancer. Front. Endocrinol. 2012, 3, 68. [Google Scholar] [CrossRef]
- Yang, Y.A.; Yu, J. Current perspectives on FOXA1 regulation of androgen receptor signaling and prostate cancer. Genes Dis. 2015, 2, 144–151. [Google Scholar] [CrossRef]
- Nollet, E.A.; Cardo-Vila, M.; Ganguly, S.S.; Tran, J.D.; Schulz, V.V.; Cress, A.; Corey, E.; Miranti, C.K. Androgen receptor-induced integrin alpha6beta1 and Bnip3 promote survival and resistance to PI3K inhibitors in castration-resistant prostate cancer. Oncogene 2020, 39, 5390–5404. [Google Scholar] [CrossRef] [PubMed]
- Lamb, L.E.; Zarif, J.C.; Miranti, C.K. The androgen receptor induces integrin alpha6beta1 to promote prostate tumor cell survival via NF-kappaB and Bcl-xL Independently of PI3K signaling. Cancer Res. 2011, 71, 2739–2749. [Google Scholar] [CrossRef]
- Schiewer, M.J.; Augello, M.A.; Knudsen, K.E. The AR dependent cell cycle: Mechanisms and cancer relevance. Mol. Cell. Endocrinol. 2012, 352, 34–45. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Attard, G.; Reid, A.H.; A’Hern, R.; Parker, C.; Oommen, N.B.; Folkerd, E.; Messiou, C.; Molife, L.R.; Maier, G.; Thompson, E.; et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J. Clin. Oncol. 2009, 27, 3742–3748. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Balbas, M.D.; Evans, M.J.; Hosfield, D.J.; Wongvipat, J.; Arora, V.K.; Watson, P.A.; Chen, Y.; Greene, G.L.; Shen, Y.; Sawyers, C.L. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife 2013, 2, e00499. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008, 68, 5469–5477. [Google Scholar] [CrossRef]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef]
- Guo, Z.; Yang, X.; Sun, F.; Jiang, R.; Linn, D.E.; Chen, H.; Chen, H.; Kong, X.; Melamed, J.; Tepper, C.G.; et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009, 69, 2305–2313. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489. [Google Scholar] [CrossRef]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef]
- Bishop, J.L.; Thaper, D.; Vahid, S.; Davies, A.; Ketola, K.; Kuruma, H.; Jama, R.; Nip, K.M.; Angeles, A.; Johnson, F.; et al. The master neural transcription factor BRN2 is an androgen receptor-suppressed driver of neuroendocrine differentiation in prostate cancer. Cancer Discov. 2017, 7, 54–71. [Google Scholar] [CrossRef]
- Blee, A.M.; He, Y.; Yang, Y.; Ye, Z.; Yan, Y.; Pan, Y.; Ma, T.; Dugdale, J.; Kuehn, E.; Kohli, M.; et al. TMPRSS2-ERG controls luminal epithelial lineage and antiandrogen sensitivity in PTEN and TP53-mutated prostate cancer. Clin. Cancer Res. 2018, 24, 4551–4565. [Google Scholar] [CrossRef]
- Blee, A.M.; Huang, H. Lineage plasticity-mediated therapy resistance in prostate cancer. Asian J. Androl. 2019, 21, 241–248. [Google Scholar] [CrossRef]
- Culig, Z.; Santer, F.R. Androgen receptor co-activators in the regulation of cellular events in prostate cancer. World J. Urol. 2012, 30, 297–302. [Google Scholar] [CrossRef]
- Heemers, H.V.; Tindall, D.J. Androgen receptor (AR) coregulators: A diversity of functions converging on and regulating the AR transcriptional complex. Endocr. Rev. 2007, 28, 778–808. [Google Scholar] [CrossRef]
- Tien, J.C.; Liu, Z.; Liao, L.; Wang, F.; Xu, Y.; Wu, Y.L.; Zhou, N.; Ittmann, M.; Xu, J. The steroid receptor coactivator-3 is required for the development of castration-resistant prostate cancer. Cancer Res. 2013, 73, 3997–4008. [Google Scholar] [CrossRef]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: A multi-institutional prospective study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Zhang, Y.Q.; Huang, J.T. Neuroendocrine cells of prostate cancer: Biologic functions and molecular mechanisms. Asian J. Androl. 2019, 21, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.J.; Mawji, N.R.; Wang, J.; Wang, G.; Haile, S.; Myung, J.K.; Watt, K.; Tam, T.; Yang, Y.C.; Banuelos, C.A.; et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 2010, 17, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Loddick, S.A.; Ross, S.J.; Thomason, A.G.; Robinson, D.M.; Walker, G.E.; Dunkley, T.P.; Brave, S.R.; Broadbent, N.; Stratton, N.C.; Trueman, D.; et al. AZD3514: A small molecule that modulates androgen receptor signaling and function in vitro and in vivo. Mol. Cancer Ther. 2013, 12, 1715–1727. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.N.; Vaishampayan, U.N.; Gordon, M.S.; Smith, D.C.; Rudsinski, E.; De Haas-Amatsaleh, A.; Thapar, N.; Perabo, F.; Montgomery, R.B. Efficacy, safety, tolerability, and pharmacokinetics of EPI-506 (ralaniten acetate), a novel androgen receptor (AR) N-terminal domain (NTD) inhibitor, in men with metastatic castration-resistant prostate cancer (mCRPC) progressing after enzalutamide and/or abiraterone. J. Clin. Oncol. 2017, 35, 5032. [Google Scholar] [CrossRef]
- Omlin, A.; Jones, R.J.; van der Noll, R.; Satoh, T.; Niwakawa, M.; Smith, S.A.; Graham, J.; Ong, M.; Finkelman, R.D.; Schellens, J.H.; et al. AZD3514, an oral selective androgen receptor down-regulator in patients with castration-resistant prostate cancer—Results of two parallel first-in-human phase I studies. Investig. New Drugs 2015, 33, 679–690. [Google Scholar] [CrossRef]
- Brand, L.J.; Olson, M.E.; Ravindranathan, P.; Guo, H.; Kempema, A.M.; Andrews, T.E.; Chen, X.; Raj, G.V.; Harki, D.A.; Dehm, S.M. EPI-001 is a selective peroxisome proliferator-activated receptor-gamma modulator with inhibitory effects on androgen receptor expression and activity in prostate cancer. Oncotarget 2015, 6, 3811–3824. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef]
- Shang, Y.; Myers, M.; Brown, M. Formation of the androgen receptor transcription complex. Mol. Cell 2002, 9, 601–610. [Google Scholar] [CrossRef]
- Fronsdal, K.; Engedal, N.; Slagsvold, T.; Saatcioglu, F. CREB binding protein is a coactivator for the androgen receptor and mediates cross-talk with AP-1. J. Biol. Chem. 1998, 273, 31853–31859. [Google Scholar] [CrossRef]
- Louie, M.C.; Yang, H.Q.; Ma, A.H.; Xu, W.; Zou, J.X.; Kung, H.J.; Chen, H.W. Androgen-induced recruitment of RNA polymerase II to a nuclear receptor-p160 coactivator complex. Proc. Natl. Acad. Sci. USA 2003, 100, 2226–2230. [Google Scholar] [CrossRef] [PubMed]
- Ianculescu, I.; Wu, D.Y.; Siegmund, K.D.; Stallcup, M.R. Selective roles for cAMP response element-binding protein binding protein and p300 protein as coregulators for androgen-regulated gene expression in advanced prostate cancer cells. J. Biol. Chem. 2012, 287, 4000–4013. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.M.; Christiaens, V.; Voulgaraki, D.; Waxman, J.; Claessens, F.; Bevan, C.L. Mechanisms of androgen receptor signalling via steroid receptor coactivator-1 in prostate. Endocr. Relat. Cancer 2004, 11, 117–130. [Google Scholar] [CrossRef]
- Estebanez-Perpina, E.; Moore, J.M.; Mar, E.; Delgado-Rodrigues, E.; Nguyen, P.; Baxter, J.D.; Buehrer, B.M.; Webb, P.; Fletterick, R.J.; Guy, R.K. The molecular mechanisms of coactivator utilization in ligand-dependent transactivation by the androgen receptor. J. Biol. Chem. 2005, 280, 8060–8068. [Google Scholar] [CrossRef]
- Heery, D.M.; Kalkhoven, E.; Hoare, S.; Parker, M.G. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 1997, 387, 733–736. [Google Scholar] [CrossRef]
- Sheppard, H.M.; Harries, J.C.; Hussain, S.; Bevan, C.; Heery, D.M. Analysis of the steroid receptor coactivator 1 (SRC1)-CREB binding protein interaction interface and its importance for the function of SRC1. Mol. Cell Biol. 2001, 21, 39–50. [Google Scholar] [CrossRef]
- Kim, M.Y.; Hsiao, S.J.; Kraus, W.L. A role for coactivators and histone acetylation in estrogen receptor alpha-mediated transcription initiation. EMBO J. 2001, 20, 6084–6094. [Google Scholar] [CrossRef]
- York, B.; O’Malley, B.W. Steroid receptor coactivator (SRC) family: Masters of systems biology. J. Biol. Chem. 2010, 285, 38743–38750. [Google Scholar] [CrossRef]
- Baek, S.H.; Ohgi, K.A.; Nelson, C.A.; Welsbie, D.; Chen, C.; Sawyers, C.L.; Rose, D.W.; Rosenfeld, M.G. Ligand-specific allosteric regulation of coactivator functions of androgen receptor in prostate cancer cells. Proc. Natl. Acad. Sci. USA 2006, 103, 3100–3105. [Google Scholar] [CrossRef]
- Li, D.; Tian, G.; Wang, J.; Zhao, L.Y.; Co, O.; Underill, Z.C.; Mymryk, J.S.; Claessens, F.; Dehm, S.M.; Daaka, Y.; et al. Inhibition of androgen receptor transactivation function by adenovirus type 12 E1A undermines prostate cancer cell survival. Prostate 2018. [Google Scholar] [CrossRef] [PubMed]
- Aarnisalo, P.; Palvimo, J.J.; Janne, O.A. CREB-binding protein in androgen receptor-mediated signaling. Proc. Natl. Acad. Sci. USA 1998, 95, 2122–2127. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Carroll, J.S.; Brown, M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol. Cell 2005, 19, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Kumari, S.; Hu, Q.; Senapati, D.; Venkadakrishnan, V.B.; Wang, D.; DePriest, A.D.; Schlanger, S.E.; Ben-Salem, S.; Valenzuela, M.M.; et al. A comprehensive analysis of coregulator recruitment, androgen receptor function and gene expression in prostate cancer. Elife 2017, 6. [Google Scholar] [CrossRef]
- Kamei, Y.; Xu, L.; Heinzel, T.; Torchia, J.; Kurokawa, R.; Gloss, B.; Lin, S.C.; Heyman, R.A.; Rose, D.W.; Glass, C.K.; et al. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell 1996, 85, 403–414. [Google Scholar] [CrossRef]
- Lavery, D.N.; Bevan, C.L. Androgen receptor signalling in prostate cancer: The functional consequences of acetylation. J. Biomed. Biotechnol. 2011, 2011, 862125. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Rao, M.; Wang, C.; Sakamaki, T.; Wang, J.; Di Vizio, D.; Zhang, X.; Albanese, C.; Balk, S.; Chang, C.; et al. Acetylation of androgen receptor enhances coactivator binding and promotes prostate cancer cell growth. Mol. Cell. Biol. 2003, 23, 8563–8575. [Google Scholar] [CrossRef] [PubMed]
- Heemers, H.V.; Sebo, T.J.; Debes, J.D.; Regan, K.M.; Raclaw, K.A.; Murphy, L.M.; Hobisch, A.; Culig, Z.; Tindall, D.J. Androgen deprivation increases p300 expression in prostate cancer cells. Cancer Res. 2007, 67, 3422–3430. [Google Scholar] [CrossRef] [PubMed]
- Comuzzi, B.; Nemes, C.; Schmidt, S.; Jasarevic, Z.; Lodde, M.; Pycha, A.; Bartsch, G.; Offner, F.; Culig, Z.; Hobisch, A. The androgen receptor co-activator CBP is up-regulated following androgen withdrawal and is highly expressed in advanced prostate cancer. J. Pathol. 2004, 204, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Wallner, L.; Dai, J.; Escara-Wilke, J.; Zhang, J.; Yao, Z.; Lu, Y.; Trikha, M.; Nemeth, J.A.; Zaki, M.H.; Keller, E.T. Inhibition of interleukin-6 with CNTO328, an anti-interleukin-6 monoclonal antibody, inhibits conversion of androgen-dependent prostate cancer to an androgen-independent phenotype in orchiectomized mice. Cancer Res. 2006, 66, 3087–3095. [Google Scholar] [CrossRef]
- Zhu, Y.; Luo, J. Regulation of androgen receptor variants in prostate cancer. Asian J. Urol. 2020, 7, 251–257. [Google Scholar] [CrossRef]
- Lu, C.; Luo, J. Decoding the androgen receptor splice variants. Transl. Androl. Urol. 2013, 2, 178–186. [Google Scholar] [CrossRef]
- Wadosky, K.M.; Koochekpour, S. Androgen receptor splice variants and prostate cancer: From bench to bedside. Oncotarget 2017, 8, 18550–18576. [Google Scholar] [CrossRef]
- Luo, J.; Attard, G.; Balk, S.P.; Bevan, C.; Burnstein, K.; Cato, L.; Cherkasov, A.; De Bono, J.S.; Dong, Y.; Gao, A.C.; et al. Role of androgen receptor variants in prostate cancer: Report from the 2017 Mission Androgen Receptor Variants Meeting. Eur. Urol. 2018, 73, 715–723. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Armstrong, A.J.; Dehm, S.M.; Luo, J. Androgen receptor variant-driven prostate cancer: Clinical implications and therapeutic targeting. Prostate Cancer Prostatic Dis. 2016, 19, 231–241. [Google Scholar] [CrossRef]
- Cato, L.; de Tribolet-Hardy, J.; Lee, I.; Rottenberg, J.T.; Coleman, I.; Melchers, D.; Houtman, R.; Xiao, T.; Li, W.; Uo, T.; et al. ARv7 represses tumor-suppressor genes in castration-resistant prostate cancer. Cancer Cell 2019, 35, 401–413.e406. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef]
- Bohrer, L.R.; Liu, P.; Zhong, J.; Pan, Y.; Angstman, J.; Brand, L.J.; Dehm, S.M.; Huang, H. FOXO1 binds to the TAU5 motif and inhibits constitutively active androgen receptor splice variants. Prostate 2013, 73, 1017–1027. [Google Scholar] [CrossRef]
- Heemers, H.V.; Debes, J.D.; Tindall, D.J. The role of the transcriptional coactivator p300 in prostate cancer progression. Adv. Exp. Med. Biol. 2008, 617, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Debes, J.D.; Sebo, T.J.; Lohse, C.M.; Murphy, L.M.; Haugen, D.A.; Tindall, D.J. p300 in prostate cancer proliferation and progression. Cancer Res. 2003, 63, 7638–7640. [Google Scholar] [PubMed]
- Gruber, M.; Ferrone, L.; Puhr, M.; Santer, F.R.; Furlan, T.; Eder, I.E.; Sampson, N.; Schafer, G.; Handle, F.; Culig, Z. p300 is upregulated by docetaxel and is a target in chemoresistant prostate cancer. Endocr. Relat. Cancer 2020, 27, 187–198. [Google Scholar] [CrossRef]
- Comuzzi, B.; Lambrinidis, L.; Rogatsch, H.; Godoy-Tundidor, S.; Knezevic, N.; Krhen, I.; Marekovic, Z.; Bartsch, G.; Klocker, H.; Hobisch, A.; et al. The transcriptional co-activator cAMP response element-binding protein-binding protein is expressed in prostate cancer and enhances androgen- and anti-androgen-induced androgen receptor function. Am. J. Pathol. 2003, 162, 233–241. [Google Scholar] [CrossRef][Green Version]
- Spriano, F.; Gaudio, E.; Cascione, L.; Tarantelli, C.; Melle, F.; Motta, G.; Priebe, V.; Rinaldi, A.; Golino, G.; Mensah, A.A.; et al. Antitumor activity of the dual BET and CBP/EP300 inhibitor NEO2734. Blood Adv. 2020, 4, 4124–4135. [Google Scholar] [CrossRef] [PubMed]
- Giles, F.; Witcher, M.; Brown, B. NEO2734: A novel potent oral dual BET and P300/CBP inhibitor. Ann. Oncol. 2018, 29, viii140–viii141. [Google Scholar] [CrossRef]
- Zhuang, M.; Calabrese, M.F.; Liu, J.; Waddell, M.B.; Nourse, A.; Hammel, M.; Miller, D.J.; Walden, H.; Duda, D.M.; Seyedin, S.N.; et al. Structures of SPOP-substrate complexes: Insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol. Cell 2009, 36, 39–50. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef]
- An, J.; Wang, C.; Deng, Y.; Yu, L.; Huang, H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep. 2014, 6, 657–669. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, D.; Zhao, Y.; Ren, S.; Gao, K.; Ye, Z.; Wang, S.; Pan, C.-W.; Zhu, Y.; Yan, Y.; et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat. Med. 2017, 23, 1055–1062. [Google Scholar] [CrossRef]
- Dai, X.; Gan, W.; Li, X.; Wang, S.; Zhang, W.; Huang, L.; Liu, S.; Zhong, Q.; Guo, J.; Zhang, J.; et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat. Med. 2017, 23, 1063–1071. [Google Scholar] [CrossRef]
- Blattner, M.; Liu, D.; Robinson, B.D.; Huang, D.; Poliakov, A.; Gao, D.; Nataraj, S.; Deonarine, L.D.; Augello, M.A.; Sailer, V.; et al. SPOP mutation drives prostate tumorigenesis in vivo through coordinate regulation of PI3K/mTOR and AR signaling. Cancer Cell 2017, 31, 436–451. [Google Scholar] [CrossRef]
- Groner, A.C.; Cato, L.; de Tribolet-Hardy, J.; Bernasocchi, T.; Janouskova, H.; Melchers, D.; Houtman, R.; Cato, A.C.B.; Tschopp, P.; Gu, L.; et al. TRIM24 is an oncogenic transcriptional activator in prostate cancer. Cancer Cell 2016, 29, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Dai, X.; Lunardi, A.; Li, Z.; Inuzuka, H.; Liu, P.; Varmeh, S.; Zhang, J.; Cheng, L.; Sun, Y.; et al. SPOP promotes ubiquitination and degradation of the ERG oncoprotein to suppress prostate cancer progression. Mol. Cell 2015, 59, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Kaochar, S.; Li, M.; Rajapakshe, K.; Fiskus, W.; Dong, J.; Foley, C.; Dong, B.; Zhang, L.; Kwon, O.J.; et al. SPOP regulates prostate epithelial cell proliferation and promotes ubiquitination and turnover of c-MYC oncoprotein. Oncogene 2017, 36, 4767–4777. [Google Scholar] [CrossRef]
- Pegg, N.; Brooks, N.; Worthington, J.; Young, B.; Prosser, A.; Lane, J.; Taddei, D.; Brown, R.; Harbottle, G.; Shannon, J.; et al. Characterisation of CCS1477: A novel small molecule inhibitor of p300/CBP for the treatment of castration resistant prostate cancer. J. Clin. Oncol. 2017, 35, 11590. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, P.S.; Barker, K.A.; Anderson, W.F. Estrogen receptor status and the future burden of invasive and in situ breast cancers in the United States. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Li, Y.; Yang, D.; Yin, X.; Zhang, X.; Huang, J.; Wu, Y.; Wang, M.; Yi, Z.; Li, H.; Li, H.; et al. Clinicopathological characteristics and breast cancer-specific survival of patients with single hormone receptor-positive breast cancer. JAMA Netw. Open 2020, 3, e1918160. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Sotiriou, C. Luminal breast cancer: From biology to treatment. Nat. Rev. Clin. Oncol. 2013, 10, 494–506. [Google Scholar] [CrossRef]
- Butt, A.J.; McNeil, C.M.; Musgrove, E.A.; Sutherland, R.L. Downstream targets of growth factor and oestrogen signalling and endocrine resistance: The potential roles of c-Myc, cyclin D1 and cyclin E. Endocr. Relat. Cancer 2005, 12 (Suppl. 1), S47–S59. [Google Scholar] [CrossRef]
- Ali, S.; Buluwela, L.; Coombes, R.C. Antiestrogens and their therapeutic applications in breast cancer and other diseases. Annu. Rev. Med. 2011, 62, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C. A century of deciphering the control mechanisms of sex steroid action in breast and prostate cancer: The origins of targeted therapy and chemoprevention. Cancer Res. 2009, 69, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Nahta, R.; Yu, D.; Hung, M.C.; Hortobagyi, G.N.; Esteva, F.J. Mechanisms of disease: Understanding resistance to HER2-targeted therapy in human breast cancer. Nat. Clin. Pract. Oncol. 2006, 3, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Zielinski, C.C. Optimal strategies for the treatment of metastatic triple-negative breast cancer with currently approved agents. Ann. Oncol. 2012, 23 (Suppl. 6), vi46–vi51. [Google Scholar] [CrossRef]
- Hudis, C.A.; Gianni, L. Triple-negative breast cancer: An unmet medical need. Oncologist 2011, 16 (Suppl. 1), 1–11. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Michmerhuizen, A.R.; Spratt, D.E.; Pierce, L.J.; Speers, C.W. ARe we there yet? Understanding androgen receptor signaling in breast cancer. NPJ Breast Cancer 2020, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Caswell-Jin, J.L.; Plevritis, S.K.; Tian, L.; Cadham, C.J.; Xu, C.; Stout, N.K.; Sledge, G.W.; Mandelblatt, J.S.; Kurian, A.W. Change in survival in metastatic breast cancer with treatment advances: Meta-analysis and systematic review. JNCI Cancer Spectr. 2018, 2, pky062. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Droog, M.; Beelen, K.; Linn, S.; Zwart, W. Tamoxifen resistance: From bench to bedside. Eur. J. Pharmacol. 2013, 717, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Yasar, P.; Ayaz, G.; User, S.D.; Gupur, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Stice, J.P.; Knowlton, A.A. Estrogen, NFkappaB, and the heat shock response. Mol. Med. 2008, 14, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Acconcia, F.; Marino, M. The effects of 17beta-estradiol in cancer are mediated by estrogen receptor signaling at the plasma membrane. Front. Physiol. 2011, 2, 30. [Google Scholar] [CrossRef]
- Dubik, D.; Dembinski, T.C.; Shiu, R.P. Stimulation of c-myc oncogene expression associated with estrogen-induced proliferation of human breast cancer cells. Cancer Res. 1987, 47, 6517–6521. [Google Scholar]
- Levin, E.R. Plasma membrane estrogen receptors. Trends Endocrinol. Metab. 2009, 20, 477–482. [Google Scholar] [CrossRef]
- Ali, S.; Rasool, M.; Chaoudhry, H.; Pushparaj, N.P.; Jha, P.; Hafiz, A.; Mahfooz, M.; Abdus Sami, G.; Azhar Kamal, M.; Bashir, S.; et al. Molecular mechanisms and mode of tamoxifen resistance in breast cancer. Bioinformation 2016, 12, 135–139. [Google Scholar] [CrossRef]
- Song, X.; Chen, J.; Zhao, M.; Zhang, C.; Yu, Y.; Lonard, D.M.; Chow, D.C.; Palzkill, T.; Xu, J.; O’Malley, B.W.; et al. Development of potent small-molecule inhibitors to drug the undruggable steroid receptor coactivator-3. Proc. Natl. Acad. Sci. USA 2016, 113, 4970–4975. [Google Scholar] [CrossRef]
- Hanstein, B.; Eckner, R.; DiRenzo, J.; Halachmi, S.; Liu, H.; Searcy, B.; Kurokawa, R.; Brown, M. p300 is a component of an estrogen receptor coactivator complex. Proc. Natl. Acad. Sci. USA 1996, 93, 11540–11545. [Google Scholar] [CrossRef]
- Acevedo, M.L.; Kraus, W.L. Mediator and p300/CBP-steroid receptor coactivator complexes have distinct roles, but function synergistically, during estrogen receptor alpha-dependent transcription with chromatin templates. Mol. Cell. Biol. 2003, 23, 335–348. [Google Scholar] [CrossRef]
- Kraus, W.L.; Kadonaga, J.T. p300 and estrogen receptor cooperatively activate transcription via differential enhancement of initiation and reinitiation. Genes Dev. 1998, 12, 331–342. [Google Scholar] [CrossRef]
- Yi, P.; Wang, Z.; Feng, Q.; Chou, C.K.; Pintilie, G.D.; Shen, H.; Foulds, C.E.; Fan, G.; Serysheva, I.; Ludtke, S.J.; et al. Structural and functional impacts of ER coactivator sequential recruitment. Mol. Cell 2017, 67, 733–743.e734. [Google Scholar] [CrossRef] [PubMed]
- Farcas, A.M.; Nagarajan, S.; Cosulich, S.; Carroll, J.S. Genome-wide estrogen receptor activity in breast cancer. Endocrinology 2021, 162. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Hu, X.; DiRenzo, J.; Lazar, M.A.; Brown, M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 2000, 103, 843–852. [Google Scholar] [CrossRef]
- Theodorou, V.; Stark, R.; Menon, S.; Carroll, J.S. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013, 23, 12–22. [Google Scholar] [CrossRef]
- Guertin, M.J.; Zhang, X.; Coonrod, S.A.; Hager, G.L. Transient estrogen receptor binding and p300 redistribution support a squelching mechanism for estradiol-repressed genes. Mol. Endocrinol. 2014, 28, 1522–1533. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.L.; Onate, S.A.; Tsai, M.J.; O’Malley, B.W. CREB binding protein acts synergistically with steroid receptor coactivator-1 to enhance steroid receptor-dependent transcription. Proc. Natl. Acad. Sci. USA 1996, 93, 8884–8888. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Cordero, K.E.; Larios, J.M.; Miller, R.S.; Johnson, M.D.; Chinnaiyan, A.M.; Lippman, M.E.; Rae, J.M. Genes regulated by estrogen in breast tumor cells in vitro are similarly regulated in vivo in tumor xenografts and human breast tumors. Genome Biol. 2006, 7, R28. [Google Scholar] [CrossRef] [PubMed]
- Frasor, J.; Danes, J.M.; Komm, B.; Chang, K.C.; Lyttle, C.R.; Katzenellenbogen, B.S. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: Insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology 2003, 144, 4562–4574. [Google Scholar] [CrossRef]
- Stossi, F.; Likhite, V.S.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Estrogen-occupied estrogen receptor represses cyclin G2 gene expression and recruits a repressor complex at the cyclin G2 promoter. J. Biol. Chem. 2006, 281, 16272–16278. [Google Scholar] [CrossRef]
- Sankar, N.; Baluchamy, S.; Kadeppagari, R.K.; Singhal, G.; Weitzman, S.; Thimmapaya, B. p300 provides a corepressor function by cooperating with YY1 and HDAC3 to repress c-Myc. Oncogene 2008, 27, 5717–5728. [Google Scholar] [CrossRef]
- Girdwood, D.; Bumpass, D.; Vaughan, O.A.; Thain, A.; Anderson, L.A.; Snowden, A.W.; Garcia-Wilson, E.; Perkins, N.D.; Hay, R.T. p300 transcriptional repression is mediated by SUMO modification. Mol. Cell 2003, 11, 1043–1054. [Google Scholar] [CrossRef]
- Kuo, H.Y.; Chang, C.C.; Jeng, J.C.; Hu, H.M.; Lin, D.Y.; Maul, G.G.; Kwok, R.P.; Shih, H.M. SUMO modification negatively modulates the transcriptional activity of CREB-binding protein via the recruitment of Daxx. Proc. Natl. Acad. Sci. USA 2005, 102, 16973–16978. [Google Scholar] [CrossRef]
- Snowden, A.W.; Anderson, L.A.; Webster, G.A.; Perkins, N.D. A novel transcriptional repression domain mediates p21(WAF1/CIP1) induction of p300 transactivation. Mol. Cell. Biol. 2000, 20, 2676–2686. [Google Scholar] [CrossRef]
- Stossi, F.; Madak-Erdogan, Z.; Katzenellenbogen, B.S. Estrogen receptor alpha represses transcription of early target genes via p300 and CtBP1. Mol. Cell. Biol. 2009, 29, 1749–1759. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, J.H.; Cho, E.J.; Kim, S.T.; Youn, H.D. CtBP represses p300-mediated transcriptional activation by direct association with its bromodomain. Nat. Struct. Mol. Biol. 2005, 12, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Dcona, M.M.; Morris, B.L.; Ellis, K.C.; Grossman, S.R. CtBP- an emerging oncogene and novel small molecule drug target: Advances in the understanding of its oncogenic action and identification of therapeutic inhibitors. Cancer Biol. Ther. 2017, 18, 379–391. [Google Scholar] [CrossRef]
- Zhao, Z.; Hao, D.; Wang, L.; Li, J.; Meng, Y.; Li, P.; Wang, Y.; Zhang, C.; Zhou, H.; Gardner, K.; et al. CtBP promotes metastasis of breast cancer through repressing cholesterol and activating TGF-beta signaling. Oncogene 2019, 38, 2076–2091. [Google Scholar] [CrossRef]
- Byun, J.S.; Park, S.; Yi, D.I.; Shin, J.H.; Hernandez, S.G.; Hewitt, S.M.; Nicklaus, M.C.; Peach, M.L.; Guasch, L.; Tang, B.; et al. Epigenetic re-wiring of breast cancer by pharmacological targeting of C-terminal binding protein. Cell Death Dis. 2019, 10, 689. [Google Scholar] [CrossRef] [PubMed]
- Merenbakh-Lamin, K.; Ben-Baruch, N.; Yeheskel, A.; Dvir, A.; Soussan-Gutman, L.; Jeselsohn, R.; Yelensky, R.; Brown, M.; Miller, V.A.; Sarid, D.; et al. D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013, 73, 6856–6864. [Google Scholar] [CrossRef] [PubMed]
- Fanning, S.W.; Mayne, C.G.; Dharmarajan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S.; et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gomez, H.; et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res. 2014, 20, 1757–1767. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shen, D.; Shao, J.; Crowder, R.; Liu, W.; Prat, A.; He, X.; Liu, S.; Hoog, J.; Lu, C.; et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013, 4, 1116–1130. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov. 2017, 7, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Guttery, D.S.; Page, K.; Hills, A.; Woodley, L.; Marchese, S.D.; Rghebi, B.; Hastings, R.K.; Luo, J.; Pringle, J.H.; Stebbing, J.; et al. Noninvasive detection of activating estrogen receptor 1 (ESR1) mutations in estrogen receptor-positive metastatic breast cancer. Clin. Chem. 2015, 61, 974–982. [Google Scholar] [CrossRef]
- Harrod, A.; Fulton, J.; Nguyen, V.T.M.; Periyasamy, M.; Ramos-Garcia, L.; Lai, C.F.; Metodieva, G.; de Giorgio, A.; Williams, R.L.; Santos, D.B.; et al. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 2017, 36, 2286–2296. [Google Scholar] [CrossRef]
- Weis, K.E.; Ekena, K.; Thomas, J.A.; Lazennec, G.; Katzenellenbogen, B.S. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol. Endocrinol. 1996, 10, 1388–1398. [Google Scholar] [CrossRef][Green Version]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell 2018, 34, 427–438.e426. [Google Scholar] [CrossRef]
- Witte, S.; Bradley, A.; Enright, A.J.; Muljo, S.A. High-density P300 enhancers control cell state transitions. BMC Genom. 2015, 16, 903. [Google Scholar] [CrossRef]
- Yang, H.; Pinello, C.E.; Luo, J.; Li, D.; Wang, Y.; Zhao, L.Y.; Jahn, S.C.; Saldanha, S.A.; Chase, P.; Planck, J.; et al. Small-molecule inhibitors of acetyltransferase p300 identified by high-throughput screening are potent anticancer agents. Mol. Cancer Ther. 2013, 12, 610–620. [Google Scholar] [CrossRef]
- Kono, M.; Fujii, T.; Lim, B.; Karuturi, M.S.; Tripathy, D.; Ueno, N.T. Androgen receptor function and androgen receptor-targeted therapies in breast cancer: A review. JAMA Oncol. 2017, 3, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Giovannelli, P.; Di Donato, M.; Galasso, G.; Di Zazzo, E.; Bilancio, A.; Migliaccio, A. The androgen receptor in breast cancer. Front. Endocrinol. 2018, 9, 492. [Google Scholar] [CrossRef] [PubMed]
- Dieci, M.V.; Tsvetkova, V.; Griguolo, G.; Miglietta, F.; Mantiero, M.; Tasca, G.; Cumerlato, E.; Giorgi, C.A.; Giarratano, T.; Faggioni, G.; et al. Androgen receptor expression and association with distant disease-free survival in triple negative breast cancer: Analysis of 263 patients treated with standard therapy for stage I–III disease. Front. Oncol. 2019, 9, 452. [Google Scholar] [CrossRef] [PubMed]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef]
- Giovannelli, P.; Di Donato, M.; Auricchio, F.; Castoria, G.; Migliaccio, A. Androgens induce invasiveness of triple negative breast cancer cells through AR/Src/PI3-K complex assembly. Sci. Rep. 2019, 9, 4490. [Google Scholar] [CrossRef] [PubMed]
- Barton, V.N.; Christenson, J.L.; Gordon, M.A.; Greene, L.I.; Rogers, T.J.; Butterfield, K.; Babbs, B.; Spoelstra, N.S.; D’Amato, N.C.; Elias, A.; et al. Androgen receptor supports an anchorage-independent, cancer stem cell-like population in triple-negative breast cancer. Cancer Res. 2017, 77, 3455–3466. [Google Scholar] [CrossRef]
- Barton, V.N.; D’Amato, N.C.; Gordon, M.A.; Lind, H.T.; Spoelstra, N.S.; Babbs, B.L.; Heinz, R.E.; Elias, A.; Jedlicka, P.; Jacobsen, B.M.; et al. Multiple molecular subtypes of triple-negative breast cancer critically rely on androgen receptor and respond to enzalutamide in vivo. Mol. Cancer Ther. 2015, 14, 769–778. [Google Scholar] [CrossRef]
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef]
- Garcia-Carpizo, V.; Ruiz-Llorente, S.; Sarmentero, J.; Gonzalez-Corpas, A.; Barrero, M.J. CREBBP/EP300 bromodomain inhibition affects the proliferation of AR-positive breast cancer cell lines. Mol. Cancer Res. 2019, 17, 720–730. [Google Scholar] [CrossRef]
- Yuan, Z.L.; Guan, Y.J.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Erion, D.M.; Yuan, Z.; Dietrich, M.; Shulman, G.I.; Horvath, T.L.; Gao, Q. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat. Cell Biol. 2009, 11, 492–500. [Google Scholar] [CrossRef]
- Lee, H.; Zhang, P.; Herrmann, A.; Yang, C.; Xin, H.; Wang, Z.; Hoon, D.S.; Forman, S.J.; Jove, R.; Riggs, A.D.; et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. USA 2012, 109, 7765–7769. [Google Scholar] [CrossRef]
- Takemaru, K.I.; Moon, R.T. The transcriptional coactivator CBP interacts with beta-catenin to activate gene expression. J. Cell Biol. 2000, 149, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Hecht, A.; Vleminckx, K.; Stemmler, M.P.; van Roy, F.; Kemler, R. The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J. 2000, 19, 1839–1850. [Google Scholar] [CrossRef] [PubMed]
- Levy, L.; Wei, Y.; Labalette, C.; Wu, Y.; Renard, C.A.; Buendia, M.A.; Neuveut, C. Acetylation of beta-catenin by p300 regulates beta-catenin-Tcf4 interaction. Mol. Cell. Biol. 2004, 24, 3404–3414. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Rodova, M.; Miska, E.A.; Calvet, J.P.; Kouzarides, T. Acetylation of beta-catenin by CREB-binding protein (CBP). J. Biol. Chem. 2002, 277, 25562–25567. [Google Scholar] [CrossRef]
- Chocarro-Calvo, A.; Garcia-Martinez, J.M.; Ardila-Gonzalez, S.; De la Vieja, A.; Garcia-Jimenez, C. Glucose-induced beta-catenin acetylation enhances Wnt signaling in cancer. Mol. Cell 2013, 49, 474–486. [Google Scholar] [CrossRef]
- Schade, B.; Lesurf, R.; Sanguin-Gendreau, V.; Bui, T.; Deblois, G.; O’Toole, S.A.; Millar, E.K.; Zardawi, S.J.; Lopez-Knowles, E.; Sutherland, R.L.; et al. beta-Catenin signaling is a critical event in ErbB2-mediated mammary tumor progression. Cancer Res. 2013, 73, 4474–4487. [Google Scholar] [CrossRef] [PubMed]
- Wend, P.; Runke, S.; Wend, K.; Anchondo, B.; Yesayan, M.; Jardon, M.; Hardie, N.; Loddenkemper, C.; Ulasov, I.; Lesniak, M.S.; et al. WNT10B/beta-catenin signalling induces HMGA2 and proliferation in metastatic triple-negative breast cancer. EMBO Mol. Med. 2013, 5, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Smid, M.; Wang, Y.; Zhang, Y.; Sieuwerts, A.M.; Yu, J.; Klijn, J.G.; Foekens, J.A.; Martens, J.W. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008, 68, 3108–3114. [Google Scholar] [CrossRef] [PubMed]
- Emami, K.H.; Nguyen, C.; Ma, H.; Kim, D.H.; Jeong, K.W.; Eguchi, M.; Moon, R.T.; Teo, J.L.; Kim, H.Y.; Moon, S.H.; et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc. Natl. Acad. Sci. USA 2004, 101, 12682–12687. [Google Scholar] [CrossRef]
- Freedman, S.J.; Sun, Z.Y.; Poy, F.; Kung, A.L.; Livingston, D.M.; Wagner, G.; Eck, M.J. Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc. Natl. Acad. Sci. USA 2002, 99, 5367–5372. [Google Scholar] [CrossRef]
- Lu, X.; Kang, Y. Hypoxia and hypoxia-inducible factors: Master regulators of metastasis. Clin. Cancer Res. 2010, 16, 5928–5935. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Vleugel, M.M.; Shvarts, D.; van der Wall, E.; van Diest, P.J. p300 and p53 levels determine activation of HIF-1 downstream targets in invasive breast cancer. Human Pathol. 2006, 37, 1085–1092. [Google Scholar] [CrossRef]
- Armache, A.; Yang, S.; Martínez de Paz, A.; Robbins, L.E.; Durmaz, C.; Cheong, J.Q.; Ravishankar, A.; Daman, A.W.; Ahimovic, D.J.; Klevorn, T.; et al. Histone H3.3 phosphorylation amplifies stimulation-induced transcription. Nature 2020. [Google Scholar] [CrossRef]
- Martire, S.; Gogate, A.A.; Whitmill, A.; Tafessu, A.; Nguyen, J.; Teng, Y.C.; Tastemel, M.; Banaszynski, L.A. Phosphorylation of histone H3.3 at serine 31 promotes p300 activity and enhancer acetylation. Nat. Genet. 2019, 51, 941–946. [Google Scholar] [CrossRef]
- Sitbon, D.; Boyarchuk, E.; Dingli, F.; Loew, D.; Almouzni, G. Histone variant H3.3 residue S31 is essential for Xenopus gastrulation regardless of the deposition pathway. Nat. Commun. 2020, 11, 1256. [Google Scholar] [CrossRef] [PubMed]
- Deevy, O.; Bracken, A.P. PRC2 functions in development and congenital disorders. Development 2019, 146. [Google Scholar] [CrossRef]
- Morgan, M.A.J.; Shilatifard, A. Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, I.; Liao, D. DAXX in cancer: Phenomena, processes, mechanisms and regulation. Nucleic Acids Res. 2019, 47, 7734–7752. [Google Scholar] [CrossRef]
- Gao, Y.; Nihira, N.T.; Bu, X.; Chu, C.; Zhang, J.; Kolodziejczyk, A.; Fan, Y.; Chan, N.T.; Ma, L.; Liu, J.; et al. Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat. Cell Biol. 2020, 22, 1064–1075. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biochemical/Functional Properties | AR | ER |
|---|---|---|
| CBP/p300 binding domain and interaction features | Direct interaction with p300 via AR NTD and AR LBD [50] | Indirectly interacts with p300 via SRC-3. p300 directly binds SRC-3 in a complex with ER and DNA [51] |
| Chromatin co-localization with CBP/p300 | 83% of androgen-induced genes with direct AR binding have overlapping p300 binding [52] | 56% (p300) [51] and 38% (CBP) [53] of binding sites are shared with ER. 69% of ER binding sites are co-occupied by CBP and p300 [53] |
| Protein stability | CBP/p300 acetylates AR and enhances AR stability [11] | CBP/p300 acetylates ER and enhances ER stability [54] |
| Tumorigenesis | CBP/p300 inhibitors downregulate AR target gene expression and inhibit PCa growth [28,52] | CBP/p300 inhibitors suppress expression of estrogen-regulated genes and block ER+ BC proliferation in vitro [55] |
| HAT Inhibitors | In Vitro Potency (IC50, nM) | Cellular Activity | In Vivo Effects |
|---|---|---|---|
| A-485 | 9.8 (p300); 2.6 (CBP) [28] | Reduces H3K27ac and H3K18ac [22,28,55]; suppresses enhancer H3K27ac; represses AR/ER target genes [28,55,85] | Blocks CRPC/MM xenograft growth [28,85]; enhances antitumor efficacy of anti-PD-L1 antibody [86] |
| iP300w | n/a | Reduces H3K27ac and H3K18ac [87] | Alters DUX4 target gene expression [87] |
| B026 | 1.8 (p300); 9.5 (CBP) [88] | Reduces H3K27ac; inhibits MYC expression; inhibits growth AR+ PCa cell lines [88] | Inhibits tumor growth of MV-4-11 AML xenografts [88] |
| 12 | 620 (p300) and 1200 (CBP) [89] | Reduces H3K27ac, H3K18ac and H3K9ac; suppresses growth of MCF-7 and other cancer cell lines [89] | n/a |
| 21 | 11 (p300) [90] | Reduces H3K27ac with an EC50 of 4 nM; inhibits LNCaP proliferation with an EC50 of 17 nM [90] | n/a |
| BD inhibitors | |||
| GNE-049 | 2.3 (p300); 1.1 (CBP) [52] | Reduces H3K27ac at enhancers [23] and represses AR/ER target genes [52,55] | Blocks CRPC PDX growth and AR target gene expression [52] |
| GNE-781 | 1.2 (p300); 0.9 (CBP) [91] | Reduces MYC expression [91] | Blocks AML xenograft growth [91] |
| CCS1477 | 19 (p300) [92] | Represses AR target genes [92] | Blocks CRPC xenograft and PDX growth; in clinical trial [92] |
| CBP/p300 PROTAC | |||
| dCBP-1 | n/a | Causes proteasomal degradation of CBP and p300; reduces enhancer H3K27ac and chromatin accessibility; downregulates MYC; suppresses in vitro proliferation of MM cells [93] | n/a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waddell, A.R.; Huang, H.; Liao, D. CBP/p300: Critical Co-Activators for Nuclear Steroid Hormone Receptors and Emerging Therapeutic Targets in Prostate and Breast Cancers. Cancers 2021, 13, 2872. https://doi.org/10.3390/cancers13122872
Waddell AR, Huang H, Liao D. CBP/p300: Critical Co-Activators for Nuclear Steroid Hormone Receptors and Emerging Therapeutic Targets in Prostate and Breast Cancers. Cancers. 2021; 13(12):2872. https://doi.org/10.3390/cancers13122872
Chicago/Turabian StyleWaddell, Aaron R., Haojie Huang, and Daiqing Liao. 2021. "CBP/p300: Critical Co-Activators for Nuclear Steroid Hormone Receptors and Emerging Therapeutic Targets in Prostate and Breast Cancers" Cancers 13, no. 12: 2872. https://doi.org/10.3390/cancers13122872
APA StyleWaddell, A. R., Huang, H., & Liao, D. (2021). CBP/p300: Critical Co-Activators for Nuclear Steroid Hormone Receptors and Emerging Therapeutic Targets in Prostate and Breast Cancers. Cancers, 13(12), 2872. https://doi.org/10.3390/cancers13122872