
Diversity of Epithelial-Mesenchymal Phenotypes in Circulating Tumour Cells from Prostate Cancer Patient-Derived Xenograft Models


Abstract
Simple Summary
Abstract
1. Introduction
2. Results
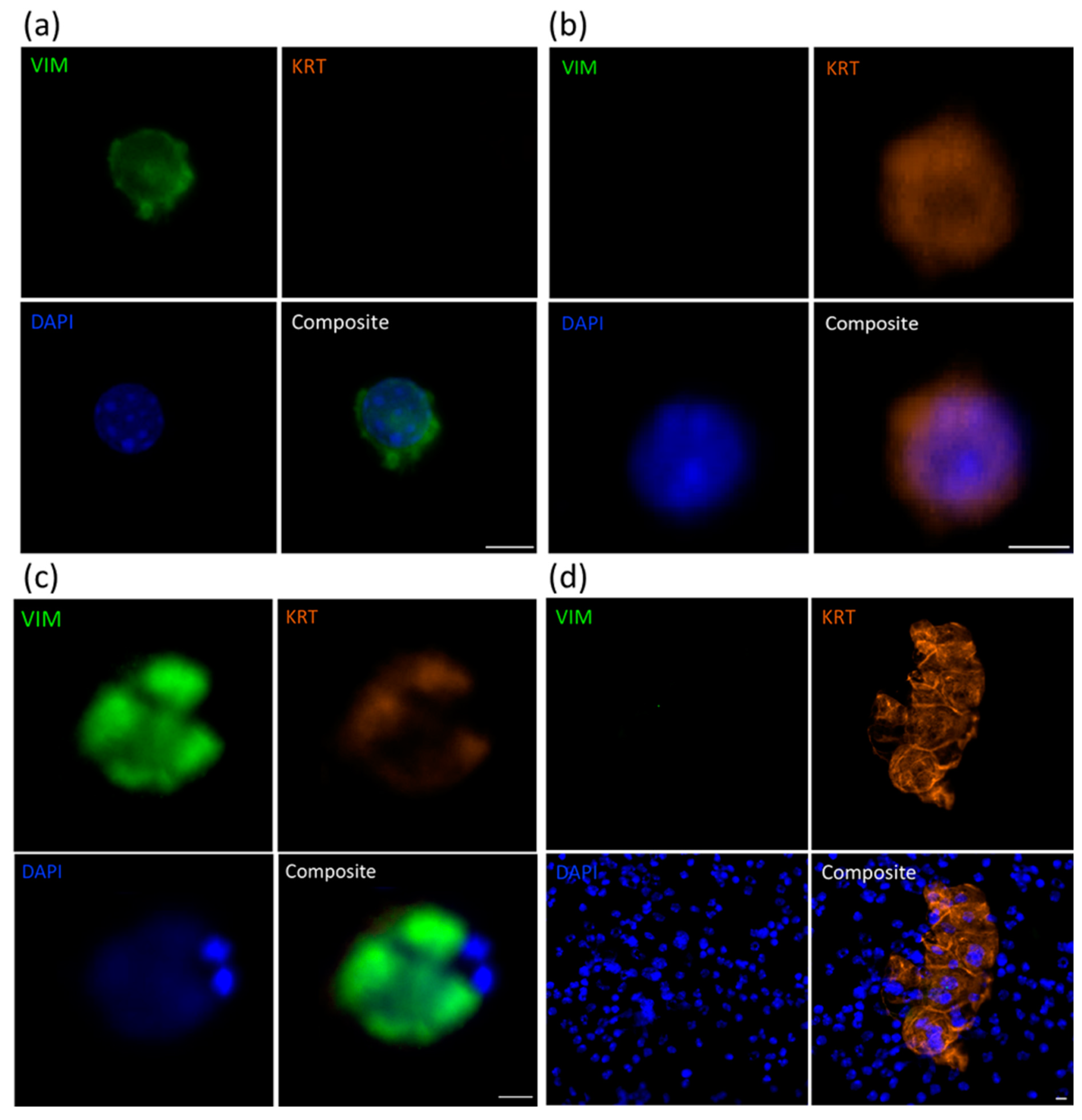
2.1. Identification of CTCs Using Immunofluorescence Analysis
2.2. RNA-Based Pre-Screening of Blood Samples for the Presence of CTCs
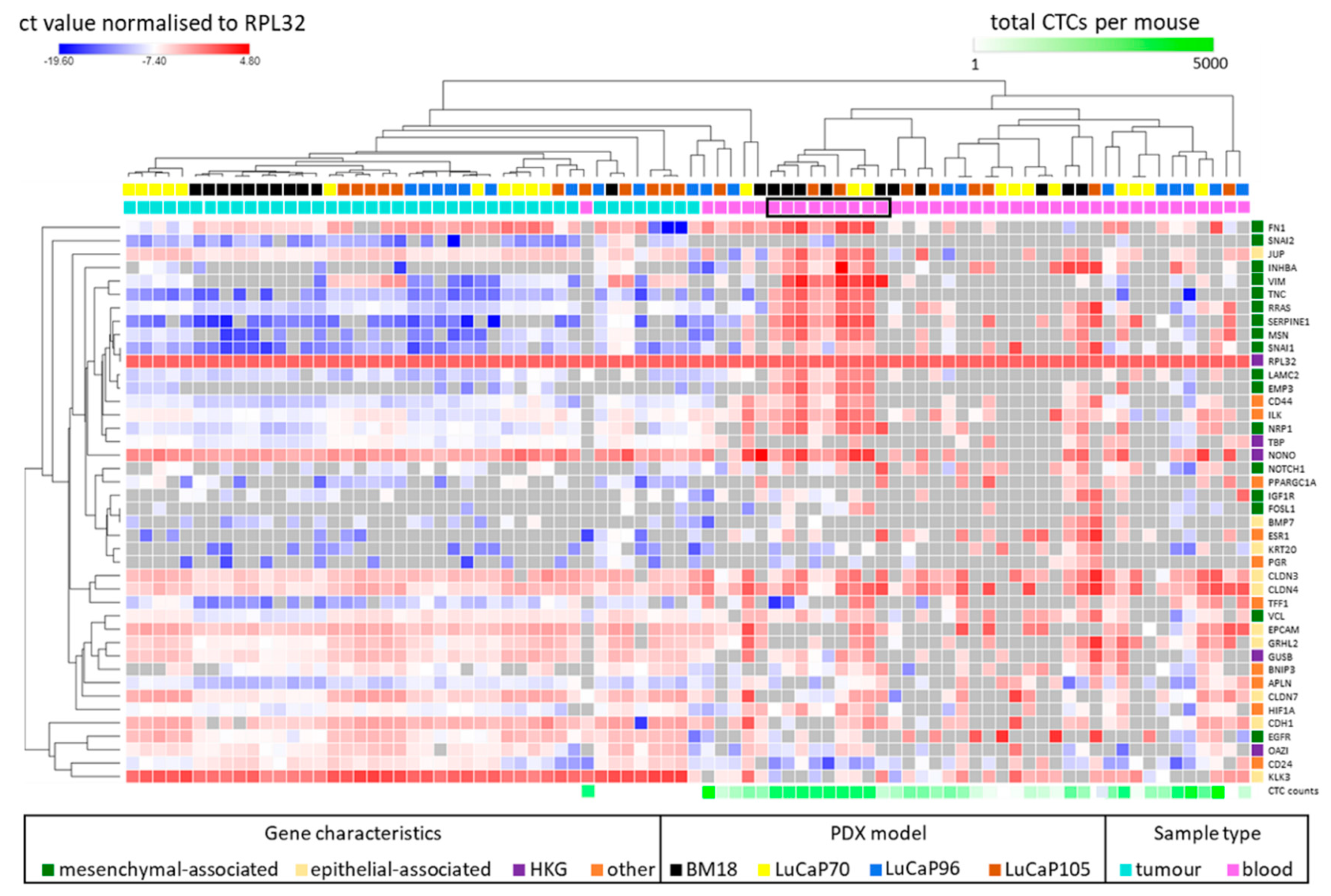
2.3. Human-Specific RT-qPCR Profiling of Paired PDX Tumour and Blood Samples
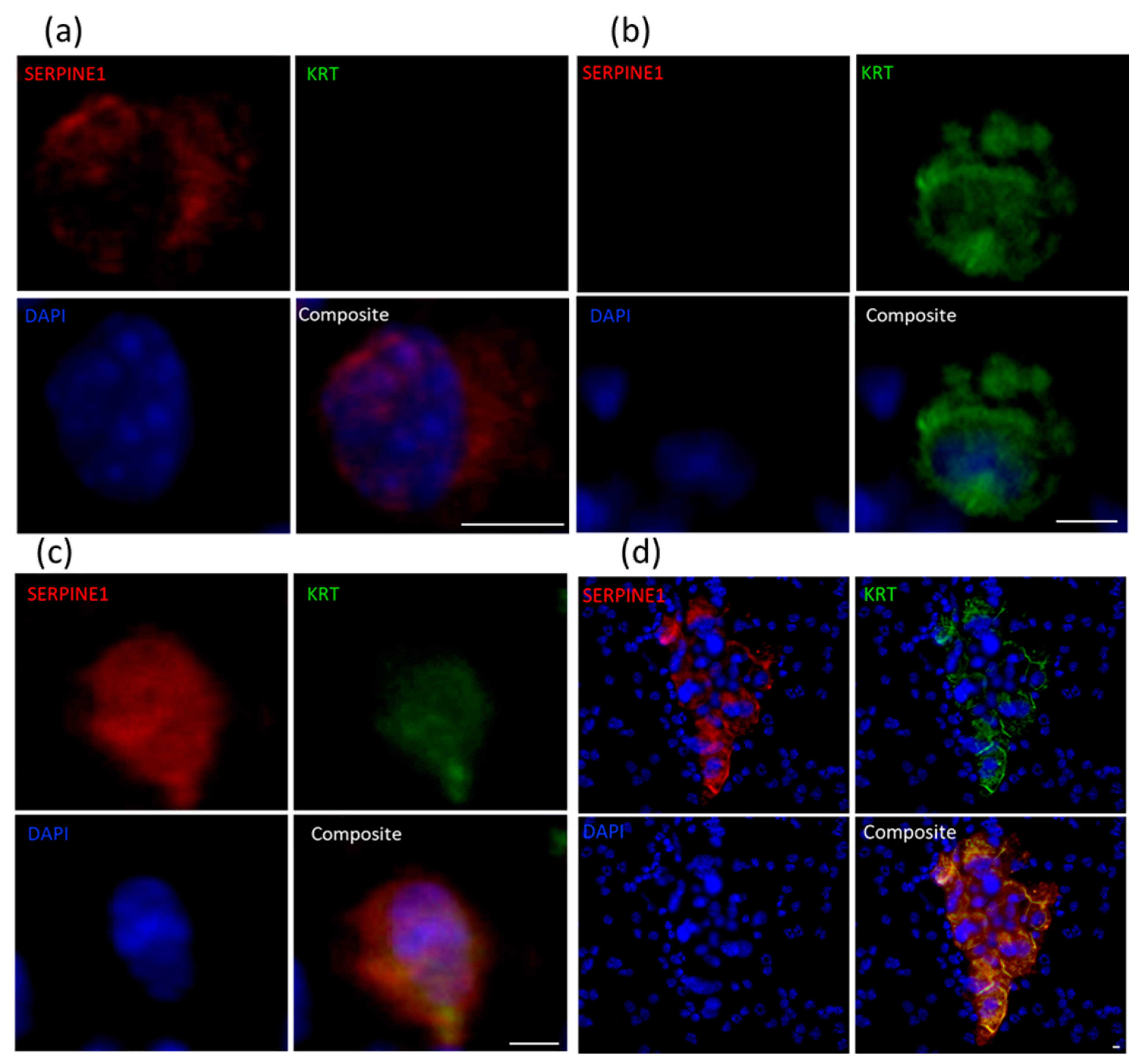
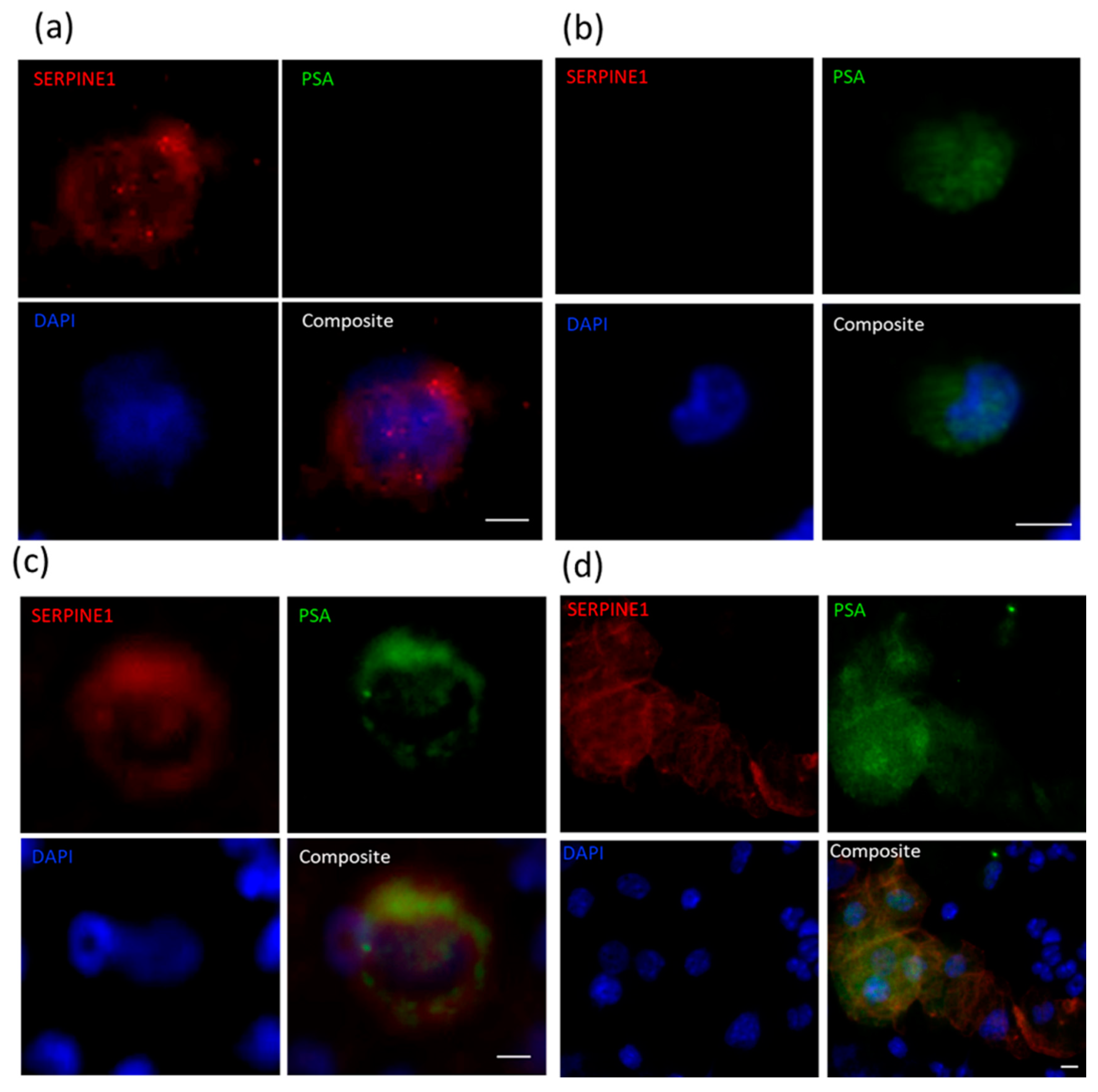
2.4. Immunocytochemical Assessment of Specific Markers
3. Discussion
4. Methodology
4.1. Mouse Blood and Tissue Collection
4.2. Sample Processing
4.3. Immunocytochemistry
4.4. Molecular Analysis Using RT-qPCR of Mouse Xenograft Samples
4.5. cDNA Synthesis from RNA Samples
4.6. cDNA Precipitation
4.7. Pre-Amplification
4.8. Tandem Nested RT-qPCR
4.9. Testing Specificity of RPL32 Primer Set
4.10. Estimation of Total Number of CTCs per Mouse
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Nicholson, C.; Vela, I.; Williams, E.D. Introduction to cancer metastasis. In Prostate Cancer Metastasis; Ahmad, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Yossepowitch, O.; Bianco, F.J.; Eggener, S.E.; Eastham, J.A.; Scher, H.I.; Scardino, P.T. The Natural History of Noncastrate Metastatic Prostate Cancer after Radical Prostatectomy. Eur. Urol. 2007, 51, 940–948. [Google Scholar] [CrossRef]
- Cieślikowski, W.A.; Budna-Tukan, J.; Świerczewska, M.; Ida, A.; Hrab, M.; Jankowiak, A.; Mazel, M.; Nowicki, M.; Milecki, P.; Pantel, K.; et al. Circulating Tumor Cells as a Marker of Disseminated Disease in Patients with Newly Diagnosed High-Risk Prostate Cancer. Cancers 2020, 12, 160. [Google Scholar] [CrossRef]
- Danila, D.C.; Heller, G.; Gignac, G.A.; Gonzalez-Espinoza, R.; Anand, A.; Tanaka, E.; Lilja, H.; Schwartz, L.; Larson, S.; Fleisher, M.; et al. Circulating Tumor Cell Number and Prognosis in Progressive Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2007, 13, 7053–7058. [Google Scholar] [CrossRef]
- Pak, S.; Suh, Y.S.; Lee, D.-E.; Kim, S.H.; Joung, J.Y.; Park, W.S.; Lee, S.-J.; Lee, K.H. Association between Postoperative Detection of Circulating Tumor Cells and Recurrence in Patients with Prostate Cancer. J. Urol. 2020, 203, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Bidard, F.-C.; Proudhon, C.; Pierga, J.-Y. Circulating tumor cells in breast cancer. Mol. Oncol. 2016, 10, 418–430. [Google Scholar] [CrossRef] [PubMed]
- Garrel, R.; Cupissol, D.; Alix-Panabieres, C. Circulating tumour cells in head and neck cancer. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2020, 137, 235. [Google Scholar] [CrossRef]
- Kulasinghe, A.; Kapeleris, J.; Cooper, C.; Warkiani, M.E.; O’Byrne, K.; Punyadeera, C. Phenotypic Characterization of Circulating Lung Cancer Cells for Clinically Actionable Targets. Cancers 2019, 11, 380. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.; Rubin, M.A.; Wei, J.T.; Chinnaiyan, A.M. Beyond PSA: The Next Generation of Prostate Cancer Biomarkers. Sci. Transl. Med. 2012, 4, 127rv3. [Google Scholar] [CrossRef]
- Pantel, K.; Hille, C.; Scher, H.I. Circulating Tumor Cells in Prostate Cancer: From Discovery to Clinical Utility. Clin. Chem. 2019, 65, 87–99. [Google Scholar] [CrossRef]
- Hugen, C.M.; Zainfeld, D.E.; Goldkorn, A. Circulating Tumor Cells in Genitourinary Malignancies: An Evolving Path to Precision Medicine. Front. Oncol. 2017, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Graf, R.P.; Schreiber, N.A.; McLaughlin, B.; Jendrisak, A.; Wang, Y.; Lee, J.; Greene, S.; Krupa, R.; Lu, D.; et al. Phenotypic Heterogeneity of Circulating Tumor Cells Informs Clinical Decisions between AR Signaling Inhibitors and Taxanes in Metastatic Prostate Cancer. Cancer Res. 2017, 77, 5687–5698. [Google Scholar] [CrossRef]
- Sepe, P.; Verzoni, E.; Miodini, P.; Claps, M.; Ratta, R.; Martinetti, A.; Mennitto, R.; Sottotetti, E.; Procopio, G.; Cappelletti, V.; et al. Could Circulating Tumor Cells and ARV7 Detection Improve Clinical Decisions in Metastatic Castration-Resistant Prostate Cancer? The Istituto Nazionale dei Tumori (INT) Experience. Cancers 2019, 11, 980. [Google Scholar] [CrossRef]
- Miller, M.C.; Doyle, G.V.; Terstappen, L.W.M.M. Significance of Circulating Tumor Cells Detected by the CellSearch System in Patients with Metastatic Breast Colorectal and Prostate Cancer. J. Oncol. 2010, 2010, 617421. [Google Scholar] [CrossRef]
- Nouri, M.; Ratther, E.; Stylianou, N.; Nelson, C.C.; Hollier, B.G.; Williams, E.D. Androgen-Targeted Therapy-Induced Epithelial Mesenchymal Plasticity and Neuroendocrine Transdifferentiation in Prostate Cancer: An Opportunity for Intervention. Front. Oncol. 2014, 4, 370. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.D.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around epithelial–mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.W.; Newgreen, D.F. Carcinoma Invasion and Metastasis: A Role for Epithelial-Mesenchymal Transition? Cancer Res. 2005, 65, 5991–5995. [Google Scholar] [CrossRef]
- Francart, M.-E.; Lambert, J.; Vanwynsberghe, A.M.; Thompson, E.W.; Bourcy, M.; Polette, M.; Gilles, C. Epithelial-mesenchymal plasticity and circulating tumor cells: Travel companions to metastases. Dev. Dyn. 2017, 247, 432–450. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Brennan, J.P.; Slavin, J.L.; Blick, T.; Thompson, E.W.; Williams, E.D. Mesenchymal-to-Epithelial Transition Facilitates Bladder Cancer Metastasis: Role of Fibroblast Growth Factor Receptor-2. Cancer Res. 2006, 66, 11271–11278. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Jolly, M.K.; Murphy, R.J.; Bhatia, S.; Whitfield, H.J.; Redfern, A.; Davis, M.J.; Thompson, E.W. Measuring and Modelling the Epithelial- Mesenchymal Hybrid State in Cancer: Clinical Implications. Cells Tissues Organs 2021, 26, 1–24. [Google Scholar] [CrossRef]
- Joshi, A.; Roberts, M.J.; Alinezhad, S.; Williams, E.D.; Vela, I. Challenges, applications and future directions of precision medicine in prostate cancer—The role of organoids and patient-derived xenografts. BJU Int. 2020, 126, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Wei, X.; Lin, S.; Qin, L.; Cheng, L.; Li, P. Current status and perspectives of patient-derived xenograft models in cancer research. J. Hematol. Oncol. 2017, 10, 106. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Li, X.; Liu, P.; Li, M.; Luo, F. Patient-derived xenograft mouse models: A high fidelity tool for individualized medicine (Review). Oncol. Lett. 2018, 17, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Tachtsidis, A.; Le, A.V.-P.; Blick, T.; Gunasinghe, D.; De Sousa, E.; Waltham, M.; Dobrovic, A.; Thompson, E.W. Human-specific RNA analysis shows uncoupled epithelial-mesenchymal plasticity in circulating and disseminated tumour cells from human breast cancer xenografts. Clin. Exp. Metastasis 2019, 36, 393–409. [Google Scholar] [CrossRef]
- Agnoletto, C.; Minotti, L.; Brulle-Soumare, L.; Pasquali, L.; Galasso, M.; Corrà, F.; Baldassari, F.; Judde, J.-G.; Cairo, S.; Volinia, S. Heterogeneous expression of EPCAM in human circulating tumour cells from patient-derived xenografts. Biomark. Res. 2018, 6, 31. [Google Scholar] [CrossRef]
- Giuliano, M.; Herrera, S.; Christiny, P.; Shaw, C.; Creighton, C.J.; Mitchell, T.; Bhat, R.; Zhang, X.; Mao, S.; Dobrolecki, L.E.; et al. Circulating and disseminated tumor cells from breast cancer patient-derived xenograft-bearing mice as a novel model to study metastasis. Breast Cancer Res. 2015, 17, 3. [Google Scholar] [CrossRef]
- Pillai, S.G.; Li, S.; Siddappa, C.M.; Ellis, M.J.; Watson, M.A.; Aft, R. Identifying biomarkers of breast cancer micrometastatic disease in bone marrow using a patient-derived xenograft mouse model. Breast Cancer Res. 2018, 20, 2. [Google Scholar] [CrossRef]
- Ramirez, A.B.; Bhat, R.; Sahay, D.; De Angelis, C.; Thangavel, H.; Hedayatpour, S.; Dobrolecki, L.E.; Nardone, A.; Giuliano, M.; Nagi, C.; et al. Circulating tumor cell investigation in breast cancer patient-derived xenograft models by automated immunofluorescence staining, image acquisition, and single cell retrieval and analysis. BMC Cancer 2019, 19, 220. [Google Scholar] [CrossRef]
- O’Connell, K.E.; Mikkola, A.M.; Stepanek, A.M.; Vernet, A.; Hall, C.D.; Sun, C.C.; Yildirim, E.; Staropoli, J.F.; Lee, J.T.; Brown, D.E. Practical Murine Hematopathology: A Comparative Review and Implications for Research. Comp. Med. 2015, 65, 96–113. [Google Scholar] [PubMed]
- Cackowski, F.C.; Taichman, R.S. Minimal Residual Disease in Prostate Cancer. Chemistry and Biology of Pteridines and Folates 2018, 1100, 47–53. [Google Scholar] [CrossRef]
- Xue, J.; Xie, V.; Wang, P.; Cui, J.; Gao, Y.; Lu, Z. Interrelationships of Circulating Tumor Cells with Metastasis and Thrombosis: Role of MicroRNAs. Curr. Pharm. Des. 2014, 20, 5298–5308. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Dicker, D.T.; Matthew, E.; El-Deiry, W.; Alpaugh, R.K. Circulating tumor cells: Silent predictors of metastasis. F1000Research 2017, 6, 1445. [Google Scholar] [CrossRef]
- He, W.; Hou, M.; Zhang, H.; Zeng, C.; He, S.; Chen, X.; Xu, M.; Sun, C.; Jiang, W.; Wang, H.; et al. Clinical significance of circulating tumor cells in predicting disease progression and chemotherapy resistance in patients with gestational choriocarcinoma. Int. J. Cancer 2019, 144, 1421–1431. [Google Scholar] [CrossRef] [PubMed]
- Lemech, C.R.; Ensell, L.; Paterson, J.C.; Eminowicz, G.; Lowe, H.; Arora, R.; Arkenau, H.-T.; Widschwendter, M.; MacDonald, N.; Olaitan, A.; et al. Enumeration and Molecular Characterisation of Circulating Tumour Cells in Endometrial Cancer. Oncol. 2016, 91, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Payne, R.E.; Wang, F.; Su, N.; Krell, J.; Zebrowski, A.; Yague, E.; Ma, X.-J.; Luo, Y.; Coombes, R.C. Viable circulating tumour cell detection using multiplex RNA in situ hybridisation predicts progression-free survival in metastatic breast cancer patients. Br. J. Cancer 2012, 106, 1790–1797. [Google Scholar] [CrossRef]
- Wang, C.; Mu, Z.; Chervoneva, I.; Austin, L.; Ye, Z.; Rossi, G.; Palazzo, J.P.; Sun, C.; Abu-Khalaf, M.; Myers, R.E.; et al. Longitudinally collected CTCs and CTC-clusters and clinical outcomes of metastatic breast cancer. Breast Cancer Res. Treat. 2017, 161, 83–94. [Google Scholar] [CrossRef]
- McDaniel, A.S.; Ferraldeschi, R.; Krupa, R.; Landers, M.; Graf, R.; Louw, J.; Jendrisak, A.; Bales, N.; Marrinucci, D.; Zafeiriou, Z.; et al. Phenotypic diversity of circulating tumour cells in patients with metastatic castration-resistant prostate cancer. BJU Int. 2016, 120, E30–E44. [Google Scholar] [CrossRef]
- Grant, C.M.; Kyprianou, N. Epithelial mesenchymal transition (EMT) in prostate growth and tumor progression. Transl. Androl. Urol. 2013, 2, 202–211. [Google Scholar] [PubMed]
- Odero-Marah, V.; Hawsawi, O.; Henderson, V.; Sweeney, J. Epithelial-Mesenchymal Transition (EMT) and Prostate Cancer. Adv. Exp. Med. Biol. 2018, 1095, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Yang, R.; Gao, W.-Q. Contributions of epithelial-mesenchymal transition and cancer stem cells to the development of castration resistance of prostate cancer. Mol. Cancer 2014, 13, 55. [Google Scholar] [CrossRef]
- Chen, C.; Mahalingam, D.; Osmulski, P.; Jadhav, R.; Wang, C.; Leach, R.J.; Chang, T.A.; Weitman, S.D.; Kumar, A.P.; Sun, L.; et al. Single-cell analysis of circulating tumor cells identifies cumulative expression patterns of EMT-related genes in metastatic prostate cancer. Prostate 2013, 73, 813–826. [Google Scholar] [CrossRef]
- Montanari, M.; Rossetti, S.; Cavaliere, C.; D’Aniello, C.; Malzone, M.G.; Vanacore, D.; Di Franco, R.; La Mantia, E.; Iovane, G.; Piscitelli, R.; et al. Epithelial-mesenchymal transition in prostate cancer: An overview. Oncotarget 2017, 8, 35376–35389. [Google Scholar] [CrossRef]
- Wade, C.A.; Kyprianou, N. Profiling Prostate Cancer Therapeutic Resistance. Int. J. Mol. Sci. 2018, 19, 904. [Google Scholar] [CrossRef]
- Park, S.; Ang, R.R.; Duffy, S.P.; Bazov, J.; Chi, K.N.; Black, P.C.; Ma, H. Morphological Differences between Circulating Tumor Cells from Prostate Cancer Patients and Cultured Prostate Cancer Cells. PLoS ONE 2014, 9, e85264. [Google Scholar] [CrossRef]
- Aceto, N. Bring along your friends: Homotypic and heterotypic circulating tumor cell clustering to accelerate metastasis. Biomed. J. 2020, 43, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nat. Cell Biol. 2019, 566, 553–557. [Google Scholar] [CrossRef]
- Sprouse, M.L.; Welte, T.; Boral, D.; Liu, H.N.; Yin, W.; Vishnoi, M.; Goswami-Sewell, D.; Li, L.; Pei, G.; Jia, P.; et al. PMN-MDSCs Enhance CTC Metastatic Properties through Reciprocal Interactions via ROS/Notch/Nodal Signaling. Int. J. Mol. Sci. 2019, 20, 1916. [Google Scholar] [CrossRef]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef]
- Duda, D.G.; Duyverman, A.M.M.J.; Kohno, M.; Snuderl, M.; Steller, E.J.A.; Fukumura, D.; Jain, R.K. Malignant cells facilitate lung metastasis by bringing their own soil. Proc. Natl. Acad. Sci. USA 2010, 107, 21677–21682. [Google Scholar] [CrossRef] [PubMed]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating Tumor Cell Clusters Are Oligoclonal Precursors of Breast Cancer Metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Castro-Giner, F.; Aceto, N. Tracking cancer progression: From circulating tumor cells to metastasis. Genome Med. 2020, 12, 31. [Google Scholar] [CrossRef]
- Liu, Y.; Chanana, P.; Davila, J.I.; Hou, X.; Zanfagnin, V.; McGehee, C.D.; Goode, E.L.; Polley, E.C.; Haluska, P.; Weroha, S.J.; et al. Gene expression differences between matched pairs of ovarian cancer patient tumors and patient-derived xenografts. Sci. Rep. 2019, 9, 6314. [Google Scholar] [CrossRef] [PubMed]
- Ting, D.; Wittner, B.S.; Ligorio, M.; Jordan, N.V.; Shah, A.M.; Miyamoto, D.T.; Aceto, N.; Bersani, F.; Brannigan, B.W.; Xega, K.; et al. Single-Cell RNA Sequencing Identifies Extracellular Matrix Gene Expression by Pancreatic Circulating Tumor Cells. Cell Rep. 2014, 8, 1905–1918. [Google Scholar] [CrossRef]
- Miyamoto, D.T.; Zheng, Y.; Wittner, B.S.; Lee, R.J.; Zhu, H.; Broderick, K.T.; Desai, R.; Fox, D.B.; Brannigan, B.W.; Trautwein, J.; et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015, 349, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, D.R.; Opeskin, K.; Thompson, E.W.; Williams, E.D. BM18: A novel androgen-dependent human prostate cancer xenograft model derived from a bone metastasis. Prostate 2005, 65, 35–43. [Google Scholar] [CrossRef]
- Nguyen, H.M.; Vessella, R.L.; Morrissey, C.; Brown, L.G.; Coleman, I.M.; Higano, C.S.; Mostaghel, E.A.; Zhang, X.; True, L.D.; Lam, H.-M.; et al. LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease and Serve as Models for Evaluating Cancer Therapeutics. Prostate 2017, 77, 654–671. [Google Scholar] [CrossRef]
- Deep, G.; Jain, A.K.; Ramteke, A.; Ting, H.; Vijendra, K.C.; Gangar, S.C.; Agarwal, C.; Agarwal, R. SNAI1 is critical for the aggressiveness of prostate cancer cells with low E-cadherin. Mol. Cancer 2014, 13, 37. [Google Scholar] [CrossRef]
- Veveris-Lowe, T.L.; Lawrence, M.G.; Collard, R.L.; Bui, L.; Herington, A.C.; Nicol, D.L.; Clements, J.A. Kallikrein 4 (hK4) and prostate-specific antigen (PSA) are associated with the loss of E-cadherin and an epithelial-mesenchymal transition (EMT)-like effect in prostate cancer cells. Endocr.-Relat. Cancer 2005, 12, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Sadacharan, S.; Su, S.; Belldegrun, A.; Persad, S.; Singh, G. Overexpression of vimentin: Role in the invasive pheno-type in an androgen-independent model of prostate cancer. Cancer Res. 2003, 63, 2306–2311. [Google Scholar] [PubMed]
- Brzozowa, M.; Michalski, M.; Wyrobiec, G.; Piecuch, A.; Dittfeld, A.; Harabin-Słowińska, M.; Boroń, D.; Wojnicz, R. The role of Snail1 transcription factor in colorectal cancer progression and metastasis. Współczesna Onkol. 2015, 4, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Kroepil, F.; Fluegen, G.; Vallböhmer, D.; Baldus, S.E.; Dizdar, L.; Raffel, A.M.; Hafner, D.; Stoecklein, N.H.; Knoefel, W.T. Snail1 expression in colorectal cancer and its correlation with clinical and pathological parameters. BMC Cancer 2013, 13, 145. [Google Scholar] [CrossRef]
- Barrallo-Gimeno, A.; Nieto, M.A. The Snail genes as inducers of cell movement and survival: Implications in development and cancer. Development 2005, 132, 3151–3161. [Google Scholar] [CrossRef] [PubMed]
- Olmeda, D.; Moreno-Bueno, G.; Flores, J.M.; Fabra, A.; Portillo, F.; Cano, A. SNAI1 Is Required for Tumor Growth and Lymph Node Metastasis of Human Breast Carcinoma MDA-MB-231 Cells. Cancer Res. 2007, 67, 11721–11731. [Google Scholar] [CrossRef] [PubMed]
- Petraki, C.D.; Papanastasiou, P.A.; Karavana, V.N.; Diamandis, E. Cellular distribution of human tissue kallikreins: Immunohistochemical localization. Biol. Chem. 2006, 387, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Veveris-Lowe, T.L.; Kruger, S.J.; Walsh, T.; Gardiner, R.A.; Clements, J.A. Seminal Fluid Characterization for Male Fertility and Prostate Cancer: Kallikrein-Related Serine Proteases and Whole Proteome Approaches. Semin. Thromb. Hemost. 2007, 33, 087–099. [Google Scholar] [CrossRef]
- Darson, M.F.; Pacelli, A.; Roche, P.; Rittenhouse, H.G.; Wolfert, R.L.; Saeid, M.S.; Young, C.Y.; Klee, G.G.; Tindall, D.J.; Bostwick, D.G. Human glandular kallikrein 2 expression in prostate adenocarcinoma and lymph node metastases. Urology 1999, 53, 939–944. [Google Scholar] [CrossRef]
- Tremblay, R.R.; Deperthes, D.; Têtu, B.; Dubé, J.Y. Immunohistochemical study suggesting a complementary role of kal-likreins hK2 and hK3 (prostate-specific antigen) in the functional analysis of human prostate tumors. Am. J. Pathol. 1997, 150, 455–459. [Google Scholar]
- Lawrence, M.G.; Veveris-Lowe, T.L.; Whitbread, A.; Nicol, D.L.; Clements, J.A. Epithelial-Mesenchymal Transition in Prostate Cancer and the Potential Role of Kallikrein Serine Proteases. Cells Tissues Organs 2007, 185, 111–115. [Google Scholar] [CrossRef]
- Isogai, C.; Laug, E.W.; Shimada, H.; Declerck, P.J.; Stins, M.F.; Durden, D.L.; Erdreich-Epstein, A.; Declerck, A.Y. Plasmino-gen activator inhibitor-1 promotes angiogenesis by stimulating endothelial cell migration toward fibronectin. Cancer Res. 2001, 61, 5587–5594. [Google Scholar]
- Li, S.; Wei, X.; He, J.; Tian, X.; Yuan, S.; Sun, L. Plasminogen activator inhibitor-1 in cancer research. Biomed. Pharmacother. 2018, 105, 83–94. [Google Scholar] [CrossRef]
- Park, J.; Wysocki, R.; Amoozgar, Z.; Maiorino, L.; Fein, M.R.; Jorns, J.; Schott, A.F.; Kinugasa-Katayama, Y.; Lee, Y.; Won, N.H.; et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med. 2016, 8, 361ra138. [Google Scholar] [CrossRef] [PubMed]
- Noubouossie, D.F.; Reeves, B.N.; Strahl, B.D.; Key, N.S. Neutrophils: Back in the thrombosis spotlight. Blood 2019, 133, 2186–2197. [Google Scholar] [CrossRef]
- Kubala, M.H.; Declerck, Y.A. The plasminogen activator inhibitor-1 paradox in cancer: A mechanistic understanding. Cancer Metastasis Rev. 2019, 38, 483–492. [Google Scholar] [CrossRef]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Kheir, T.B.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Herrera, K.N.G.; Wikman-Kocher, H.; Pantel, K.; Haigis, M.C.; De Carvalho, F.M.; Damascena, A.; Chinen, L.T.D.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef]
- Yun, E.-J.; Zhou, J.; Lin, C.-J.; Hernandez, E.; Fazli, L.; Gleave, M.; Hsieh, J.-T. Targeting Cancer Stem Cells in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Jaggupilli, A.; Elkord, E. Significance of CD44 and CD24 as Cancer Stem Cell Markers: An Enduring Ambiguity. Clin. Dev. Immunol. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zöller, M. CD44: Can a cancer-initiating cell profit from an abundantly expressed molecule? Nat. Rev. Cancer 2011, 11, 254–267. [Google Scholar] [CrossRef]
- De Wit, S.; Manicone, M.; Rossi, E.; Lampignano, R.; Yang, L.; Zill, B.; Rengel-Puertas, A.; Ouhlen, M.; Crespo, M.; Berghuis, A.M.S.; et al. EpCAMhigh and EpCAMlow circulating tumor cells in metastatic prostate and breast cancer patients. Oncotarget 2018, 9, 35705–35716. [Google Scholar] [CrossRef] [PubMed]
- Navone, N.M.; Van Weerden, W.M.; Vessella, R.L.; Williams, E.D.; Wang, Y.; Isaacs, J.T.; Bs, H.M.N.; Culig, Z.; Van Der Pluijm, G.; Rentsch, C.A.; et al. Movember GAP1 PDX project: An international collection of serially transplantable prostate cancer patient-derived xenograft (PDX) models. Prostate 2018, 78, 1262–1282. [Google Scholar] [CrossRef] [PubMed]
- Aslakson, C.J.; Miller, F.R. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992, 52, 1399–1405. [Google Scholar]
- Sambrook, J.; Russell, D.W. Molecular Cloning—A Laboratory Manual, 3rd ed.; Cold Spring Harbor Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Riches, A.C.; Sharp, J.G.; Thomas, D.B.; Smith, S.V. Blood volume determination in the mouse. J. Physiol. 1973, 228, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Kowalik, A.; Kowalewska, M.; Góźdź, S. Current approaches for avoiding the limitations of circulating tumor cells detection methods—implications for diagnosis and treatment of patients with solid tumors. Transl. Res. 2017, 185, 58–84.e15. [Google Scholar] [CrossRef]
- Chen, W.; Weng, S.; Zhang, F.; Allen, S.; Li, X.; Bao, L.; Lam, R.H.W.; Macoska, J.A.; Merajver, S.D.; Fu, J. Nanoroughened Surfaces for Efficient Capture of Circulating Tumor Cells without Using Capture Antibodies. ACS Nano 2013, 7, 566–575. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDX ID | Total Detected (CTCs/mL) | Clusters (Clusters/mL) | VIM+KRT− (%) | VIM−KRT+ (%) | VIM+KRT+ Hybrid (%) |
|---|---|---|---|---|---|
| BM18: 20-3 | 4 | 0 | 50 | 50 | 0 |
| BM18: 20-28 | 4 | 0 | 0 | 25 | 75 |
| BM18: 20-132 | 400 | 38 | 29 | 49 | 22 |
| LuCaP70: 20-6 | 901 | 5 | 2 | 98 | 0 |
| LuCaP70: 20-105 | 5 | 0 | 0 | 100 | 0 |
| LuCaP70: 20-112 | 1294 | 100 | 45 | 52 | 3 |
| LuCaP96: 20-7 | 3 | 0 | 0 | 0 | 100 |
| LuCaP96: 20-33 | 114 | 13 | 69 | 18 | 13 |
| LuCaP96: 20-125 | 21 | 0 | 84 | 11 | 5 |
| LuCaP105: 19-183 | 7 | 0 | 0 | 100 | 0 |
| LuCaP105: 20-102 | 1 | 0 | 100 | 0 | 0 |
| PDX | Total Samples (n) | Positive Samples # (n) | Percentage Positive (%) |
|---|---|---|---|
| BM18 | 22 | 11 | 50 |
| LuCaP70 | 16 | 11 | 69 |
| LuCaP96 | 13 | 10 | 77 |
| LuCaP105 | 20 | 10 | 50 |
| GENE | BM18 | LuCaP70 | LuCaP96 | LuCaP105 |
|---|---|---|---|---|
| SNAI1 | 0.0000985 | 0.0011992 | NS | 0.0000715 |
| VIM | NS | NS | NS | NS |
| NOTCH1 | 0.0388021 | NS | NS | NS |
| EGFR | NS | NS | NS | NS |
| FN1 | NS | NS | NS | NS |
| SERPINE1 | 0.0000001 | 0.0005100 | NS | 0.0003550 |
| SNAI2 | NS | NS | NS | |
| VCL | 0.0011664 | NS | NS | NS |
| IGF1R | 0.0152763 | NS | NS | NS |
| RRAS | 0.0001359 | NS | NS | 0.0000048 |
| FOSL1 | NS | NS | NS | |
| MSN | 0.0000247 | 0.0008278 | NS | 0.0068664 |
| NRP1 | 0.0000003 | 0.0000273 | NS | 0.0184160 |
| LAMC2 | 0.0000368 | 0.0225527 | NS | 0.0036031 |
| TNC | 0.0000312 | NS | NS | 0.0000017 |
| INHBA | 0.0120652 | NS | NS | NS |
| EMP3 | 0.0000003 | NS | NS | |
| JUP | NS | NS | NS | 0.0164246 |
| KRT20 | 0.0303967 | NS | NS | NS |
| CDH1 | NS | NS | NS | 0.0000830 |
| GRHL2 | 0.0218429 | 0.0001708 | NS | NS |
| EPCAM | NS | NS | NS | NS |
| BMP7 | 0.0070227 | NS | NS | NS |
| CLDN3 | 0.0044251 | NS | NS | NS |
| CLDN4 | 0.0019099 | 0.0008343 | 0.0004490 | NS |
| CLDN7 | NS | NS | NS | 0.0083468 |
| KLK3 | 0.0001113 | 0.0001525 | 0.0009885 | 0.0000037 |
| CD24 | 0.0029071 | NS | NS | 0.0135173 |
| CD44 | 0.0000004 | 0.0077010 | NS | 0.0000067 |
| APLN | NS | 0.0081341 | NS | 0.0310010 |
| HIF1A | 0.0190586 | 0.0027857 | NS | NS |
| BNIP3 | NS | NS | NS | NS |
| PPARGC1A | 0.0073689 | NS | NS | 0.0000008 |
| ILK | 0.0000058 | 0.0175717 | NS | 0.0083933 |
| ESR1 | 0.0273697 | NS | NS | NS |
| TFF1 | NS | 0.0001196 | 0.0001279 | |
| PGR | 0.0759697 | NS | NS | |
| TBP | 0.0009111 | 0.0183997 | NS | 0.0010770 |
| GUSB | NS | NS | NS | NS |
| NONO | 0.0160096 | 0.0133378 | NS | NS |
| OAZ1 | 0.0226327 | NS | NS | NS |
| PDX ID | Total Detected (CTCs/mL) | Clusters (Clusters/mL) | SERPINE1+ KRT− (%) | SERPINE1− KRT+ (%) | SERPINE1+ KRT+ Hybrid (%) |
|---|---|---|---|---|---|
| BM18: 20-3 | 11 | 0 | 0 | 60 | 40 |
| BM18: 20-28 | 23 | 0 | 9 | 91 | 0 |
| BM18: 20-132 | 694 | 68 | 0 | 0 | 100 |
| LuCaP70: 20-6 | 779 | 29 | 0 | 100 | 0 |
| LuCaP70: 20-105 | 0 | 0 | 0 | 0 | 0 |
| LuCaP70: 20-112 | 950 | 56 | 0 | 1 | 99 |
| LuCaP96: 20-7 | 26 | 0 | 0 | 11 | 89 |
| LuCaP96: 20-33 | 178 | 13 | 14 | 28 | 58 |
| LuCaP96: 20-125 | 82 | 6 | 38 | 19 | 43 |
| LuCaP105: 19-183 | 0 | 0 | 0 | 0 | 0 |
| LuCaP105: 20-102 | 0 | 0 | 0 | 0 | 0 |
| PDX ID | Total Detected (CTCs/mL) | Clusters (Clusters/mL) | SERPINE1+PSA− (%) | VIM+PSA− (%) | SERPINE1−PSA+ or VIM−PSA+ (%) | SERPINE1+PSA+ or VIM+PSA+ Hybrid (%) |
|---|---|---|---|---|---|---|
| BM18: 20-3 | 71 | 4 | 13 | 75 | 13 | |
| BM18: 20-28 | 8 | 1 | 13 | 0 | 88 | |
| BM18: 20-132 | 635 | 24 | 35 | 3 | 62 | |
| LuCaP70: 20-6 | 13 | 0 | 70 | 0 | 30 | |
| LuCaP70: 20-105 | 8 | 0 | 88 | 0 | 13 | |
| LuCaP70: 20-112 | 1166 | 70 | 3 | 49 | 48 | |
| LuCaP96: 20-7 | 17 | 0 | 33 | 0 | 67 | |
| LuCaP96: 20-33 | 31 | 4 | 32 | 0 | 68 | |
| LuCaP96: 20-125 | 24 | 2 | 23 | 5 | 73 | |
| LuCaP105: 19-183 | 9 | 1 | 75 | 13 | 13 | |
| LuCaP105: 20-102 | 3 | 1 | 67 | 0 | 33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, S.; Blick, T.; Thompson, E.W.; Williams, E.D. Diversity of Epithelial-Mesenchymal Phenotypes in Circulating Tumour Cells from Prostate Cancer Patient-Derived Xenograft Models. Cancers 2021, 13, 2750. https://doi.org/10.3390/cancers13112750
Hassan S, Blick T, Thompson EW, Williams ED. Diversity of Epithelial-Mesenchymal Phenotypes in Circulating Tumour Cells from Prostate Cancer Patient-Derived Xenograft Models. Cancers. 2021; 13(11):2750. https://doi.org/10.3390/cancers13112750
Chicago/Turabian StyleHassan, Sara, Tony Blick, Erik W. Thompson, and Elizabeth D. Williams. 2021. "Diversity of Epithelial-Mesenchymal Phenotypes in Circulating Tumour Cells from Prostate Cancer Patient-Derived Xenograft Models" Cancers 13, no. 11: 2750. https://doi.org/10.3390/cancers13112750
APA StyleHassan, S., Blick, T., Thompson, E. W., & Williams, E. D. (2021). Diversity of Epithelial-Mesenchymal Phenotypes in Circulating Tumour Cells from Prostate Cancer Patient-Derived Xenograft Models. Cancers, 13(11), 2750. https://doi.org/10.3390/cancers13112750