
A Novel Benzopyrane Derivative Targeting Cancer Cell Metabolic and Survival Pathways

 , , , , , , ,
, , , , , , ,
Abstract
Simple Summary
Abstract
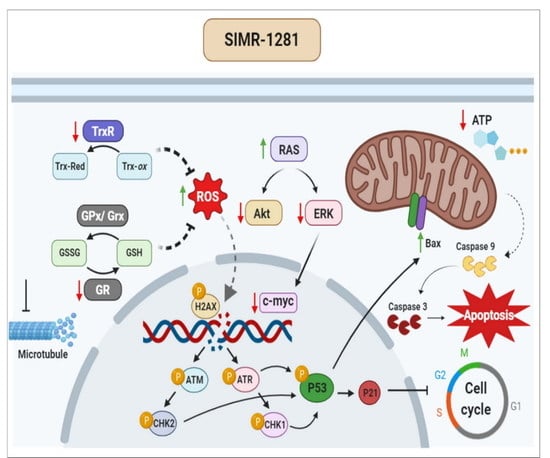
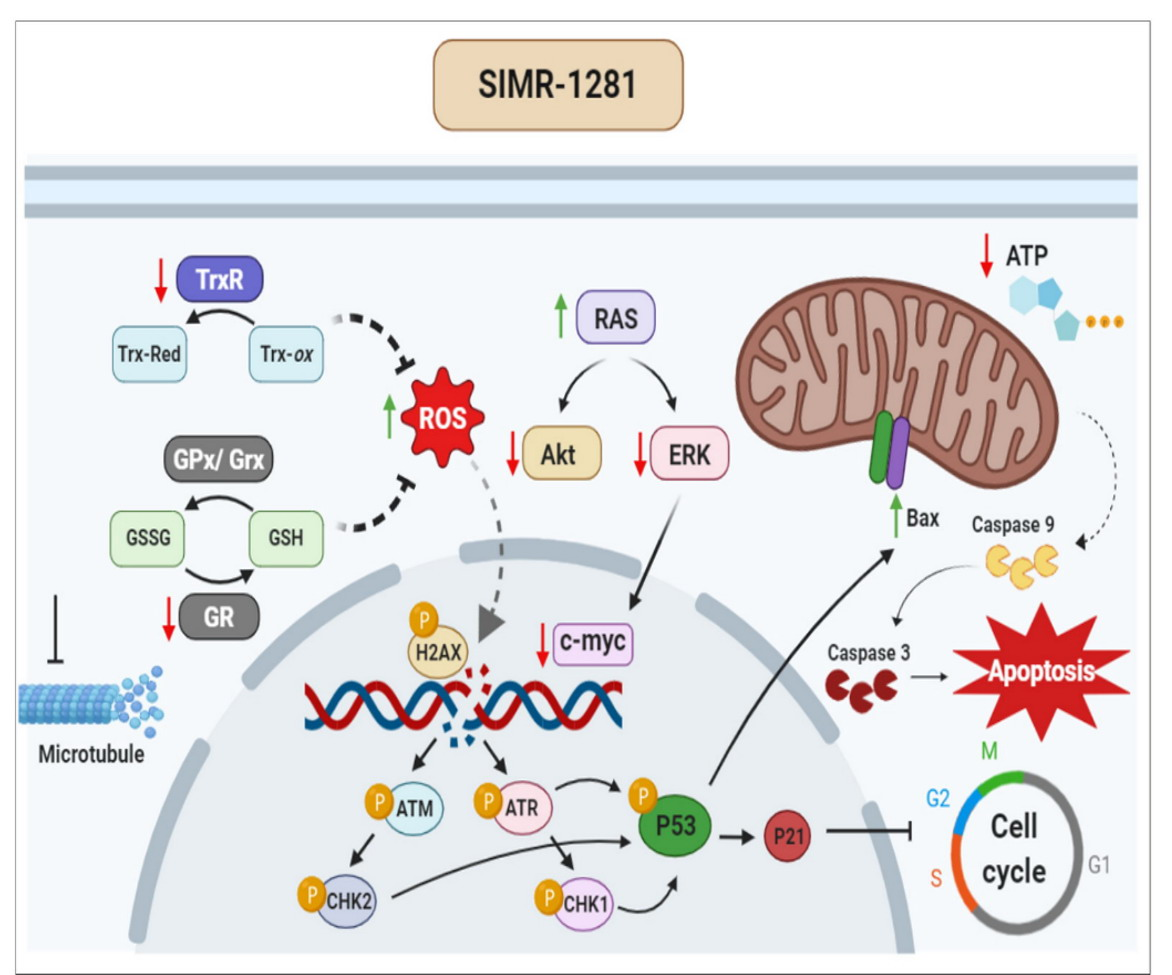{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Analysis
2.3. Western Blot Analysis
2.4. Annexin V Staining Assay
2.5. Cell Cycle Analysis
2.6. Immunofluorescence Assay
2.7. In Vitro Tubulin Polymerization Assay
2.8. DARTS Assay
2.9. Mitochondrial Membrane Potential
2.10. Analysis of ATP Content Assay
2.11. Analysis of Cellular Redox Potential
2.12. Thioredoxin Reductase and Glutathione Reductase Inhibition Assays
Ethics Statement
2.13. Mice Models
2.14. In Vivo Safety Studies
2.15. Tumor Xenograft Mice Model Implantation
2.16. Hematoxylin and Eosin Staining and Immunohistochemistry
2.17. Data and Statistical Analysis
3. Results
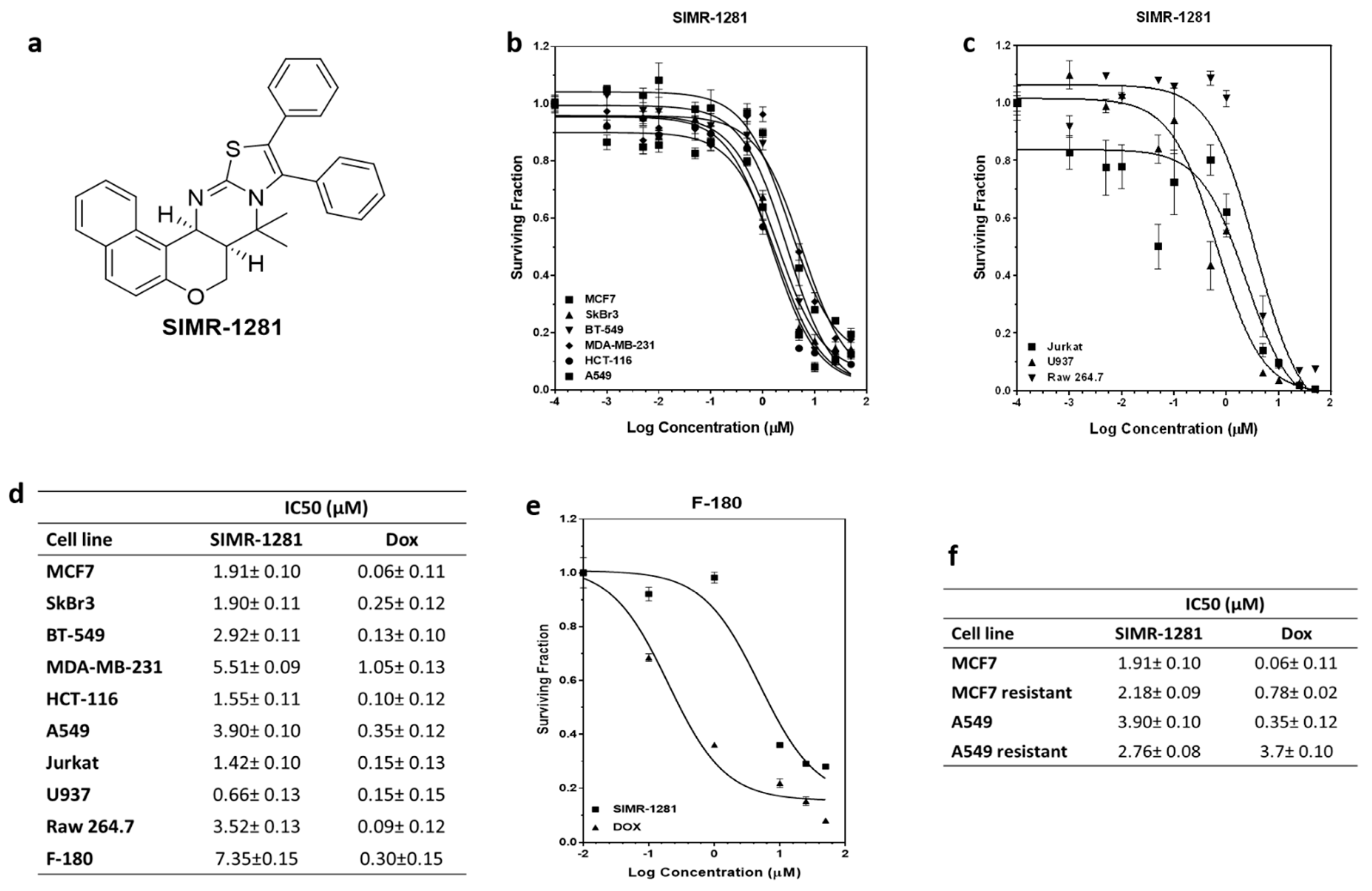
3.1. SIMR1281 Antiproliferative Effect on a Panel of Cancer Cell Lines
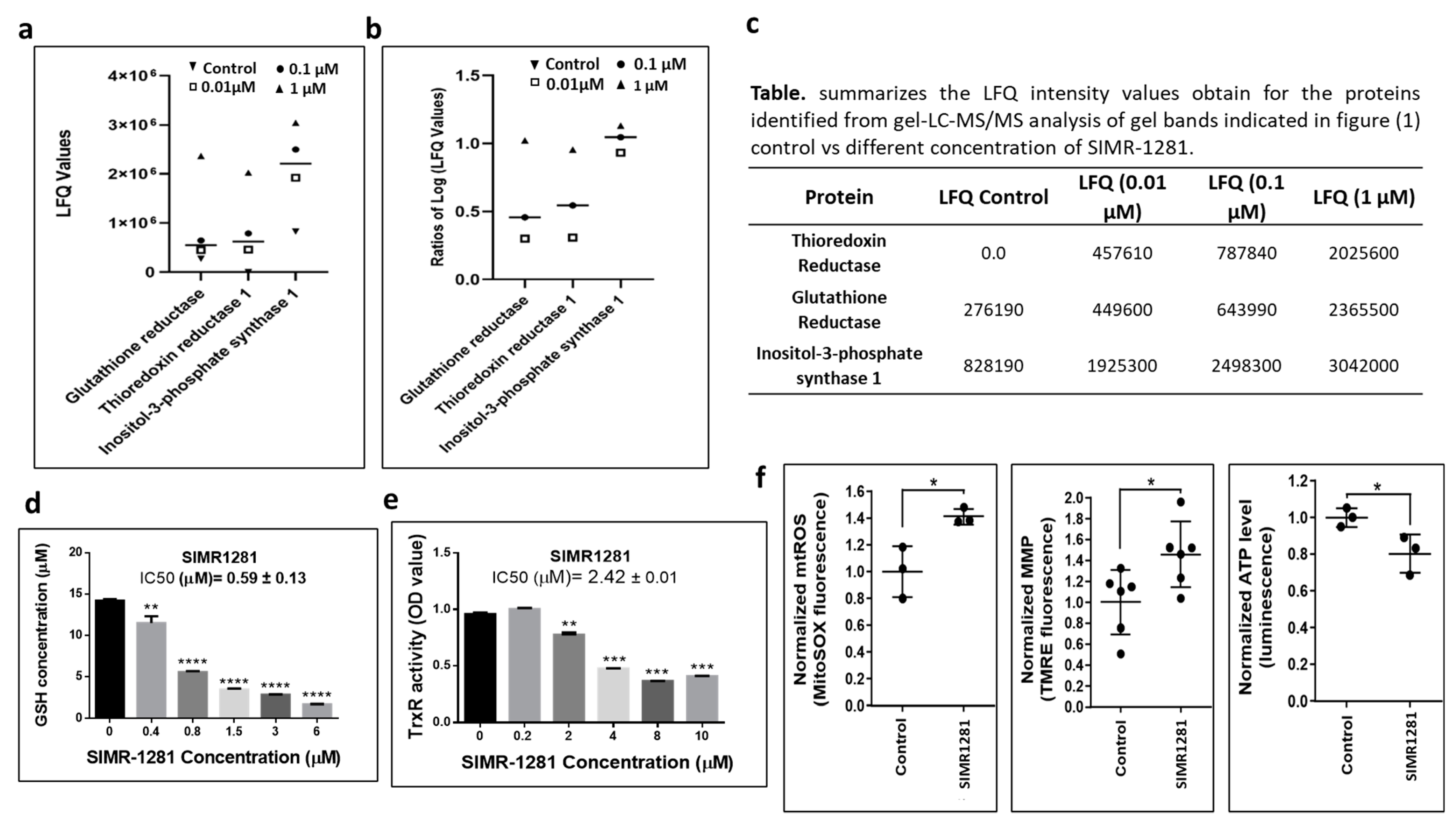
3.2. SIMR1281 Modulates the Functions of Key Proteins and Mitochondria
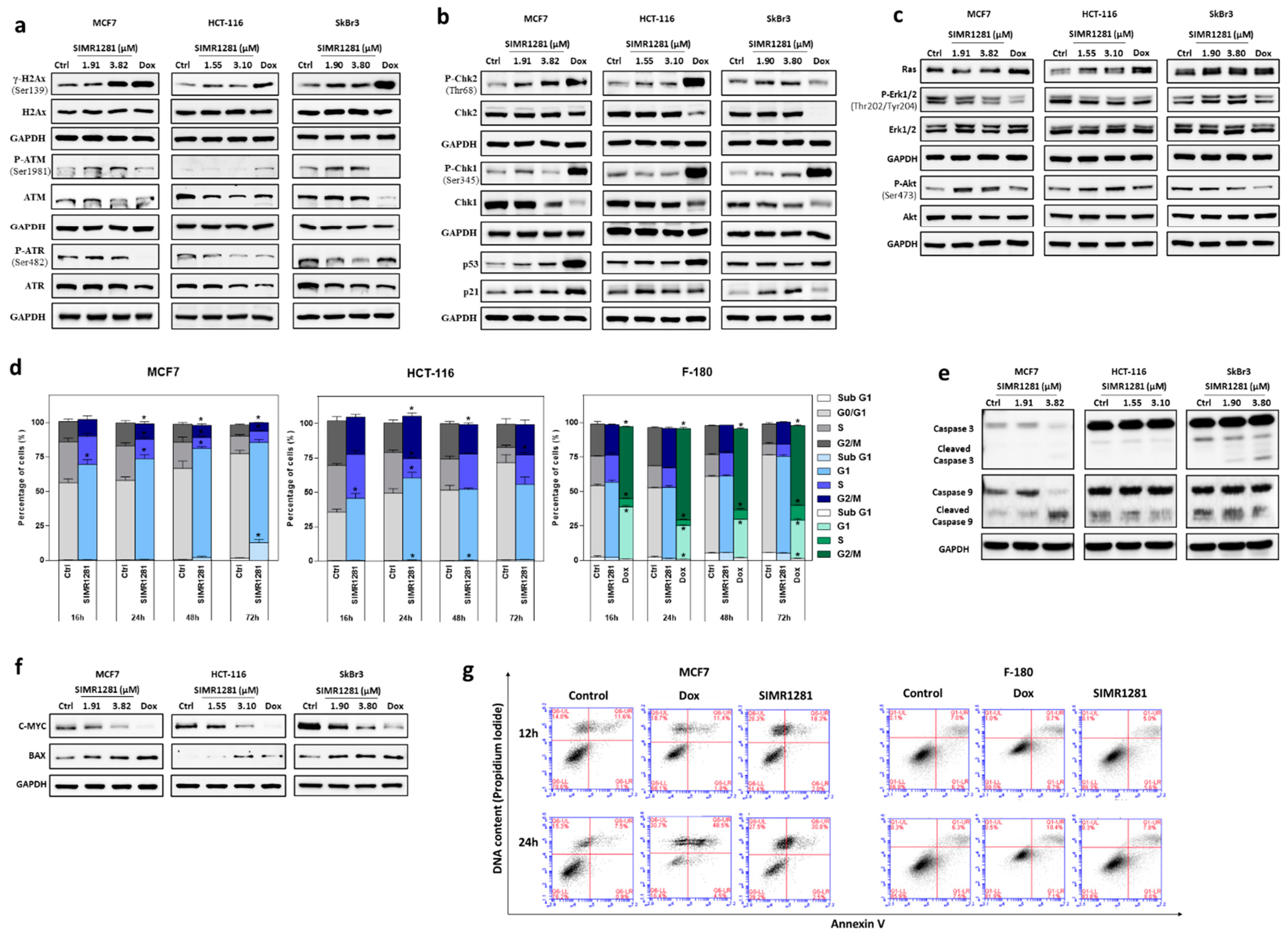
3.3. SIMR1281 Induces DNA Damage and Regulates Checkpoint Kinases, Ras/ERK and PI3K/Akt Pathways
3.4. SIMR1281 Induces Apoptosis and Cell Cycle Arrest
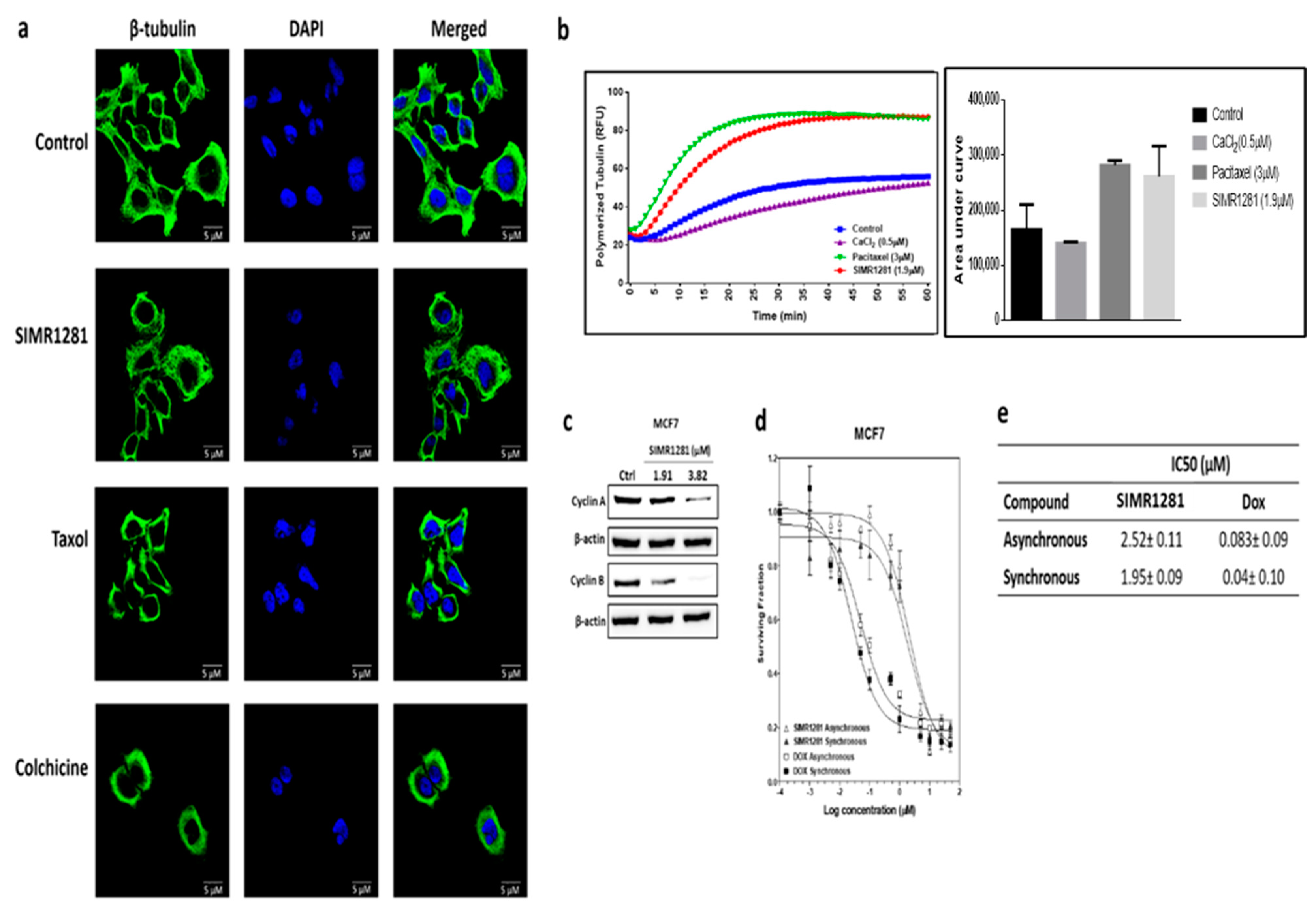
3.5. SIMR1281 Disrupts Tubulin Polymerization
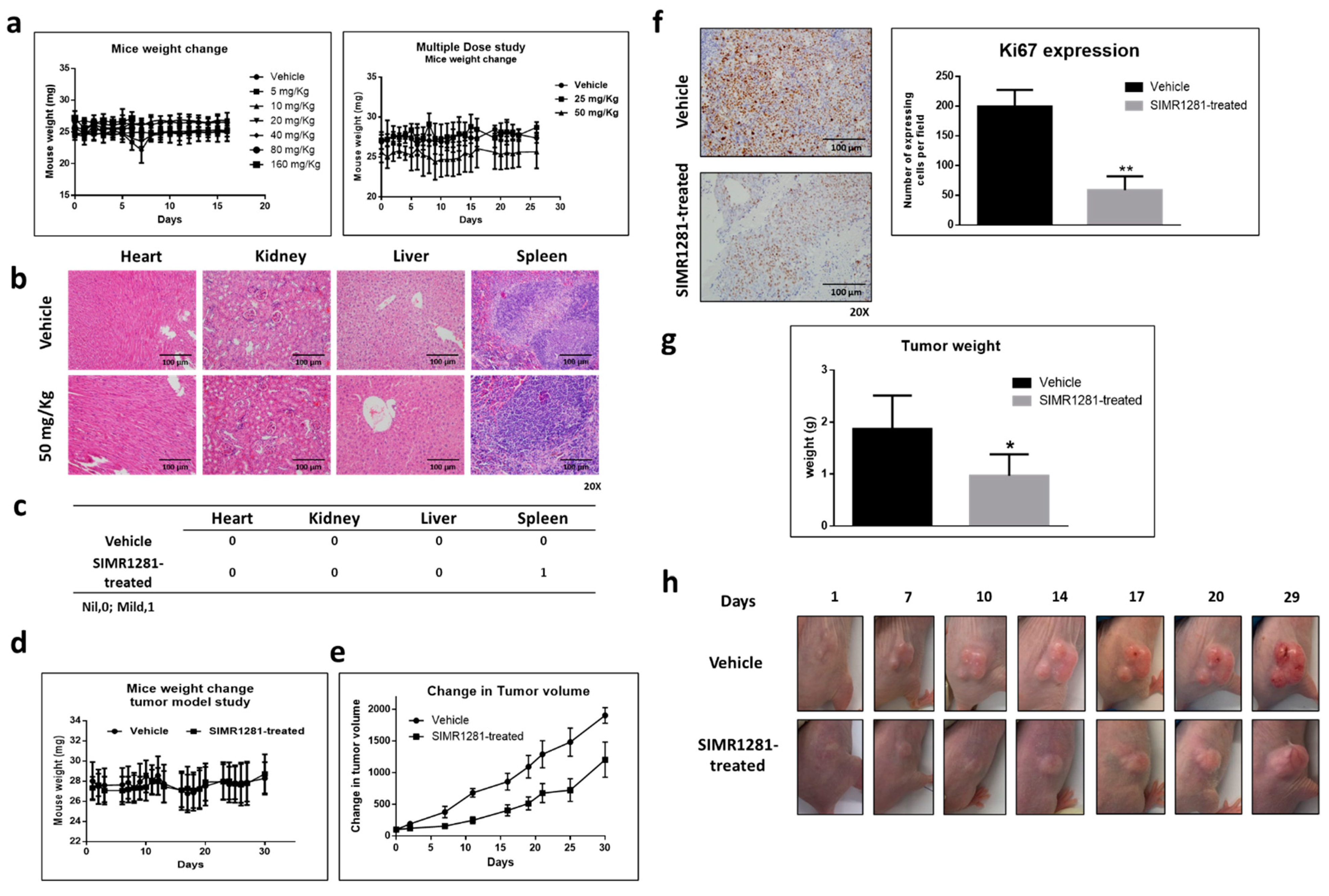
3.6. Safety Profile of SIMR1281 in SJL/J Mice
3.7. SIMR1281 Significantly Attenuates the Proliferation of HCT116 Xenograft Model
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kamb, A.; Wee, S.; Lengauer, C. Why is cancer drug discovery so difficult? Nat. Rev. Drug. Discov. 2007, 6, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Shang, E.; Yuan, Y.; Chen, X.; Liu, Y.; Pei, J.; Lai, L. De novo design of multitarget ligands with an iterative fragment-growing strategy. J. Chem. Inf. Model. 2014, 54, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Anastasio, T.J. Editorial: Computational and experimental approaches in multi-target pharmacology. Front. Pharmacol. 2017, 8, 443. [Google Scholar] [CrossRef]
- Bolognesi, M.L. Harnessing polypharmacology with medicinal chemistry. ACS Med. Chem. Lett. 2019, 10, 273–275. [Google Scholar] [CrossRef]
- Lincoln, D.T.; Ali Emadi, E.M.; Tonissen, K.F.; Clarke, F.M. The thioredoxin-thioredoxin reductase system: Over-expression in human cancer. Anticancer Res. 2003, 23, 2425–2433. [Google Scholar]
- Benhar, M.; Shytaj, I.L.; Stamler, J.S.; Savarino, A. Dual targeting of the thioredoxin and glutathione systems in cancer and HIV. J. Clin. Investig. 2016, 126, 1630–1639. [Google Scholar] [CrossRef]
- George, V.C.; Dellaire, G.; Rupasinghe, H.P.V. Plant flavonoids in cancer chemoprevention: Role in genome stability. J. Nutr. Biochem. 2017, 45, 1–14. [Google Scholar] [CrossRef]
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and molecular targeting therapy in cancer. BioMed. Res. Int. 2014, 2014, 150845. [Google Scholar] [CrossRef]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Hersi, F.; Omar, H.A.; Al-Qawasmeh, R.A.; Ahmad, Z.; Jaber, A.M.; Zaher, D.M.; Al-Tel, T.H. Design and synthesis of new energy restriction mimetic agents: Potent anti-tumor activities of hybrid motifs of aminothiazoles and coumarins. Sci. Rep. 2020, 10, 2893. [Google Scholar] [CrossRef]
- Omar, H.A.; Zaher, D.M.; Srinivasulu, V.; Hersi, F.; Tarazi, H.; Al-Tel, T.H. Design, synthesis and biological evaluation of new pyrrolidine carboxamide analogues as potential chemotherapeutic agents for hepatocellular carcinoma. Eur. J. Med. Chem. 2017, 139, 804–814. [Google Scholar] [CrossRef]
- Rivera, M.; Ramos, Y.; Rodríguez-Valentín, M.; López-Acevedo, S.; Cubano, L.A.; Zou, J.; Zhang, Q.; Wang, G.; Boukli, N.M. Targeting multiple pro-apoptotic signaling pathways with curcumin in prostate cancer cells. PLoS ONE 2017, 12, e0179587. [Google Scholar] [CrossRef]
- Pozarowski, P.; Darzynkiewicz, Z. Analysis of cell cycle by flow cytometry. Methods Mol. Biol. 2004, 281, 301–311. [Google Scholar]
- Semenova, I.; Rodionov, V. Fluorescence microscopy of microtubules in cultured cells. Methods Mol. Med. 2007, 137, 93–102. [Google Scholar] [PubMed]
- Barrero, C.A.; Perez-Leal, O.; Aksoy, M.; Moncada, C.; Ji, R.; Lopez, Y.; Mallilankaraman, K.; Madesh, M.; Criner, G.J.; Kelsen, S.G.; et al. Histone 3.3 participates in a self-sustaining cascade of apoptosis that contributes to the progression of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2013, 188, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Homko, C.J.; Barrero, C.A.; Stein, T.P.; Chen, X.; Cheung, P.; Fecchio, C.; Koller, S.; Merali, S. Excessive caloric intake acutely causes oxidative stress, GLUT4 carbonylation, and insulin resistance in healthy men. Sci. Transl. Med. 2015, 7, 304re7. [Google Scholar] [CrossRef] [PubMed]
- Van den Heuvel, M.J.; Dayan, A.D.; Shillaker, R.O. Evaluation of the BTS approach to the testing of substances and preparations for their acute toxicity. Hum. Toxicol. 1987, 6, 279–291. [Google Scholar] [CrossRef]
- Stallard, N.; Whitehead, A. Reducing animal numbers in the fixed-dose procedure. Hum. Exp. Toxicol. 1995, 14, 315–323. [Google Scholar] [CrossRef]
- Tolba, M.F.; El-Serafi, A.T.; Omar, H.A. Caffeic acid phenethyl ester protects against glucocorticoid-induced osteoporosis in vivo: Impact on oxidative stress and RANKL/OPG signals. Toxicol. Appl. Pharmacol. 2017, 324, 26–35. [Google Scholar] [CrossRef]
- Srinivasulu, V.; Shehadeh, I.; Khanfar, M.A.; Malik, O.G.; Tarazi, H.; Abu-Yousef, I.A.; Sebastian, A.; Baniowda, N.; O’Connor, M.J.; Al-Tel, T.H. One-pot synthesis of diverse collections of benzoxazepine and indolopyrazine fused to heterocyclic systems. J. Org. Chem. 2019, 84, 934–948. [Google Scholar] [CrossRef]
- Srinivasulu, V.; Schilf, P.; Ibrahim, S.; Khanfar, M.A.; Sieburth, S.M.; Omar, H.; Sebastian, A.; AlQawasmeh, R.A.; O’Connor, M.J.; Al-Tel, T.H. Multidirectional desymmetrization of pluripotent building block en route to diastereoselective synthesis of complex nature-inspired scaffolds. Nat. Commun. 2018, 9, 4989. [Google Scholar] [CrossRef] [PubMed]
- Srinivasulu, V.; Sieburth, S.M.; El-Awady, R.; Kariem, N.M.; Tarazi, H.; O’Connor, M.J.; Al-Tel, T.H. Post-Ugi cascade transformations for accessing diverse chromenopyrrole collections. Org. Lett. 2018, 20, 836–839. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, H.; Park, S.B. Privileged structures: Efficient chemical “navigators” toward unexplored biologically relevant chemical spaces. J. Am. Chem. Soc. 2014, 136, 14629–14638. [Google Scholar] [CrossRef] [PubMed]
- Taleb, H.; Altel, R.A.E.-A.; Srinivasulu, V.; Cijo, G.V.; Hany, A.O. Novel Heterocyclic Systems And Pharmaceutical Compositions Thereof. US Patent Patent Application No. 15/935155, 26 September 2019. [Google Scholar]
- Srinivasulu, V.; Reddy, A.; Mazitschek, R.; Lukens, A.K.; Wirth, D.F.; Li, L.; Naumov, P.; O’Connor, M.J.; Al-Tel, T.H.; Vunnam, S. Intramolecular diaza-diels-alder protocol: A new diastereoselective and modular one-step synthesis of constrained polycyclic frameworks. Chemistry 2017, 23, 4137–4148. [Google Scholar] [CrossRef]
- Pai, M.Y.; Lomenick, B.; Hwang, H.; Schiestl, R.; McBride, W.; Loo, J.A.; Huang, J. Drug affinity responsive target stability (DARTS) for small-molecule target identification. Methods Mol. Biol. 2015, 1263, 287–298. [Google Scholar]
- Cox, J.; Hein, M.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef]
- Logan, A.; Pell, V.R.; Shaffer, K.J.; Evans, C.; Stanley, N.J.; Robb, E.L.; Prime, T.A.; Chouchani, E.T.; Cochemé, H.M.; Fearnley, I.M.; et al. Assessing the Mitochondrial Membrane Potential in Cells and In Vivo using Targeted Click Chemistry and Mass Spectrometry. Cell Metab. 2016, 23, 379–385. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Yaffe, M.B. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell. Biol. 2009, 21, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Ravingerova, T.; Barancik, M.; Strniskova, M. Mitogen-activated protein kinases: A new therapeutic target in cardiac pathology. Mol. Cell Biochem. 2003, 247, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Cheng, J.C.; Chang, H.M.; Leung, P.C. COX2 and PGE2 mediate EGF-induced E-cadherin-independent human ovarian cancer cell invasion. Endocr. Relat. Cancer. 2014, 21, 533–543. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ahn, I.E.; Ju, J.H.; Lee, S.Y.; Park, J.S.; Oh, H.J.; Kim, H.R.; Lee, S.H.; Park, S.H.; Kim, H.Y.; Cho, M.L. Upregulation of stromal cell-derived factor by IL-17 and IL-18 via a phosphatidylinositol 3-kinase-dependent pathway. Scand. J. Immunol. 2012, 76, 433–439. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, Y.R.; Park, J.; Kim, S. Inositol polyphosphate multikinase signaling in the regulation of metabolism. Ann. N. Y. Acad. Sci. 2012, 1271, 68–74. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef]
- Emami, S.; Dadashpour, S. Current developments of coumarin-based anticancer agents in medicinal chemistry. Eur. J. Med. Chem. 2015, 102, 611–630. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, J.; Peng, S.; Liu, R.; Li, X.; Hou, Y.; Han, X.; Fang, J. Thioredoxin reductase inhibitors: A patent review. Expert Opin. Ther. Pat. 2017, 27, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M.; McGrath, K.L.; Di Trapani, G.; Charoentong, P.; Shah, F.; King, M.M.; Clarke, F.M.; Tonissen, K.F. The thioredoxin system in breast cancer cell invasion and migration. Redox Biol. 2016, 8, 68–78. [Google Scholar] [CrossRef]
- Kiebala, M.; Skalska, J.; Casulo, C.; Brookes, P.; Peterson, D.R.; Hilchey, S.P.; Dai, Y.; Grant, S.; Maggirwar, S.B.; Bernstein, S.H. Dual targeting of the thioredoxin and glutathione antioxidant systems in malignant B cells: A novel synergistic therapeutic approach. Exp. Hematol. 2015, 43, 89–99. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef]
- Bizzarri, M.; Dinicola, S.; Bevilacqua, A.; Cucina, A. Broad spectrum anticancer activity of myo-inositol and inositol hexakisphosphate. Int. J. Endocrinol. 2016, 2016, 5616807. [Google Scholar] [CrossRef] [PubMed]
- Frej, A.D.; Clark, J.; Le Roy, C.I.; Lilla, S.; Thomason, P.; Otto, G.; Churchill, G.; Insall, R.; Claus, S.P.; Hawkins, P.; et al. The inositol-3-phosphate synthase biosynthetic enzyme has distinct catalytic and metabolic roles. Mol. Cell. Biol. 2016, 36, 1464–1479. [Google Scholar] [CrossRef]
- Maag, D.; Maxwell, M.J.; Hardesty, D.A.; Boucher, K.L.; Choudhari, N.; Hanno, A.G.; Ma, J.F.; Snowman, A.S.; Pietropaoli, J.W.; Xu, R.; et al. Inositol polyphosphate multikinase is a physiologic PI3-kinase that activates Akt/PKB. Proc. Natl. Acad. Sci. USA 2011, 108, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Ravanat, J.-L.; TavernaPorro, M.; Menoni, H.; Angelov, D. Oxidatively generated complex DNA damage: Tandem and clustered lesions. Cancer Lett. 2012, 327, 5–15. [Google Scholar] [CrossRef]
- El-Awady, R.A.; Semreen, M.H.; Saber-Ayad, M.M.; Cyprian, F.; Menon, V.; Al-Tel, T.H. Modulation of DNA damage response and induction of apoptosis mediates synergism between doxorubicin and a new imidazopyridine derivative in breast and lung cancer cells. DNA Repair 2016, 37, 1–11. [Google Scholar] [CrossRef] [PubMed]
- El-Awady, R.A.; Ali, M.M.; Saleh, E.M.; Ghaleb, F.M. Apoptosis is the most efficient death-pathway in tumor cells after topoisomerase II inhibition. Saudi Med. J. 2008, 29, 558–564. [Google Scholar]
- Lim, J.K.M.; Leprivier, G. The impact of oncogenic RAS on redox balance and implications for cancer development. Cell Death Dis. 2019, 10, 955. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Ju, S.; Greenberg, M.L. 1D-myo-inositol 3-phosphate synthase: Conservation, regulation, and putative target of mood stabilizers. Clin. Neurosci. Res. 2004, 4, 181–187. [Google Scholar] [CrossRef]
- Sales, G.R.; Vagnarelli, P. Ki-67: More hidden behind a ‘classic proliferation marker’. Trends Biochem. Sci. 2018, 43, 747–748. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaher, D.M.; Ramadan, W.S.; El-Awady, R.; Omar, H.A.; Hersi, F.; Srinivasulu, V.; Hachim, I.Y.; Al-Marzooq, F.I.; Vazhappilly, C.G.; Merali, S.; et al. A Novel Benzopyrane Derivative Targeting Cancer Cell Metabolic and Survival Pathways. Cancers 2021, 13, 2840. https://doi.org/10.3390/cancers13112840
Zaher DM, Ramadan WS, El-Awady R, Omar HA, Hersi F, Srinivasulu V, Hachim IY, Al-Marzooq FI, Vazhappilly CG, Merali S, et al. A Novel Benzopyrane Derivative Targeting Cancer Cell Metabolic and Survival Pathways. Cancers. 2021; 13(11):2840. https://doi.org/10.3390/cancers13112840
Chicago/Turabian StyleZaher, Dana M., Wafaa S. Ramadan, Raafat El-Awady, Hany A. Omar, Fatema Hersi, Vunnam Srinivasulu, Ibrahim Y. Hachim, Farah I. Al-Marzooq, Cijo G. Vazhappilly, Salim Merali, and et al. 2021. "A Novel Benzopyrane Derivative Targeting Cancer Cell Metabolic and Survival Pathways" Cancers 13, no. 11: 2840. https://doi.org/10.3390/cancers13112840
APA StyleZaher, D. M., Ramadan, W. S., El-Awady, R., Omar, H. A., Hersi, F., Srinivasulu, V., Hachim, I. Y., Al-Marzooq, F. I., Vazhappilly, C. G., Merali, S., Merali, C., Soares, N. C., Schilf, P., Ibrahim, S. M., & Al-Tel, T. H. (2021). A Novel Benzopyrane Derivative Targeting Cancer Cell Metabolic and Survival Pathways. Cancers, 13(11), 2840. https://doi.org/10.3390/cancers13112840