Simple Summary
The assessment of risk of recurrence following surgery in patients with localized colorectal cancer (CRC) is crucial to indicate systemic adjuvant therapy. The presence of circulating tumor (ct)DNA in the plasma of patients after treatment with curative intent has recently been defined as minimal residual disease (MRD). Detection of MRD is a powerful prognostic biomarker which reflects the presence of micrometastasis and can potentially guide the need of systemic treatment before becoming clinically evident. The aim of this review was to highlight and explore the current situation of MRD detection in CRC cancer and its potential impact in routine clinical practice.
Abstract
Currently, the standard treatment for patients with localized colorectal cancer (CRC) includes surgical resection followed by adjuvant chemotherapy based on clinicopathological features. Recurrence risk stratification in those patients is of utmost importance to guide clinicians to avoid both under- and overtreatment. Recently, the concept of minimal residual disease (MRD) has emerged as the detection of circulating tumor DNA (ctDNA) carrying tumor-specific genomic or epigenomic alterations in the bloodstream of patients after surgery. Emerging studies described how the detection of MRD is a powerful prognostic biomarker to identify patients at higher risk of recurrence and who will potentially benefit the most from a systemic adjuvant treatment. Based on that unprecedented finding, several clinical trials involving stage II and III CRC patients are ongoing evaluating the impact of ctDNA guided treatment by escalating or deescalating adjuvant chemotherapy based on ctDNA MRD detection. This review provides a critical overview of current perspectives of liquid biopsy in early-stage CRC including technical, biological, and clinical key points, as well as ongoing ctDNA-based clinical trials that ultimately aim to improve clinical outcomes of patients with CRC.
1. Introduction
Current Perspectives of Liquid Biopsy in Colorectal Cancer (CRC)
Colorectal cancer (CRC) is the third most frequent cancer in men and the second in women. The prognosis depends not only on the stage at diagnosis, but also the surgical alternatives and the systemic treatment received. Due to the implementation of screening programs, the introduction of novel systemic therapies, and the advanced surgical procedures, the oncological outcomes have dramatically improved in the last years. However, CRC is still the second leading cause of cancer-related mortality worldwide [1].
In patients with localized CC, the assessment of risk of recurrence following surgery is a crucial point to indicate systemic adjuvant therapy. This assessment is based on tumor TNM staging and other clinicopathological characteristics including CEA status or lymphovascular and perineural invasion [2]. Currently, standard fluoropyrimidine-based therapies estimate an increase in overall survival of approximately 5% in stage II patients and 20% in stage III patients when adding oxaliplatin [3,4]. However, at least 50% of patients with stages I, II, and III treated with curative-intent surgery receives unnecessary adjuvant chemotherapy [5,6]. On the other hand, there is a small proportion of patients with stages I and II who do not receive adjuvant treatment and recur [5,6].
Treatment paradigm is slightly different in locally advanced rectal cancer (LARC). Patients with clinical stage T3/4 or node-positive tumors are treated with neoadjuvant chemo-radiotherapy (CRT) or total neoadjuvant treatment (TNT) [7,8] followed by surgery. For those patients who received neoadjuvant CRT, the role of adjuvant chemotherapy is controversial, and FOLFOX only demonstrated benefit in stage III pathological tumors [9,10].
Moreover, it is essential to consider that these treatments are not exempt from adverse effects, such as digestive toxicity or palmoplantar erythrodysesthesia which can be fatal in a small subset of patients carrying a DPYD polymorphism [11], or peripheral neuropathy caused by oxaliplatin that limits day-to-day activity and is permanent in at least 10% of patients [12,13].
Thus, determining the presence or absence of minimal residual disease (MRD) is of utmost importance to guide clinicians to avoid both under- and overtreatment in the adjuvant setting. Circulating cell-free DNA (cfDNA) is a highly fragmented DNA mainly derived from apoptotic cells, predominantly apoptotic leukocytes, found in the blood. cfDNA concentrations in healthy individuals range between 1 and 10 ng mL−1 in plasma [14,15]. Circulating tumor DNA (ctDNA) is a fraction of cfDNA characterized by the presence of tumor-specific genomic alterations with a half-life of a few hours allowing for a real-time, non-invasive characterization of the tumor molecular profile.
ctDNA allows for a better and less aggressive characterization of the spatial tumor molecular heterogeneity and its temporal evolution compared to the traditional use of tissue tumor biopsy. ctDNA, although generally obtained from peripheral blood, can also be isolated from other fluids such as urine, saliva, or cerebrospinal fluid [16,17,18,19,20]. In the metastatic setting, a high concordance between detection of mutations in tissue tumor compared to ctDNA has been reported [21,22,23]. However, ctDNA represents between 0.005% and 11.7% of the whole cfDNA shed in the bloodstream [24] depending on the tumor size, tumor growth rate, and cell turnover [25]. During the early stages of cancer, the total amount of ctDNA might be <1% of the total cfDNA concentration according to some studies [24,26,27]. These extremely low concentrations of ctDNA have been one of the main challenges of liquid biopsies in early-stage disease.
Recently, there have been several advances in the development of new technologies for the detection of ctDNA. Current techniques allow for the detection of genomic molecular alterations (including point mutations, short insertions and deletions, copy number alterations, and fusions), as well as epigenomic changes (i.e., methylation) and cfDNA fragmentation pattern identification. The increased sensitivity in the detection of these biomarkers is crucial for an accurate detection of MRD in CRC cancer patients.
Several ongoing clinical trials are evaluating the use of ctDNA to guide adjuvant therapeutic strategies in CRC patients after curative-intent surgery, and will ultimately shed light onto whether ctDNA is a useful tool to improve outcomes of localized CRC patients. This review highlights and explores the current situation on this topic and its potential application in routine clinical practice.
2. Technical Approaches in Detecting MRD
The molecular landscape of colorectal cancer has been well characterized in the past decades, including chromosomal aberrations such as copy numbers alterations (CNAs), inversions, translocations, insertions, and deletions, as well as single nucleotide point mutations [28]. Epigenomics, referring to covalent modifications of DNA that result in a change in its function or in the regulation of the affected genes without altering the primary sequence, has also been well described in colorectal cancer. These molecular alterations are highly specific to cancer, and, thus, their detection in an individual’s blood potentially indicates the presence of cancer. The extremely low concentration of ctDNA (approximately 0.01% of total cfDNA) especially in early stages of the disease makes its detection challenging [29,30,31].
Currently, two main strategies are used to study tumor genomic material in ctDNA for MRD detection after curative-intent surgery. On the one hand, techniques based on the detection of one or several mutations previously found in the primary tissue tumor. These techniques include sensitive qPCR based methods, such as ARMS or COLD-PCR as well as digital PCR-based methods, such as ddPCR or BEAMing among others [32]. This strategy has the main disadvantage of needing detailed information of the primary tumor (mutation needs to be known ahead of time) and that only a limited number of known mutations can be tracked in the blood (although multiplexing assay is possible). However, it is a very sensitive (0.01%) and specific technique in addition to being rapid and cost-effective [33,34,35].
The second strategy, the so-called deep next-generation sequencing (NGS), allows for the detection of multiple genetic alterations in one sample. NGS conducts a non-directed scan by analyzing the entire genome to detect CNAs or point mutations through whole genome sequencing (WGS) or exome sequencing (WES) [36]. The technique can be PCR or capture-based which allows for the reading of short fragments (around 150 bp). NGS has a high false discovery rate that requires pre-sequencing barcoding and post-sequencing bioinformatics for error suppression. In order to obtain a high sensitivity (0.1%), it is necessary to perform DNA barcodes or UMIs (unique molecular identifiers), initial DNA molecules with 10–12 random bases, such that the barcode is amplified and sequenced together with the DNA. Thus, if we find an alteration, and it is present in all the sequences of the same patient with the same UMI, it will be considered a real alteration, whereas if it is only found in one of the sequences it will be considered as a sequencing failure (Figure 1) [37,38,39].
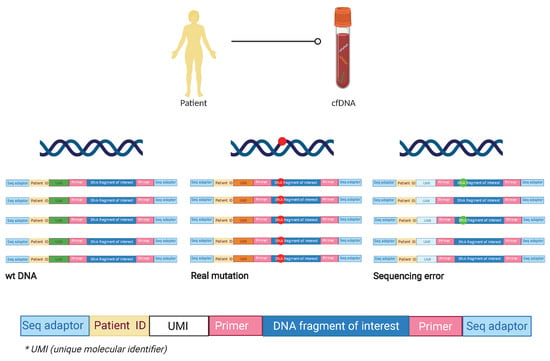
Figure 1.
Preparation of sequencing libraries using ID barcodes and unique molecular identifiers (UMI). After the area of interest is selected, the ID barcodes specific to each patient are added to enable to analyze multiple patients in the same assay. After that, UMIs are added to each molecule during library preparation so that sequencing reads originating from the same starting molecule of each patient included in the library can be identified. This approach increases the assay sensitivity in terms of detecting sequencing errors. In the right sequence corresponding to the blue UMI, the alteration is objectified in only 2 of the amplifications, so it is assumed a sequencing error. In the middle sequence corresponding to the orange UMI, the mutation is found in all the amplifications so it is assumed a real mutation. In the left sequence, there is no mutation founded in any of the reads, so it is a wt ctDNA molecule. cfDNA: cell free DNA; wt: wild type; ID: identification; * UMI: unique molecular identifier.
NGS has several advantages, including the ability to detect molecular alterations that emerge during treatment, and the fact that it is not necessary to have molecular information on the primary tumor. Potential limitations include the requirement for higher amounts of cfDNA [40] (20–50 ng of cfDNA) to decrease the presence of false negative results. Another challenge is the process known as clonal hematopoiesis (CH). Healthy individuals, with age, acquire new mutations in hematopoietic cells [41] and, when lysed, can release cfDNA carrying somatic mutations. This can be incorrectly interpreted as the presence of ctDNA from tumor cells. Razavi et al. [42] sought to develop a protocol for the distinction between mutations of tumor origin and those of hematological origin. They analyzed 124 patients with different types of metastatic tumor and in all cases parallelly sequenced a primary tumor tissue sample, cfDNA, and white blood cells. The authors found that half of the mutations identified in cfDNA of cancer patients were originated in the hematological compartment as a result of clonal hematopoiesis and were therefore not derived from the primary tumor. This study shows the need to sequence the leukocyte fraction in order to discriminate the origin of the mutation detected in cfDNA. All of this is of utmost relevance in the detection of MRD and in cancer screening in which a false positive result may have important consequences.
Currently, new strategies are being studied to increase the sensitivity of diagnostic techniques. These include the analysis of methylation tumor profile via nucleosomal positioning or epigenomic alterations at transcription factor binding sites [43,44]. Methylation patterns that inhibit gene expression are related to the tissue to which they originally belong and therefore reflect the origin of circulating DNA. When a gene is methylated, it does so in various positions of the promoter, thus increasing the opportunity to detect these regions and thus increasing the sensitivity of the technique. This approach has been recently explored by detecting ctDNA using two methylation markers (WIFI and NPY) by ddPCR in CRC patients [45,46].
New NGS strategies are arising by combining different methods to improve ctDNA detection. Such is the case of LUNAR-1 technology (Guardant RevealTM Guardant Health), which integrates assessment of somatic alterations with an epigenomic cancer signature without a priori knowledge of tumor mutation. Sequencing data files are analyzed using a proprietary bioinformatics pipeline software to exclude common sources of interference such as CH of indeterminate potential [47]. Novel NGS assays are increasing the sensitivity to detect ctDNA in MRD. However, the need of bioinformatics analysis to exclude eventual false negative results increases the complexity and subsequently its costs and turnaround times.
Regarding fragmentation, it is well described that the fragmentation cut-off points of circulating DNA are not random and are different for ctDNA compared to cfDNA from normal cells. While the mechanisms of this different fragmentation pattern are yet unknown, they help to characterize whether cfDNA is of tumoral origin or not (Figure 2).
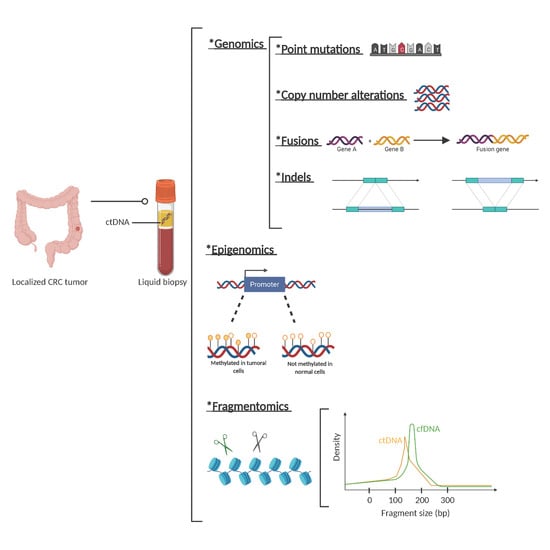
Figure 2.
Circulating tumor (ct)DNA features. Description of genetic, epigenetic, and fragmentomic alterations that can be found in plasma cell free DNA analysis. *: The asterisks are placed as a way of listing the different techniques.
Emerging data suggest that elucidating nucleosome positioning opens promising new perspectives to identify the tissue source of origin of cancer from cfDNA, with an important clinical value to classify cancers and, to a further extent, to characterize, for example, cancers of unknown origin [48].
Single biomarkers in liquid biopsy often do not accurately predict disease status due to heterogeneity between individuals. To address this challenge, investigators are combining multiplexed measurements of different biomarkers that together define robust signatures for specific disease states. Machine learning is a useful tool to automatically discover and detect these signatures, especially as new technologies output increasing quantities of molecular data. Machine learning approaches have been proposed to classify the cell of origin based on somatic mutation profiles in the genome of solid tissue biopsies. Therefore, it is crucial to investigate the applicability of sparse somatic mutation profiles in the identification of ‘cell of origin’ and explore potential improvements of the data analysis and prediction models to overcome sparsity [49].
With only few years, liquid biopsy has rapidly evolved from detecting point mutations in known given genes, to large genome sequencing and the detection of methylations and fragmentomics. This dramatic change has allowed us to improve ctDNA detection sensitivity and even avoid tissue information and carry out molecular studies only on liquid biopsy. However, this entails an increase in the complexity of the analysis and therefore in cost and time to results. Despite these technical improvements, liquid biopsies still have some biological limitations. In the metastatic CRC setting, the discordance in the finding of mutations between tissue and plasma has been shown to be related to the location of the metastasis (pulmonary or peritoneal) and the histology of the tumor (predominantly mucinous) [22,50,51].
3. ctDNA Detection in Localized CRC
The presence of ctDNA after curative-intent surgery in patients with localized disease has been very consistently associated with a high risk of recurrence in different tumor types [52,53,54,55]. Several observational studies have shown that ctDNA detects MRD and is associated with recurrence in patients with localized CRC that have undergone surgery of the primary tumor. In these studies, ctDNA detection typically precedes the appearance of both clinical and radiological recurrence by an average of 3–5 months [56,57,58,59]. In addition, ctDNA has been shown to also be a useful tool for patients with metastatic CRC receiving multi-therapy with curative intention [24]. Interestingly, subjects with detectable ctDNA after surgery generally relapsed within 1 year, while patients who completely negativized ctDNA after surgery have a much lower probability of recurrence and eventual relapse tends to occur later in time.
3.1. Stage II Colon Cancer
Up to 25% of newly diagnosed CRC cases are stage II. Data from clinical trials show that up to 80% of patients who receive surgery with curative intent do not recur [60] and are therefore cured. While TNM staging remains the most relevant criteria for risk assessment after surgery, in stage II CC other clinicopathological parameters need consideration to decide the need for adjuvant chemotherapy [2]. Major prognostic factors include the pT4 stage and perforation and lymph nodes sampling < 12. Minor prognostic parameters for stage II risk assessment include high histological grade, lymphatic, vascular, or perineural invasion, tumor presentation with obstruction or high preoperative CEA levels [2,61,62]. MMR/MSI status is also a validated prognostic marker. MSI/MMR status defines, in stage II CC, a subgroup of patients with a better prognosis and less expected benefit from chemotherapy [63,64,65]. Adjuvant systemic therapy is recommended in stage II CC with at least one risk factor. However, a survival benefit of adjuvant chemotherapy in high-risk stage II CC has not been conclusively demonstrated [66,67]. Identification of an accurate prognostic biomarker would help for a better selection of patients that benefit from adjuvant systemic therapy, and spare chemotherapy in cured patients.
With the aim to use ctDNA as a prognostic biomarker, Tie et al. [56] studied the impact of post-operative ctDNA detection in stage II colon cancer patients.
A total of 250 patients with stage II CC were included, in which a blood sample with ctDNA and CEA analysis at 4–10 weeks post-surgery (post-IQ) and subsequently every 3 months for 2 years was performed. Patients were given chemotherapy at the investigator’s criteria and a clinical follow-up was performed every 3 months including CT imaging tests every 6 months for 2 years. During the follow-up, 34 patients out of 230 (14.8%) had radiological recurrence, including 27 of 178 (15%) patients not treated with chemotherapy and 7 of 52 (13%) patients treated with adjuvant chemotherapy.
Seventy-eight percent (11 out of 14 patients) of patients who did not receive adjuvant treatment and had post-surgery positive ctDNA had radiological recurrence, whereas only 16 out of 164 (9.8%) ctDNA negative patients recurred. CEA was elevated in 7.4% of recurrent patients (2 of 27 cases) and none of the patients with positive ctDNA had an increase in CEA after surgery. Recurrence-free survival (RFS) was estimated to be 0% at 3 years in patients with post-surgery positive ctDNA, compared to 90% in patients with post-surgery negative ctDNA (HR 18; 95% CI 7.9 to 40; p = 2.6 × 10−12). Post-surgery ctDNA was found to be the most significant prognostic independent factor associated with RFS. Finally, it was observed that the time between the detection of ctDNA in peripheral blood and radiological recurrence was on average 167 days, much longer than in the case of CEA which was 61 days.
The use of ctDNA was shown to be superior to the currently used clinicopathological risk parameters to decide on adjuvant treatment. On the other hand, the probability of recurrence was very low for patients with ctDNA negative, defining a subset of patients that could avoid adjuvant chemotherapy and potential associated toxicities.
Combining the use of ctDNA with imaging tests in the follow-up of these patients may help in the early detection of disease recurrence and potentially ultimately lead to better results in terms of survival [68,69].
3.2. Stage III Colon Cancer
Randomized phase III clinical trials have shown that the use of adjuvant chemotherapy with oxaliplatin plus a fluoropyrimidine (FOLFOX or XELOX regimens) improves the overall survival of patients with stage III CC [70]. Most of these patients receive chemotherapy, although up to 50% of them are cured by surgery, and chemotherapy could be potentially be spared [71,72]. Among patients receiving chemotherapy, approximately 30% will have recurrence of the disease and are potential candidates for other systemic treatments [73,74].
Retrospective subgroup analysis of large randomized clinical trials supported a different duration of adjuvant FOLFOX/XELOX treatment depending on pathological risk factors. Thus, in patients considered to be at low risk (≤T3 and N1) the duration of the adjuvant treatment of 3 months is considered appropriate, whereas in the case of patients with high-risk tumors (T4 and/or N2), the duration of treatment is recommended to be extended to 6 months [75].
Reinert and colleagues [76] carried out a prospective and observational study analyzing ctDNA as a biomarker in patients with stage I to III CRC (mostly stage III) with the aim of demonstrating that the presence of post-surgery ctDNA is related to a high probability of recurrence. The study enrolled 130 patients and plasma samples were collected before surgery, after 30 days, and then every 3 months up to 3 years. The recurrence rate was 70% (7 out of 10 patients) in patients with positive ctDNA after surgery, compared to 11.9% (10 patients out of 84) for patients with no ctDNA detection. After surgery, patients with ctDNA were 7 times more likely to relapse than ctDNA-negative patients (HR 7.2; 95% CI 2.7–19.0; p < 0.001). After a multivariate analysis in which known clinical-pathological risk factors, such as staging and lymphovascular invasion, were included, the detection of ctDNA was the only statistically significant prognostic factor associated with RFS.
In line with the aforementioned, the Australian team led by Tie [77] has published the results of a multicentric clinical trial in which 100 patients with a diagnosis of CRC stage III with the provision of administering adjuvant treatment with chemotherapy for 6 months were consecutively recruited. Samples were collected from the primary tumor and later peripheral blood samples for ctDNA determination 4–10 weeks post-surgery and later after completion of adjuvant treatment. The aim of the study was to find whether the determination of ctDNA post-surgery and after completion of adjuvant chemotherapy treatment may give information on minimal residual disease, the efficacy of adjuvant treatment, and recurrence in patients with stage III CRC.
ctDNA was detected in 20 out of 96 (21%) post-surgery patients and was associated with a decrease in RFS (HR 3.8; 95% CI 2.4–21.0; p < 0.001). On the other hand, ctDNA was detected in 10 out of 66 patients (15%) after finishing treatment with adjuvant chemotherapy. Detection of ctDNA after completion of adjuvant treatment was significantly associated with recurrence free interval (RFI). RFI at 3 years was 30% in patients with detectable ctDNA and 77% in patients with negative ctDNA after chemotherapy (HR 6.8; 95% CI, 11.0–157.0; p < 0.001). ctDNA status was the prognostic factor most strongly associated with RFI.
Recently, the same group published a pooled analysis of three cohort studies including 485 stage II–III CRC patients with long term follow-up of 5-years after-surgery ctDNA collection [78]. The authors describe the association of post-surgery ctDNA detection and higher risk of recurrence (38.6% vs. 85.5%; p < 0.001) and poorer OS (64.6% vs. 89.4%; p < 0.001). Furthermore, post-surgery ctDNA status was more accurate in predicting recurrence than individual clinical-pathological risk features such as tumor differentiation, T stage, N stage, lymphovascular invasion, and post-surgery CEA.
All published studies are consistent regarding the clinical impact of ctDNA to detect MRD in stage III CC, not only in identifying patients at high risk of recurrence, but also guiding clinical trials to explore new adjuvant approaches for patients with detectable ctDNA after surgical resection.
Table 1 summarizes the main published studies assessing ctDNA prognostic role in localized CC.

Table 1.
ctDNA as prognostic marker in early-stage colorectal cancer.
3.3. Locally Advanced Rectal Cancer (LARC)
As previously mentioned, CRC is a health problem of global importance. Among patients diagnosed with CRC, 30% have a rectal location [1]. Patients with high-risk LARC (defined by either clinical stage T3/4 or node-positive disease) are usually treated by neoadjuvant CRT or TNT followed by total mesorectal excision (TME) surgery (a mutilating procedure with significant alteration in the quality of life of patients). Moreover, the main site of recurrence in LARC patients is not local but distant metastasis which causes greater morbidity and mortality [10].
Currently, the main prognostic marker in LARC patients is pathological complete response (pCR) as assessed in the rectal surgical specimen after neoadjuvant treatment [81,82].
The need to avoid such mutilating surgery with the associated morbidity has led to an increased interest in the search for prognostic factors to select candidate patients for organ preservation [83]. Currently, watch-and-wait strategies are a possibility in patients with clinical and radiological complete response after TNT. In this setting, the use of ctDNA may potentially help in better identifying patients that are cured after total neoadjuvant therapy and that are potential candidates for a watch-and-wait strategy.
The Australian team led by Tie [84] published a prospective multicenter study that included 159 patients with LARC treated with neoadjuvant CRT followed by TME. Samples were collected at baseline, after completing CRT and 4–10 weeks after surgery. ctDNA was detectable in 77%, 8.3%, and 12% of pretreatment, post-CRT, and post-surgery plasma samples, respectively. Patients with detectable ctDNA after CRT or after surgery had a significantly worse RFS irrespective of receiving or not adjuvant CT (HR 6.6; 95% CI 2.6–17; p < 0.001 and HR 13.0; 95% CI 5.5–31; p < 0.001) respectively. No association was found between post-CRT ctDNA status and pCR.
In the same year, Khakoo and colleagues [85] investigated the use of ctDNA combined with MRI as an early indicator of response in 47 patients with LARC treated with neoadjuvant CRT followed by TME. Blood samples for ctDNA detection were obtained before treatment, during CRT, after completion of CRT and after-surgery. Metastatic disease was observed in 70% of patients with positive ctDNA after completing CRT and in 100% of patients with post-surgery positive ctDNA. Furthermore, metastasis-free survival (MFS) was significantly lower in patients with persistent ctDNA after completing CRT compared with patients with undetectable or non-persistent ctDNA (HR 7.1 95% CI 2.4–21.5, p < 0.001). However, no correlation was found between post-CRT ctDNA and pCR rate.
Accordingly, Zhou et al. [86] recently published a prospective multicentric study with the aim of analyzing the value of ctDNA in predicting response to neoadjuvant CRT. ctDNA from 104 patients was extracted and analyzed by NGS at four time points: baseline, during neoadjuvant CRT treatment, before surgery, and after surgery. With a median follow-up of 18.8 months, 12.5% of patients developed distant metastases. Positive ctDNA at all four time points was associated with decreased MFS. Moreover, variant allele frequency (VAF) of baseline ctDNA mutations was found to be a significant independent predictor of MFS (HR, 1.27; p < 0.001).
Murahashi et al. [87] studied plasma at baseline and after CRT from 85 patients and found that variations in ctDNA was an independent predictor of complete response to preoperative therapy (p = 0.0276).
Similarly, Pazdirek et al. [88] investigated changes in ctDNA levels of 36 patients with LARC undergoing neoadjuvant CRT and their relationship to treatment response. Positive ctDNA at baseline was associated with lower DFS and OS at 1.47 and 1.41 years respectively (p = 0.015 and p = 0.010, respectively).
Table 2 summarizes the main published studies assessing ctDNA prognostic role in LARC.
Table 2.
ctDNA as prognostic marker in locally advanced rectal cancer.
In contrast to the previously described studies, Vidal et al. [89] have recently published a study in which the concept of MMD (minimal metastatic disease) was introduced. This term refers to the detection of ctDNA following TNT and before surgery in patients that are likely to recur at distant sites. Plasma samples were collected at baseline and after TNT within 48 h before surgery (pre-surgery). Patients with pre-surgery positive ctDNA had an increased risk of recurrence compared to patients with negative ctDNA (HR 4.029; 95% CI, 1.004–16.16 p = 0.033) and a marked reduced survival (HR 23; 95% CI, 2.4–212 p < 0.0001). Interestingly, ctDNA was able to predict distant recurrence involving the liver more than metastasis to the peritoneum only or the lung only. In line with previous works in LARC, no correlation was found between pre-surgery ctDNA and pCR.
Globally, in LARC there are three timepoints where ctDNA has been mostly interrogated: baseline, after neoadjuvant treatment, and post-surgery. Data are inconclusive regarding the utility of baseline ctDNA. The prognostic role of post-surgery ctDNA is consistent with previous studies in CC and confirms the value of ctDNA to detect MRD. This finding is important because the clinical benefit of adjuvant therapy in patients with rectal cancer receiving neoadjuvant CRT has not yet been established, and adjuvant clinical trials based on post-operative ctDNA may help to answer this crucial question. Finally, pre-surgery ctDNA analysis after neoadjuvant treatment (either CRT or TNT) detects minimal metastatic disease (MMD), systemic recurrence and death. Prospective clinical trials are needed to validate these finding by assessing systemic treatment intensification in pre-surgery ctDNA positive patients. Interestingly, no association between ctDNA and pCR rates was found, potentially limiting the use of ctDNA alone to select for an organ-preserving approach.
4. ctDNA as a Post-Treatment Surveillance Strategy
Routine follow-up up of localized CRC includes clinical examination, serial CT-scan, blood tests, and CEA. However, when recurrence is detected, it is usually too late to give a curative treatment. ctDNA may potentially monitor adjuvant chemotherapy efficacy or detect resistance after treatment before clinical or radiological evidence of relapse.
In the study by Tie et al. [56] in patients with CRC stage II, assessment of ctDNA at the end of adjuvant therapy showed that a positive result was predictive of disease recurrence (HR 11, 95% CI 1.8–68). Similarly, in the study by Reinert et al. [76], ctDNA levels at the end of CT were correlated with recurrence in patients with CRC stages I–III. In this study, ctDNA after adjuvant chemotherapy was associated with disease recurrence with a sensitivity of 88% and specificity of 98%, whereas CEA had a sensitivity of 69% and specificity of 64%. ctDNA was analyzed every 3 months during follow-up. During surveillance, ctDNA-positive patients were 43 times more likely to experience disease recurrence than ctDNA-negative patients (HR 43.5; 95% CI 9.8–193.5; p < 0.001). The time from detection of ctDNA to detection of recurrence by imaging test was 8.7 months, as opposed to CEA, which rose at the same time as imaging recurrence was detected. At the time of radiological recurrence, an increase in the VAF of ctDNA from all patients, up to 300-fold, was also observed. Tarazona et al. [79] also monitored ctDNA after adjuvant treatment every 4 months for up to 5 years. ctDNA positivity after completion of CT was associated with poorer DFS (HR 10.02; CI 9.202–307.3; p < 0.0001) and detection of ctDNA preceded radiological relapse by a median of 11.5 months.
Interestingly, Reinert et al. [76] investigated whether ctDNA detects the presence of driver mutations to potentially guide administration of targeted personalized treatment in patients with detectable ctDNA after adjuvant therapy. Driver mutations were detected in 81.8% of the post-adjuvant ctDNA positive patients, concluding that ctDNA may serve as a biomarker to detect actionable mutations.
The detection of ctDNA, after the completion of adjuvant treatment, identifies patients that are refractory to standard adjuvant therapy and have poorer prognosis. This reflects the presence of molecular metastatic disease that is not yet identified in imaging tests. Compared to other currently used tests, such as CEA or imaging tests (i.e., computed tomography (CT)), ctDNA monitoring has shown an earlier detection of recurrence (median 9 months) [90,91]. Future studies should aim to determine whether a switch in chemotherapy regimen or administration of targeted therapy at the time of ctDNA detection ultimately impacts the clinical outcome of patients.
5. ctDNA Based Clinical Trials
Clinical implementation of liquid biopsies in localized CRC need prospective randomized clinical trials that show a meaningful impact in clinical outcome in the use of ctDNA to guide adjuvant treatment. Different study designs need to evaluate different clinical uses of ctDNA, including [92]: (a) ctDNA as a tool to detect MRD after curative-intent surgery and guide personalization of adjuvant therapy (intensification versus watch and wait strategy), (b) serial longitudinal ctDNA extractions to monitor and detect early recurrence of the disease (molecular relapse (MR)), (c) ctDNA to guide systemic therapy (i.e., chemotherapy, targeted therapy) in ctDNA positive patients after completion of adjuvant standard therapy (molecular metastatic disease) (Figure 3). Of note, the best time frame for the extraction of ctDNA samples (both in the search for MRD and for MR) has not been established yet.

Figure 3.
ctDNA detection of Minimal residual disease in colorectal cancer. (a) Design proposal for adjuvant clinical trials based on ctDNA analysis to detect MRD after surgery in localized colorectal cancer. (b) Graphical representation of early detection of MRD thanks to ctDNA compared to commonly surveillance used methods (CT scan, CEA). Sx: surgery; CRC: colorectal cancer; dPCR: digital PCR; NGS: next-generation sequencing; MRD: minimal residual disease; CT: computerized tomography; CEA: carcinoembryonic antigen.
There are several challenges in the design of ctDNA-based clinical trials in adjuvant CRC. A major challenge is the requirement of a high number of patients in order to observe a significant clinical effect. A second challenge is the lack of standardization of ctDNA detection techniques [93], with different sensitivity and specificity. Specificity can be affected by multiple factors. A short follow-up can be related to false negative results. Other factors include clonal hematopoiesis or the presence of another primary tumor [94] which can lead to false positive results. Sensitivity is also highly variable among studies [24,80], and may be due to multiple factors including study design, temporal differences in the extraction of the first post-treatment sample, and differences in patient follow-up. A determining factor is the low amount of ctDNA in patients with localized disease [56]. Sensitivity has been shown to increase with serial sample extraction compared to one time-point extraction [55,80,95]. Figure 4 shows the main causes of false positive and false negative results and some proposed solutions.
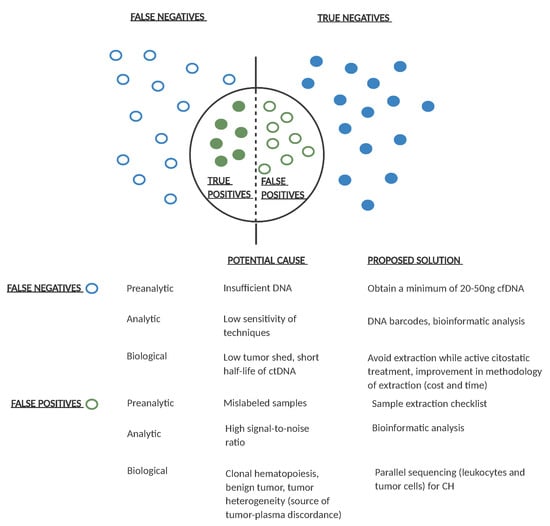
Figure 4.
Potential causes of false positive and false negative results in ctDNA analysis and proposed solutions. Empty dots represent false positive/negative results and filled dots represent true positive/negative ctDNA results. For each pre-analytical, analytical, or biological potential false positive/negative result, a possible solution is proposed. Ng: nanograms; cfDNA: cell free DNA; CH: clonal hematopoiesis.
Retrospective analysis of the impact of ctDNA in large randomized clinical trials would help define the clinical utility of ctDNA in the adjuvant setting. In this regard, a retrospective analysis of the French IDEA study showed that ctDNA-positive patients benefited from 6 months of chemotherapy compare to 3 months [46]. Prospective trials for the detection of MRD and its use to guide therapeutic decisions are ongoing. Currently, most of ongoing trials are designed to evaluate ctDNA as a tool to detect MRD after surgery and guide adjuvant therapy, while some of them are also addressing the question of chemo-resistant clones or persistent MRD following adjuvant chemotherapy (Table 3).
Table 3.
ctDNA−guided ongoing clinical trials in early-stage colorectal cancer.
To sum up, in order to incorporate the use of ctDNA into routine clinical practice, clinical trials with robust and standardized methodologies are needed. The detection of MRD and MR by ctDNA needs to be proven of clinical utility to increase survival and/or reduce toxicities without affecting their long-term survival.
6. Conclusions
ctDNA-based liquid biopsy is changing the paradigm in the diagnosis and monitoring of CRC patients. Initially developed in the metastatic setting, the potential applications in MRD detection can change the treatment of patients with localized CRC. Studies carried out in recent years have validated the role of liquid biopsy as a powerful tool to detect MRD and a prognostic biomarker in patients with early-stage CRC. Thus, the detection of ctDNA after surgery correlates with a high risk of recurrence. Of note, most studies so far have been designed based on plasma testing of point mutations previously found in tumor tissue. The complexity of this approach may limit the implementation of ctDNA for MRD in clinical practice. With recent improvements in ctDNA technology, not only genomic but also epigenetic changes or fragmentomic can be used to detect ctDNA. This increases sensitivity and specificity in ctDNA detection, which is of utmost importance in the post-surgery clinical scenario where minimum amount of ctDNA is circulating in the bloodstream.
Results from ongoing ctDNA-guided prospective clinical trials in the adjuvant setting are eagerly awaited. Multiple efforts by cooperative research groups are being made to demonstrate the clinical impact of treatment intervention based on post-surgery ctDNA risk-stratification which will be essential to change the way we treat stage II and III CRC, as well as LARC.
Author Contributions
Conceptualization, L.M., J.V., and C.M.; writing—original draft preparation, L.M. and J.V.; writing—review and editing, C.M. and C.F.-R.; funding acquisition, C.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
J.V. received honoraria for advisory role, speaker or travel grants from Amgen, Hoffman La-Roche, Merck-Serono, Novartis, and Sanofi. C.M. received honoraria for advisory role, speaker or research grants (past 5 years): Amgen, Biocartis, Bristol Myers Squibb, Guardant-Health, Merck-Serono, Hoffman La-Roche, Sanofi Aventis. C.F.-R. received honoraria for speaker from Merck and consultancy from Roche. L.M. declares no conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Argilés, G.; Tabernero, J.; Labianca, R.; Hochhauser, D.; Salazar, R.; Iveson, T.; Laurent-Puig, P.; Quirke, P.; Yoshino, T.; Taieb, J.; et al. Localised Colon Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2020, S0923753420399324. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Atkin, W.; Lenz, H.-J.; Lynch, H.T.; Minsky, B.; Nordlinger, B.; Starling, N. Colorectal Cancer. Lancet 2010, 375, 18. [Google Scholar] [CrossRef]
- Schmoll, H.-J.; Twelves, C.; Sun, W.; O’Connell, M.J.; Cartwright, T.; McKenna, E.; Saif, M.; Lee, S.; Yothers, G.; Haller, D. Effect of Adjuvant Capecitabine or Fluorouracil, with or without Oxaliplatin, on Survival Outcomes in Stage III Colon Cancer and the Effect of Oxaliplatin on Post-Relapse Survival: A Pooled Analysis of Individual Patient Data from Four Randomised Controlled Trials. Lancet Oncol. 2014, 15, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Zaniboni, A.; Labianca, R.; Marsoni, S.; Torri, V.; Mosconi, P.; Grilli, R.; Apolone, G.; Cifani, S.; Tinazzi, A. A Randomized Trial of Adjuvant 5-Fluorouracil and Folinic Acid Administered to Patients with Colon Carcinoma—Long Term Results and Evaluation of the Indicators of Health-Related Quality of Life. Cancer 1998, 82, 2135–2144. [Google Scholar] [CrossRef]
- International Multicentre Pooled Analysis of Colon Cancer Trials (IMPACT) Investigators. Efficacy of Adjuvant Fluorouracil and Folinic Acid in Colon Cancer. Lancet 1995, 345, 939–944. [Google Scholar] [CrossRef]
- Bahadoer, R.R.; Dijkstra, E.A.; van Etten, B.; Marijnen, C.A.M.; Putter, H.; Kranenbarg, E.M.-K.; Roodvoets, A.G.H.; Nagtegaal, I.D.; Beets-Tan, R.G.H.; Blomqvist, L.K.; et al. Short-Course Radiotherapy Followed by Chemotherapy before Total Mesorectal Excision (TME) versus Preoperative Chemoradiotherapy, TME, and Optional Adjuvant Chemotherapy in Locally Advanced Rectal Cancer (RAPIDO): A Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2021, 22, 29–42. [Google Scholar] [CrossRef]
- Conroy, T.; Lamfichekh, N.; Etienne, P.-L.; Rio, E.; Francois, E.; Mesgouez-Nebout, N.; Vendrely, V.; Artignan, X.; Bouché, O.; Gargot, D.; et al. Total Neoadjuvant Therapy with MFOLFIRINOX versus Preoperative Chemoradiation in Patients with Locally Advanced Rectal Cancer: Final Results of PRODIGE 23 Phase III Trial, a UNICANCER GI Trial. J. Clin. Oncol. 2020, 38, 4007. [Google Scholar] [CrossRef]
- Hong, Y.S.; Kim, S.Y.; Lee, J.S.; Nam, B.-H.; Kim, K.; Kim, J.E.; Park, Y.S.; Park, J.O.; Baek, J.Y.; Kim, T.-Y.; et al. Oxaliplatin-Based Adjuvant Chemotherapy for Rectal Cancer After Preoperative Chemoradiotherapy (ADORE): Long-Term Results of a Randomized Controlled Trial. J. Clin. Oncol. 2019, 37, 3111–3123. [Google Scholar] [CrossRef] [PubMed]
- Breugom, A.J.; Swets, M.; Bosset, J.-F.; Collette, L.; Sainato, A.; Cionini, L.; Glynne-Jones, R.; Counsell, N.; Bastiaannet, E.; van den Broek, C.B.M.; et al. Adjuvant Chemotherapy after Preoperative (Chemo)Radiotherapy and Surgery for Patients with Rectal Cancer: A Systematic Review and Meta-Analysis of Individual Patient Data. Lancet Oncol. 2015, 16, 200–207. [Google Scholar] [CrossRef]
- Henricks, L.M.; Lunenburg, C.A.T.C.; de Man, F.M.; Meulendijks, D.; Frederix, G.W.J.; Kienhuis, E.; Creemers, G.-J.; Baars, A.; Dezentjé, V.O.; Imholz, A.L.T.; et al. DPYD Genotype-Guided Dose Individualisation of Fluoropyrimidine Therapy in Patients with Cancer: A Prospective Safety Analysis. Lancet Oncol. 2018, 19, 1459–1467. [Google Scholar] [CrossRef]
- Argyriou, A.A.; Polychronopoulos, P.; Iconomou, G.; Chroni, E.; Kalofonos, H.P. A Review on Oxaliplatin-Induced Peripheral Nerve Damage. Cancer Treat. Rev. 2008, 34, 368–377. [Google Scholar] [CrossRef]
- Grothey, A. Oxaliplatin-Safety Profile: Neurotoxicity. Semin. Oncol. 2003, 30, 5–13. [Google Scholar] [CrossRef]
- Mouliere, F.; Robert, B.; Arnau Peyrotte, E.; Del Rio, M.; Ychou, M.; Molina, F.; Gongora, C.; Thierry, A.R. High Fragmentation Characterizes Tumour-Derived Circulating DNA. PLoS ONE 2011, 6, e23418. [Google Scholar] [CrossRef]
- Mouliere, F.; El Messaoudi, S.; Pang, D.; Dritschilo, A.; Thierry, A.R. Multi-Marker Analysis of Circulating Cell-Free DNA toward Personalized Medicine for Colorectal Cancer. Mol. Oncol. 2014, 8, 927–941. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Durkee, K.H.; Moore, K.J.; Goodman, S.N.; Shuber, A.P.; Kinzler, K.W.; Vogelstein, B. Analysis of Mutations in DNA Isolated from Plasma and Stool of Colorectal Cancer Patients. Gastroenterology 2008, 135, 489–498.e7. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martínez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal Fluid-Derived Circulating Tumour DNA Better Represents the Genomic Alterations of Brain Tumours than Plasma. Nat. Commun. 2015, 6, 8839. [Google Scholar] [CrossRef]
- Kimura, H.; Fujiwara, Y.; Sone, T.; Kunitoh, H.; Tamura, T.; Kasahara, K.; Nishio, K. EGFR Mutation Status in Tumour-Derived DNA from Pleural Effusion Fluid Is a Practical Basis for Predicting the Response to Gefitinib. Br. J. Cancer 2006, 95, 1390–1395. [Google Scholar] [CrossRef]
- Wang, Y.; Springer, S.; Mulvey, C.L.; Silliman, N.; Schaefer, J.; Sausen, M.; James, N.; Rettig, E.M.; Guo, T.; Pickering, C.R.; et al. Detection of Somatic Mutations and HPV in the Saliva and Plasma of Patients with Head and Neck Squamous Cell Carcinomas. Sci. Transl. Med. 2015, 7, 293ra104. [Google Scholar] [CrossRef]
- Reckamp, K.L.; Melnikova, V.O.; Karlovich, C.; Sequist, L.V.; Camidge, D.R.; Wakelee, H.; Perol, M.; Oxnard, G.R.; Kosco, K.; Croucher, P.; et al. A Highly Sensitive and Quantitative Test Platform for Detection of NSCLC EGFR Mutations in Urine and Plasma. J. Thorac. Oncol. 2016, 11, 1690–1700. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef]
- Vidal, J.; Muinelo, L.; Dalmases, A.; Jones, F.; Edelstein, D.; Iglesias, M.; Orrillo, M.; Abalo, A.; Rodríguez, C.; Brozos, E.; et al. Plasma CtDNA RAS Mutation Analysis for the Diagnosis and Treatment Monitoring of Metastatic Colorectal Cancer Patients. Ann. Oncol. 2017, 28, 1325–1332. [Google Scholar] [CrossRef]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating Mutant DNA to Assess Tumor Dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Avanzini, S. A Mathematical Model of CtDNA Shedding Predicts Tumor Detection Size_Science Advances 2020. Sci. Adv. 2020, 6, eabc4308. [Google Scholar] [CrossRef]
- Holdhoff, M.; Schmidt, K.; Donehower, R.; Diaz, L.A. Analysis of Circulating Tumor DNA to Confirm Somatic KRAS Mutations. JNCI J. Natl. Cancer Inst. 2009, 101, 1284–1285. [Google Scholar] [CrossRef]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and Quantification of Mutations in the Plasma of Patients with Colorectal Tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Stoehlmacher, J.; Pantel, K.; Goekkurt, E. Detection and Monitoring of Cell-Free DNA in Blood of Patients with Colorectal Cancer. Ann. N. Y. Acad. Sci. 2008, 1137, 190–196. [Google Scholar] [CrossRef]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.Y.; Kaper, F.; Dawson, S.-J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive Identification and Monitoring of Cancer Mutations by Targeted Deep Sequencing of Plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef]
- Kennedy, S.R.; Schmitt, M.W.; Fox, E.J.; Kohrn, B.F.; Salk, J.J.; Ahn, E.H.; Prindle, M.J.; Kuong, K.J.; Shen, J.-C.; Risques, R.-A.; et al. Detecting Ultralow-Frequency Mutations by Duplex Sequencing. Nat. Protoc. 2014, 9, 2586–2606. [Google Scholar] [CrossRef] [PubMed]
- Czeiger, D.; Shaked, G.; Eini, H.; Vered, I.; Belochitski, O.; Avriel, A.; Ariad, S.; Douvdevani, A. Measurement of Circulating Cell-Free DNA Levels by a New Simple Fluorescent Test in Patients with Primary Colorectal Cancer. Am. J. Clin. Pathol. 2011, 135, 264–270. [Google Scholar] [CrossRef]
- Cristofanilli, M. Circulating Tumor Cells Revisited. JAMA 2010, 303, 1092. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jiang, J.; Mostert, B.; Sieuwerts, A.; Martens, J.W.M.; Sleijfer, S.; Foekens, J.A.; Wang, Y. Allele-Specific, Non-Extendable Primer Blocker PCR (AS-NEPB-PCR) for DNA Mutation Detection in Cancer. J. Mol. Diagn. 2013, 15, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Freidin, M.B.; Freydina, D.V.; Leung, M.; Montero Fernandez, A.; Nicholson, A.G.; Lim, E. Circulating Tumor DNA Outperforms Circulating Tumor Cells for KRAS Mutation Detection in Thoracic Malignancies. Clin. Chem. 2015, 61, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Glenn, T.C. Field Guide to Next-Generation DNA Sequencers: Field Guide to Next-Gen Sequencers. Mol. Ecol. Resour. 2011, 11, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Meddeb, R.; Pisareva, E.; Thierry, A.R. Guidelines for the Preanalytical Conditions for Analyzing Circulating Cell-Free DNA. Clin. Chem. 2019, 65, 623–633. [Google Scholar] [CrossRef]
- Van Dessel, L.F.; Beije, N.; Helmijr, J.C.A.; Vitale, S.R.; Kraan, J.; Look, M.P.; de Wit, R.; Sleijfer, S.; Jansen, M.P.H.M.; Martens, J.W.M.; et al. Application of Circulating Tumor DNA in Prospective Clinical Oncology Trials—Standardization of Preanalytical Conditions. Mol. Oncol. 2017, 11, 295–304. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Chabner, B.A. Application of Cell-Free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765. [Google Scholar] [CrossRef]
- Elazezy, M.; Joosse, S.A. Techniques of Using Circulating Tumor DNA as a Liquid Biopsy Component in Cancer Management. Comput. Struct. Biotechnol. J. 2018, 16, 370–378. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal Hematopoiesis of Indeterminate Potential and Its Distinction from Myelodysplastic Syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef]
- Razavi, P.; Li, B.T.; Brown, D.N.; Jung, B.; Hubbell, E.; Shen, R.; Abida, W.; Juluru, K.; De Bruijn, I.; Hou, C.; et al. High-Intensity Sequencing Reveals the Sources of Plasma Circulating Cell-Free DNA Variants. Nat. Med. 2019, 25, 1928–1937. [Google Scholar] [CrossRef]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-Free DNA Comprises an In Vivo Nucleosome Footprint That Informs Its Tissues-Of-Origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive Human Cell-Type Methylation Atlas Reveals Origins of Circulating Cell-Free DNA in Health and Disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef]
- Garrigou, S.; Perkins, G.; Garlan, F.; Normand, C.; Didelot, A.; Le Corre, D.; Peyvandi, S.; Mulot, C.; Niarra, R.; Aucouturier, P.; et al. A Study of Hypermethylated Circulating Tumor DNA as a Universal Colorectal Cancer Biomarker. Clin. Chem. 2016, 62, 1129–1139. [Google Scholar] [CrossRef]
- Taieb, J.; Taly, V.; Vernerey, D.; Bourreau, C.; Bennouna, J.; Faroux, R.; Desrame, J.; Bouche, O.; Borg, C.; Egreteau, J.; et al. Analysis of Circulating Tumour DNA (CtDNA) from Patients Enrolled in the IDEA-FRANCE Phase III Trial: Prognostic and Predictive Value for Adjuvant Treatment Duration. Ann. Oncol. 2019, 30, v867. [Google Scholar] [CrossRef]
- Artieri, C.; Axelrod, H.; Baca, A.; Burke, J.; Chudova, D.; Dahdouli, M.; Ghadiri, F.; Hartwig, A.; He, Y.; Hite, D.; et al. Analytical Validation of a Tissue Agnostic CtDNA MRD Assay Using Tumor Specific Methylation and Somatic Variant Profiles in Early-Stage CRC. J. Clin. Oncol. 2020, 38, e15549. [Google Scholar] [CrossRef]
- Keller, L.; Belloum, Y.; Wikman, H.; Pantel, K. Clinical Relevance of Blood-Based CtDNA Analysis: Mutation Detection and Beyond. Br. J. Cancer 2021, 124, 345–358. [Google Scholar] [CrossRef]
- Danyi, A.; Jager, M.; de Ridder, J. Cancer Type Classification in Liquid Biopsies Based on Sparse Mutational Profiles Enabled through Data Augmentation and Integration. Bioinformatics 2021, in press. [Google Scholar]
- Bachet, J.B.; Bouché, O.; Taieb, J.; Dubreuil, O.; Garcia, M.L.; Meurisse, A.; Normand, C.; Gornet, J.M.; Artru, P.; Louafi, S.; et al. RAS Mutation Analysis in Circulating Tumor DNA from Patients with Metastatic Colorectal Cancer: The AGEO RASANC Prospective Multicenter Study. Ann. Oncol. 2018, 29, 1211–1219. [Google Scholar] [CrossRef]
- García-Foncillas, J.; Tabernero, J.; Élez, E.; Aranda, E.; Benavides, M.; Camps, C.; Jantus-Lewintre, E.; López, R.; Muinelo-Romay, L.; Montagut, C.; et al. Prospective Multicenter Real-World RAS Mutation Comparison between OncoBEAM-Based Liquid Biopsy and Tissue Analysis in Metastatic Colorectal Cancer. Br. J. Cancer 2018, 119, 1464–1470. [Google Scholar] [CrossRef]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; et al. Mutation Tracking in Circulating Tumor DNA Predicts Relapse in Early Breast Cancer. Sci. Transl. Med. 2015, 7, 302ra133. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An Ultrasensitive Method for Quantitating Circulating Tumor DNA with Broad Patient Coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Kruger, S.; Heinemann, V.; Ross, C.; Diehl, F.; Nagel, D.; Ormanns, S.; Liebmann, S.; Prinz-Bravin, I.; Westphalen, C.B.; Haas, M.; et al. Repeated MutKRAS CtDNA Measurements Represent a Novel and Promising Tool for Early Response Prediction and Therapy Monitoring in Advanced Pancreatic Cancer. Ann. Oncol. 2018, 29, 2348–2355. [Google Scholar] [CrossRef]
- Lee, R.J.; Gremel, G.; Marshall, A.; Myers, K.A.; Fisher, N.; Dunn, J.A.; Dhomen, N.; Corrie, P.G.; Middleton, M.R.; Lorigan, P.; et al. Circulating Tumor DNA Predicts Survival in Patients with Resected High-Risk Stage II/III Melanoma. Ann. Oncol. 2018, 29, 490–496. [Google Scholar] [CrossRef]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.-L.; Christie, M.; et al. Circulating Tumor DNA Analysis Detects Minimal Residual Disease and Predicts Recurrence in Patients with Stage II Colon Cancer. Sci. Transl. Med. 2016, 8, 346ra92. [Google Scholar] [CrossRef]
- The TRACERx Consortium; The PEACE Consortium; Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; et al. Phylogenetic CtDNA Analysis Depicts Early-Stage Lung Cancer Evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Dudley, J.C.; Schroers-Martin, J.; Lazzareschi, D.V.; Shi, W.Y.; Chen, S.B.; Esfahani, M.S.; Trivedi, D.; Chabon, J.J.; Chaudhuri, A.A.; Stehr, H.; et al. Detection and Surveillance of Bladder Cancer Using Urine Tumor DNA. Cancer Discov. 2019, 9, 500–509. [Google Scholar] [CrossRef]
- Wang, Y.; Li, L.; Cohen, J.D.; Kinde, I.; Ptak, J.; Popoli, M.; Schaefer, J.; Silliman, N.; Dobbyn, L.; Tie, J.; et al. Prognostic Potential of Circulating Tumor DNA Measurement in Postoperative Surveillance of Nonmetastatic Colorectal Cancer. JAMA Oncol. 2019, 5, 1118. [Google Scholar] [CrossRef]
- Edge, S.B.; Compton, C.C. The American Joint Committee on Cancer: The 7th Edition of the AJCC Cancer Staging Manual and the Future of TNM. Ann. Surg. Oncol. 2010, 17, 1471–1474. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.D.; Delorenzi, M.; Tejpar, S.; Yan, P.; Klingbiel, D.; Fiocca, R.; d’Ario, G.; Cisar, L.; Labianca, R.; Cunningham, D.; et al. Integrated Analysis of Molecular and Clinical Prognostic Factors in Stage II/III Colon Cancer. JNCI J. Natl. Cancer Inst. 2012, 104, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Wells, K.O.; Hawkins, A.T.; Krishnamurthy, D.M.; Dharmarajan, S.; Glasgow, S.C.; Hunt, S.R.; Mutch, M.G.; Wise, P.; Silviera, M.L. Omission of Adjuvant Chemotherapy Is Associated with Increased Mortality in Patients With T3N0 Colon Cancer With Inadequate Lymph Node Harvest. Dis. Colon Rectum 2017, 60, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Benatti, P.; Gafà, R.; Barana, D.; Marino, M.; Scarselli, A.; Pedroni, M.; Maestri, I.; Guerzoni, L.; Roncucci, L.; Menigatti, M.; et al. Microsatellite Instability and Colorectal Cancer Prognosis. Clin. Cancer Res. 2005, 11, 8332–8340. [Google Scholar] [CrossRef] [PubMed]
- Jover, R.; Zapater, P.; Castells, A.; Llor, X.; Andreu, M.; Cubiella, J.; Balaguer, F.; Sempere, L.; Xicola, R.M.; Bujanda, L.; et al. The Efficacy of Adjuvant Chemotherapy with 5-Fluorouracil in Colorectal Cancer Depends on the Mismatch Repair Status. Eur. J. Cancer 2009, 45, 365–373. [Google Scholar] [CrossRef]
- Carethers, J.M.; Smith, E.J.; Behling, C.A.; Nguyen, L.; Tajima, A.; Doctolero, R.T.; Cabrera, B.L.; Goel, A.; Arnold, C.A.; Miyai, K.; et al. Use of 5-Fluorouracil and Survival in Patients with Microsatellite-Unstable Colorectal Cancer. Gastroenterology 2004, 126, 394–401. [Google Scholar] [CrossRef]
- Quah, H.-M.; Chou, J.F.; Gonen, M.; Shia, J.; Schrag, D.; Landmann, R.G.; Guillem, J.G.; Paty, P.B.; Temple, L.K.; Wong, W.D.; et al. Identification of Patients with High-Risk Stage II Colon Cancer for Adjuvant Therapy. Dis. Colon. Rectum 2008, 51, 503–507. [Google Scholar] [CrossRef]
- Niedzwiecki, D.; Bertagnolli, M.M.; Warren, R.S.; Compton, C.C.; Kemeny, N.E.; Benson, A.B.; Eckhardt, S.G.; Alberts, S.; Porjosh, G.N.; Kerr, D.J.; et al. Documenting the Natural History of Patients With Resected Stage II Adenocarcinoma of the Colon After Random Assignment to Adjuvant Treatment With Edrecolomab or Observation: Results From CALGB 9581. J. Clin. Oncol. 2011, 29, 3146–3152. [Google Scholar] [CrossRef]
- Bozic, I.; Reiter, J.G.; Allen, B.; Antal, T.; Chatterjee, K.; Shah, P.; Moon, Y.S.; Yaqubie, A.; Kelly, N.; Le, D.T.; et al. Evolutionary Dynamics of Cancer in Response to Targeted Combination Therapy. eLife 2013, 2, e00747. [Google Scholar] [CrossRef]
- Montagut, C.; Siravegna, G.; Bardelli, A. Liquid Biopsies to Evaluate Early Therapeutic Response in Colorectal Cancer. Ann. Oncol. 2015, 26, 1525–1527. [Google Scholar] [CrossRef]
- Thierry, A.; Corrado, B.; Lamia, M.-B.; Matilde, N.; Josep, T.; Tamas, H.; Clare, T.; Marta, Z.; Philip, C.; John, B.; et al. Oxaliplatin, Fluorouracil, and Leucovorin as Adjuvant Treatment for Colon Cancer. N. Engl. J. Med. 2004, 350, 2343–2351. [Google Scholar]
- Påhlman, L.A.; Hohenberger, W.M.; Matzel, K.; Sugihara, K.; Quirke, P.; Glimelius, B. Should the Benefit of Adjuvant Chemotherapy in Colon Cancer Be Re-Evaluated? J. Clin. Oncol. 2016, 34, 1297–1299. [Google Scholar] [CrossRef]
- Böckelman, C.; Engelmann, B.E.; Kaprio, T.; Hansen, T.F.; Glimelius, B. Risk of Recurrence in Patients with Colon Cancer Stage II and III: A Systematic Review and Meta-Analysis of Recent Literature. Acta Oncol. 2015, 54, 5–16. [Google Scholar] [CrossRef]
- Lash, T.L.; Riis, A.H.; Ostenfeld, E.B.; Erichsen, R.; Vyberg, M.; Ahern, T.P.; Thorlacius-Ussing, O. Associations of Statin Use With Colorectal Cancer Recurrence and Mortality in a Danish Cohort. Am. J. Epidemiol. 2017, 186, 679–687. [Google Scholar] [CrossRef]
- Osterman, E.; Glimelius, B. Recurrence Risk After Up-to-Date Colon Cancer Staging, Surgery, and Pathology: Analysis of the Entire Swedish Population. Dis. Colon Rectum 2018, 61, 1016–1025. [Google Scholar] [CrossRef]
- Grothey, A.; Sobrero, A.F.; Shields, A.F.; Yoshino, T.; Paul, J.; Taieb, J.; Souglakos, J.; Shi, Q.; Kerr, R.; Labianca, R.; et al. Duration of Adjuvant Chemotherapy for Stage III Colon Cancer. N. Engl. J. Med. 2018, 378, 1177–1188. [Google Scholar] [CrossRef]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.-T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients with Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Wang, Y.; Christie, M.; Simons, K.; Lee, M.; Wong, R.; Kosmider, S.; Ananda, S.; McKendrick, J.; et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncol. 2019, 5, 1710. [Google Scholar] [CrossRef]
- Tie, J. Prognostic Significance of Postsurgery CtDNA in Nonmetastatic CRC Individual Patient Pooled Analysis of Three Cohort Studies. Int. J. Cancer 2020, 148, 1014–1026. [Google Scholar] [CrossRef]
- Tarazona, N.; Gimeno-Valiente, F.; Gambardella, V.; Zuñiga, S.; Rentero-Garrido, P.; Huerta, M.; Roselló, S.; Martinez-Ciarpaglini, C.; Carbonell-Asins, J.A.; Carrasco, F.; et al. Targeted Next-Generation Sequencing of Circulating-Tumor DNA for Tracking Minimal Residual Disease in Localized Colon Cancer. Ann. Oncol. 2019, 30, 1804–1812. [Google Scholar] [CrossRef]
- Schøler, L.V.; Reinert, T.; Ørntoft, M.-B.W.; Kassentoft, C.G.; Árnadóttir, S.S.; Vang, S.; Nordentoft, I.; Knudsen, M.; Lamy, P.; Andreasen, D.; et al. Clinical Implications of Monitoring Circulating Tumor DNA in Patients with Colorectal Cancer. Clin. Cancer Res. 2017, 23, 5437–5445. [Google Scholar] [CrossRef]
- Maas, M.; Nelemans, P.J.; Valentini, V.; Das, P.; Rödel, C.; Kuo, L.-J.; Calvo, F.A.; García-Aguilar, J.; Glynne-Jones, R.; Haustermans, K.; et al. Long-Term Outcome in Patients with a Pathological Complete Response after Chemoradiation for Rectal Cancer: A Pooled Analysis of Individual Patient Data. Lancet Oncol. 2010, 11, 835–844. [Google Scholar] [CrossRef]
- Janjan, N.A.; Khoo, V.S.; Abbruzzese, J.; Pazdur, R.; Dubrow, R.; Cleary, K.R.; Allen, P.K.; Lynch, P.M.; Glober, G.; Wolff, R.; et al. Tumor Downstaging and Sphincter Preservation with Preoperative Chemoradiation in Locally Advanced Rectal Cancer: The M. D. Anderson Cancer Center Experience. Int. J. Radiat. Oncol. 1999, 44, 1027–1038. [Google Scholar] [CrossRef]
- Hendren, S.K.; O’Connor, B.I.; Liu, M.; Asano, T.; Cohen, Z.; Swallow, C.J.; MacRae, H.M.; Gryfe, R.; McLeod, R.S. Prevalence of Male and Female Sexual Dysfunction Is High Following Surgery for Rectal Cancer. Ann. Surg. 2005, 242, 212–223. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Wang, Y.; Li, L.; Christie, M.; Simons, K.; Elsaleh, H.; Kosmider, S.; Wong, R.; Yip, D.; et al. Serial Circulating Tumour DNA Analysis during Multimodality Treatment of Locally Advanced Rectal Cancer: A Prospective Biomarker Study. Gut 2019, 68, 663–671. [Google Scholar] [CrossRef]
- Khakoo, S.; Carter, P.D.; Brown, G.; Valeri, N.; Picchia, S.; Bali, M.A.; Shaikh, R.; Jones, T.; Begum, R.; Rana, I.; et al. MRI Tumor Regression Grade and Circulating Tumor DNA as Complementary Tools to Assess Response and Guide Therapy Adaptation in Rectal Cancer. Clin. Cancer Res. 2020, 26, 183–192. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, C.; Lin, G.; Xiao, Y.; Jia, W.; Xiao, G.; Liu, Q.; Wu, B.; Wu, A.; Qiu, H.; et al. Serial Circulating Tumor DNA in Predicting and Monitoring the Effect of Neoadjuvant Chemoradiotherapy in Patients with Rectal Cancer: A Prospective Multicenter Study. Clin. Cancer Res. 2021, 27, 301–310. [Google Scholar] [CrossRef]
- Murahashi, S.; Akiyoshi, T.; Sano, T.; Fukunaga, Y.; Noda, T.; Ueno, M.; Zembutsu, H. Serial Circulating Tumour DNA Analysis for Locally Advanced Rectal Cancer Treated with Preoperative Therapy: Prediction of Pathological Response and Postoperative Recurrence. Br. J. Cancer 2020, 123, 803–810. [Google Scholar] [CrossRef]
- Pazdirek, F.; Minarik, M.; Benesova, L.; Halkova, T.; Belsanova, B.; Macek, M.; Stepanek, L.; Hoch, J. Monitoring of Early Changes of Circulating Tumor DNA in the Plasma of Rectal Cancer Patients Receiving Neoadjuvant Concomitant Chemoradiotherapy: Evaluation for Prognosis and Prediction of Therapeutic Response. Front. Oncol. 2020, 10, 1028. [Google Scholar] [CrossRef]
- Vidal, J.; Casadevall, D.; Bellosillo, B.; Pericay, C.; Garcia-Carbonero, R.; Losa, F.; Layos, L.; Alonso, V.; Capdevila, J.; Gallego, J.; et al. Clinical Impact of Presurgery Circulating Tumor DNA after Total Neoadjuvant Treatment in Locally Advanced Rectal Cancer: A Biomarker Study from the GEMCAD 1402 Trial. Clin. Cancer Res. 2021, 27. [Google Scholar] [CrossRef]
- Snyder, R.A.; Hu, C.-Y.; Cuddy, A.; Francescatti, A.B.; Schumacher, J.R.; Van Loon, K.; You, Y.N.; Kozower, B.D.; Greenberg, C.C.; Schrag, D.; et al. Association Between Intensity of Posttreatment Surveillance Testing and Detection of Recurrence in Patients with Colorectal Cancer. JAMA 2018, 319, 2104. [Google Scholar] [CrossRef]
- Wille-Jørgensen, P.; Syk, I.; Smedh, K.; Laurberg, S.; Nielsen, D.T.; Petersen, S.H.; Renehan, A.G.; Horváth-Puhó, E.; Påhlman, L.; Sørensen, H.T.; et al. Effect of More vs Less Frequent Follow-up Testing on Overall and Colorectal Cancer–Specific Mortality in Patients With Stage II or III Colorectal Cancer: The COLOFOL Randomized Clinical Trial. JAMA 2018, 319, 2095. [Google Scholar] [CrossRef] [PubMed]
- Coakley, M.; Garcia-Murillas, I.; Turner, N.C. Molecular Residual Disease and Adjuvant Trial Design in Solid Tumors. Clin. Cancer Res. 2019, 25, 6026–6034. [Google Scholar] [CrossRef] [PubMed]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Lindeman, N.; Lockwood, C.M.; Rai, A.J.; Schilsky, R.L.; et al. Circulating Tumor DNA Analysis in Patients with Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J. Clin. Oncol. 2018, 36, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.J.; Abascal, F.; Hall, M.W.J.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic Mutant Clones Colonize the Human Esophagus with Age. Science 2018, 362, 911–917. [Google Scholar] [CrossRef]
- Reinert, T.; Schøler, L.V.; Thomsen, R.; Tobiasen, H.; Vang, S.; Nordentoft, I.; Lamy, P.; Kannerup, A.-S.; Mortensen, F.V.; Stribolt, K.; et al. Analysis of Circulating Tumour DNA to Monitor Disease Burden Following Colorectal Cancer Surgery. Gut 2016, 65, 625–634. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).