
Comparison of Resistance Spectra after First and Second Line Osimertinib Treatment Detected by Liquid Biopsy


 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. Liquid Biopsy
3. Results
3.1. Patient Characteristics
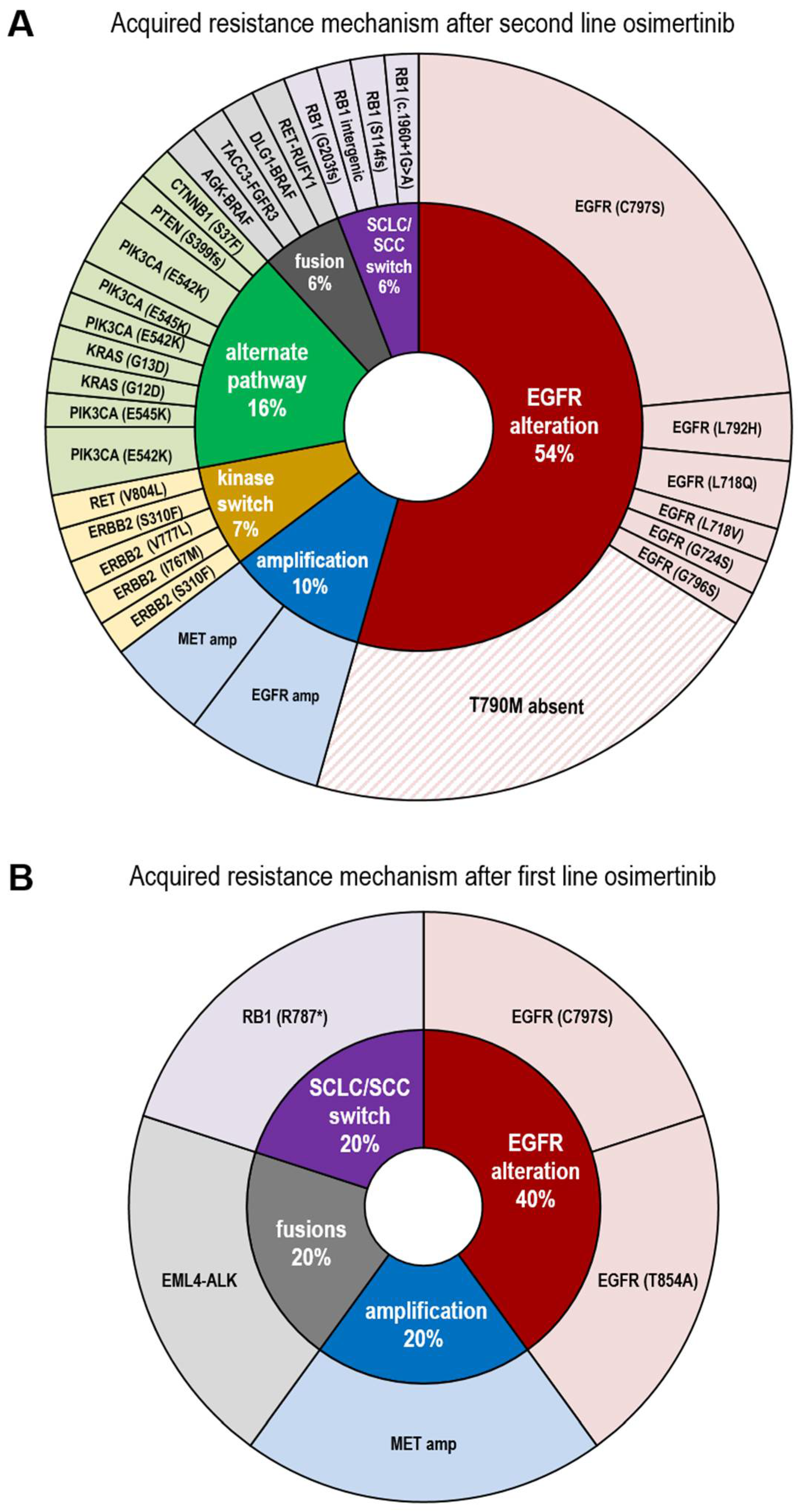
3.2. Resistance Spectrum after First Line Osimertinib Treatment
3.3. Resistance Spectrum after Second Line Osimertinib Treatment
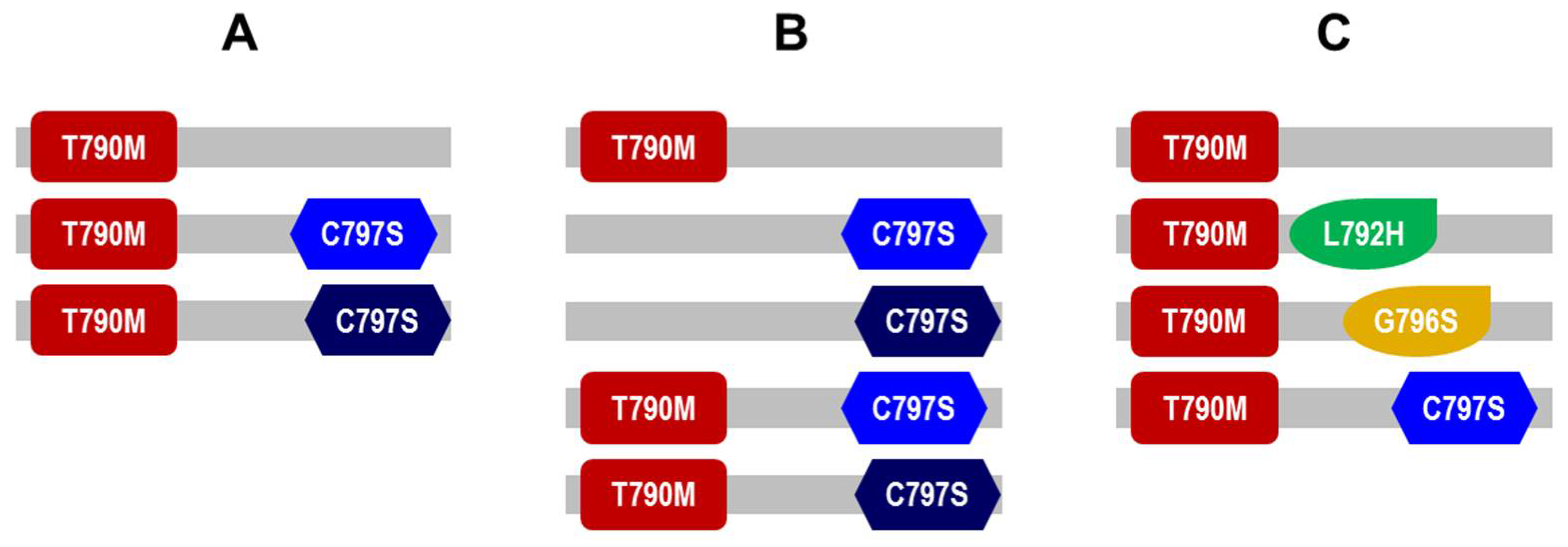
3.4. Presence of Diverse EGFR and PIK3CA Molecular Subclones of in Patients’ Plasma ctDNA after Second Line Osimertinib
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.-L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of MET amplification in EGFR Mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef]
- Wang, W.; Wang, H.; Lu, P.; Yu, Z.; Xu, C.; Zhuang, W.; Song, Z. Crizotinib with or without an EGFR-TKI in treating EGFR-mutant NSCLC patients with acquired MET amplification after failure of EGFR-TKI therapy: A multicenter retrospective study. J. Transl. Med. 2019, 17, 1–9. [Google Scholar] [CrossRef]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef]
- Wang, S.; Tsui, S.T.; Liu, C.; Song, Y.; Liu, D. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 59. [Google Scholar] [CrossRef]
- Ercan, D.; Choi, H.G.; Yun, C.-H.; Capelletti, M.; Xie, T.; Eck, M.J.; Gray, N.S.; Jänne, P.A. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin. Cancer Res. 2015, 21, 3913–3923. [Google Scholar] [CrossRef]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.P.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef]
- Menon, R.; Müller, J.; Schneider, P.; Lakis, S.; Thress, K.; Wolf, J.; Heukamp, L.; Heuckmann, J.M.; Griesinger, F. A Novel EGFR C797 Variant Detected in a Pleural Biopsy Specimen from an Osimertinib-Treated Patient Using a Comprehensive Hybrid Capture–Based Next-Generation Sequencing Assay. J. Thorac. Oncol. 2016, 11, e105–e107. [Google Scholar] [CrossRef]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, J.-J.; Huang, J.; Ye, J.-Y.; Zhang, X.-C.; Tu, H.-Y.; Han-Zhang, H.; Wu, Y.-L. Lung Adenocarcinoma Harboring EGFR T790M and In Trans C797S Responds to Combination Therapy of First- and Third-Generation EGFR TKIs and Shifts Allelic Configuration at Resistance. J. Thorac. Oncol. 2017, 12, 1723–1727. [Google Scholar] [CrossRef]
- Lee, J.; Shim, J.H.; Park, W.-Y.; Kim, H.K.; Sun, J.-M.; Lee, S.-H.; Ahn, J.S.; Park, K.; Ahn, M.-J. Rare Mechanism of Acquired Resistance to Osimertinib in Korean Patients with EGFR-mutated Non-small Cell Lung Cancer. Cancer Res. Treat. 2019, 51, 408–412. [Google Scholar] [CrossRef]
- Schrock, A.B.; Zhu, V.W.; Hsieh, W.-S.; Madison, R.; Creelan, B.; Silberberg, J.; Costin, D.; Bharne, A.; Bonta, I.; Bosemani, T.; et al. Receptor Tyrosine Kinase Fusions and BRAF Kinase Fusions are Rare but Actionable Resistance Mechanisms to EGFR Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2018, 13, 1312–1323. [Google Scholar] [CrossRef]
- Le, X.; Puri, S.; Negrao, M.V.; Nilsson, M.B.; Robichaux, J.P.; A Boyle, T.; Hicks, J.K.; Lovinger, K.L.; Roarty, E.B.; Rinsurongkawong, W.; et al. Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC. Clin. Cancer Res. 2018, 24, 6195–6203. [Google Scholar] [CrossRef]
- Rolfo, C.; Mack, P.C.; Scagliotti, G.V.; Baas, P.; Barlesi, F.; Bivona, T.G.; Herbst, R.S.; Mok, T.S.; Peled, N.; Pirker, R.; et al. Liquid Biopsy for Advanced Non-Small Cell Lung Cancer (NSCLC): A Statement Paper from the IASLC. J. Thorac. Oncol. 2018, 13, 1248–1268. [Google Scholar] [CrossRef]
- Lindeman, N.I.; Cagle, P.T.; Aisner, D.L.; Arcila, M.E.; Beasley, M.B.; Bernicker, E.H.; Colasacco, C.; Dacic, S.; Hirsch, F.R.; Kerr, K.; et al. Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment With Targeted Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2018, 13, 323–358. [Google Scholar] [CrossRef]
- Mellert, H.; Foreman, T.; Jackson, L.; Maar, D.; Thurston, S.; Koch, K.; Weaver, A.; Cooper, S.; Dupuis, N.; Sathyanarayana, U.G.; et al. Development and Clinical Utility of a Blood-Based Test Service for the Rapid Identification of Actionable Mutations in Non–Small Cell Lung Carcinoma. J. Mol. Diagn. 2017, 19, 404–416. [Google Scholar] [CrossRef]
- Kit, A.H.; Mazaleyrat, N.; Daunay, A.; Nielsen, H.M.; Terris, B.; Tost, J. Sensitive Detection of KRAS Mutations Using Enhanced-ice -COLD-PCR Mutation Enrichment and Direct Sequence Identification. Hum. Mutat. 2013, 34, 1568–1580. [Google Scholar] [CrossRef]
- Heuckmann, J.M.; Thomas, R.K. A new generation of cancer genome diagnostics for routine clinical use: Overcoming the roadblocks to personalized cancer medicine. Ann. Oncol. 2015, 26, 1830–1837. [Google Scholar] [CrossRef]
- Müller, J.N.; Falk, M.; Talwar, J.; Neemann, N.; Mariotti, E.; Bertrand, M.; Zacherle, T.; Lakis, S.; Menon, R.; Gloeckner, C.; et al. Concordance between Comprehensive Cancer Genome Profiling in Plasma and Tumor Specimens. J. Thorac. Oncol. 2017, 12, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Lim, S.H.; An, H.J.; Kim, K.H.; Park, K.U.; Kang, E.J.; Choi, Y.H.; Ahn, M.S.; Lee, M.H.; Sun, J.-M.; et al. Osimertinib for Patients With Non–Small-Cell Lung Cancer Harboring Uncommon EGFR Mutations: A Multicenter, Open-Label, Phase II Trial (KCSG-LU15-09). J. Clin. Oncol. 2020, 38, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.-H.; Yang, C.-T.; Shih, J.-Y.; Huang, M.-S.; Su, W.-C.; Lai, R.-S.; Wang, C.-C.; Hsiao, S.-H.; Lin, Y.-C.; Ho, C.-L.; et al. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Treatment Response in Advanced Lung Adenocarcinomas with G719X/L861Q/S768I Mutations. J. Thorac. Oncol. 2015, 10, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Offin, M.; Kubota, D.; Yu, H.A.; Wilhelm, C.; Toyooka, S.; Somwar, R.; Kris, M.G.; Ladanyi, M. Allele-Specific Role of ERBB2 in the Oncogenic Function of EGFR L861Q in EGFR-Mutant Lung Cancers. J. Thorac. Oncol. 2021, 16, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Chiba, M.; Togashi, Y.; Bannno, E.; Kobayashi, Y.; Nakamura, Y.; Hayashi, H.; Terashima, M.; De Velasco, M.A.; Sakai, K.; Fujita, Y.; et al. Efficacy of irreversible EGFR-TKIs for the uncommon secondary resistant EGFR mutations L747S, D761Y, and T854A. BMC Cancer 2017, 17, 281. [Google Scholar] [CrossRef] [PubMed]
- Demierre, N.; Zoete, V.; Michielin, O.; Stauffer, E.; Zimmermann, D.R.; Betticher, D.C.; Peters, S. A dramatic lung cancer course in a patient with a rare EGFR germline mutation exon 21 V843I: Is EGFR TKI resistance predictable? Lung Cancer 2013, 80, 81–84. [Google Scholar] [CrossRef]
- Matsushima, S.; Ohtsuka, K.; Ohnishi, H.; Fujiwara, M.; Nakamura, H.; Morii, T.; Kishino, T.; Goto, H.; Watanabe, T. V843I, a Lung Cancer Predisposing EGFR Mutation, Is Responsible for Resistance to EGFR Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2014, 9, 1377–1384. [Google Scholar] [CrossRef]
- Zhao, S.; Li, X.; Zhao, C.; Jiang, T.; Jia, Y.; Shi, J.; He, Y.; Li, J.; Zhou, F.; Gao, G.; et al. Loss of T790M mutation is associated with early progression to osimertinib in Chinese patients with advanced NSCLC who are harboring EGFR T790M. Lung Cancer 2019, 128, 33–39. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.; Wu, Y.-L.; Han, J.-Y.; Ahn, M.-J.; Ramalingam, S.; John, T.; Okamoto, I.; Yang, J.-H.; Bulusu, K.; Laus, G.; et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann. Oncol. 2018, 29, viii741. [Google Scholar] [CrossRef]
- Nakagawa, K.; Garon, E.B.; Seto, T.; Nishio, M.; Aix, S.P.; Paz-Ares, L.; Chiu, C.-H.; Park, K.; Novello, S.; Nadal, E.; et al. Ramucirumab plus erlotinib in patients with untreated, EGFR-mutated, advanced non-small-cell lung cancer (RELAY): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 1655–1669. [Google Scholar] [CrossRef]
- Lazzari, C.; Gregorc, V.; Karachaliou, N.; Rosell, R.; Santarpia, M. Mechanisms of resistance to osimertinib. J. Thorac. Dis. 2020, 12, 2851–2858. [Google Scholar] [CrossRef]
- Ribeiro, M.F.S.A.; Knebel, F.H.; Bettoni, F.; Saddi, R.; Sacardo, K.P.; Canedo, F.S.N.A.; Alessi, J.V.M.; Shimada, A.K.; Marin, J.F.G.; Camargo, A.A.; et al. Impressive response to dabrafenib, trametinib, and osimertinib in a metastatic EGFR-mutant/BRAF V600E lung adenocarcinoma patient. NPJ Precis. Oncol. 2021, 5, 1–7. [Google Scholar] [CrossRef]
- Nagano, T.; Tachihara, M.; Nishimura, Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells 2018, 7, 212. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Ward, R.A.; Doig, P.; Argyrou, A. Insight into the therapeutic selectivity of the irreversible EGFR tyrosine kinase inhibitor osimertinib through enzyme kinetic studies. Biochemistry 2020, 59, 1428–1441. [Google Scholar] [CrossRef] [PubMed]
- Lim, Z.-F.; Ma, P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019, 12, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Syn, N.L.; Cho, B.C.; Soo, R.A. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat. Rev. 2018, 65, 1–10. [Google Scholar] [CrossRef]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Variable | Characteristics | Categories | Number (N) and Value (%) | |
|---|---|---|---|---|
| N = 56 | Age at diagnosis (years) | Median | 63 | |
| Mean (±SD) | 60 (±12) | |||
| Interquartile range | 49.5–71.5 | |||
| <65 years | 30 | (54%) | ||
| ≥65 years | 26 | (46%) | ||
| Sex | Female | 36 | (64%) | |
| Male | 20 | (36%) | ||
| Smoking status | Current smoker | 2 | (4%) | |
| Never smoker | 31 | (55%) | ||
| Ex-smoker | 17 | (30%) | ||
| n.d. | 6 | (11%) | ||
| Histology | Adenocarcinoma | 54 | (96%) | |
| Adeno-squamous | 1 | (2%) | ||
| Large-cell carcinoma | 1 | (2%) | ||
| UICC stage | IV | 55 | (98%) | |
| n.d. | 1 | (2%) | ||
| Primary EGFR mutation type | Exon 19 deletions | 40 | (71%) | |
| L858R | 13 | (23%) | ||
| L861Q | 1 | (2%) | ||
| E709G, G719S | 1 | (2%) | ||
| E709V, G719C | 1 | (2%) | ||
| TKI sequence | sequential therapy | 47 | (84%) | |
| osimertinib first line | 9 | (16%) | ||
| N = 41 | EGFR dependent resistance to first and second generation TKI | T790M | 36 | (88%) |
| N = 56 | Primary EGFR mutation confirmed by liquid biopsy | yes | 48 | (86%) |
| no (after sequential therapy) | 6 | (11%) | ||
| no (previously treated with osimertinib first line) | 2 | (4%) | ||
| N = 41 | Patients resistance under sequential therapy | resistance alteration acquired | 33 | (80%) |
| no resistance alteration | 8 | (20%) | ||
| absolute number of detected resistance mechanisms | 68 | |||
| N = 7 | Patients resistance under first line osimertinib | resistance alteration acquired | 3 | (43%) |
| no resistance alteration | 4 | (57%) | ||
| absolute number of detected resistance mechanisms | 5 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jóri, B.; Schatz, S.; Kaller, L.; Kah, B.; Roeper, J.; Ramdani, H.O.; Diehl, L.; Hoffknecht, P.; Grohé, C.; Griesinger, F.; et al. Comparison of Resistance Spectra after First and Second Line Osimertinib Treatment Detected by Liquid Biopsy. Cancers 2021, 13, 2861. https://doi.org/10.3390/cancers13122861
Jóri B, Schatz S, Kaller L, Kah B, Roeper J, Ramdani HO, Diehl L, Hoffknecht P, Grohé C, Griesinger F, et al. Comparison of Resistance Spectra after First and Second Line Osimertinib Treatment Detected by Liquid Biopsy. Cancers. 2021; 13(12):2861. https://doi.org/10.3390/cancers13122861
Chicago/Turabian StyleJóri, Balázs, Stefanie Schatz, Len Kaller, Bettina Kah, Julia Roeper, Hayat O. Ramdani, Linda Diehl, Petra Hoffknecht, Christian Grohé, Frank Griesinger, and et al. 2021. "Comparison of Resistance Spectra after First and Second Line Osimertinib Treatment Detected by Liquid Biopsy" Cancers 13, no. 12: 2861. https://doi.org/10.3390/cancers13122861
APA StyleJóri, B., Schatz, S., Kaller, L., Kah, B., Roeper, J., Ramdani, H. O., Diehl, L., Hoffknecht, P., Grohé, C., Griesinger, F., Tiemann, M., Heukamp, L. C., & Falk, M. (2021). Comparison of Resistance Spectra after First and Second Line Osimertinib Treatment Detected by Liquid Biopsy. Cancers, 13(12), 2861. https://doi.org/10.3390/cancers13122861