
Clinical Application of Next-Generation Sequencing of Plasma Cell-Free DNA for Genotyping Untreated Advanced Non-Small Cell Lung Cancer

 ,
,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Study Outcomes
2.3. Plasma and Tumor Tissue Genotyping
2.4. Statistical Analysis
3. Results
3.1. Patient’s Characteristics, Disease Extension, and Tumor Burden
3.2. Tissue and ctDNA Genotyping Results and ctDNA Test Performance
3.3. Determinants of ctDNA Positivity
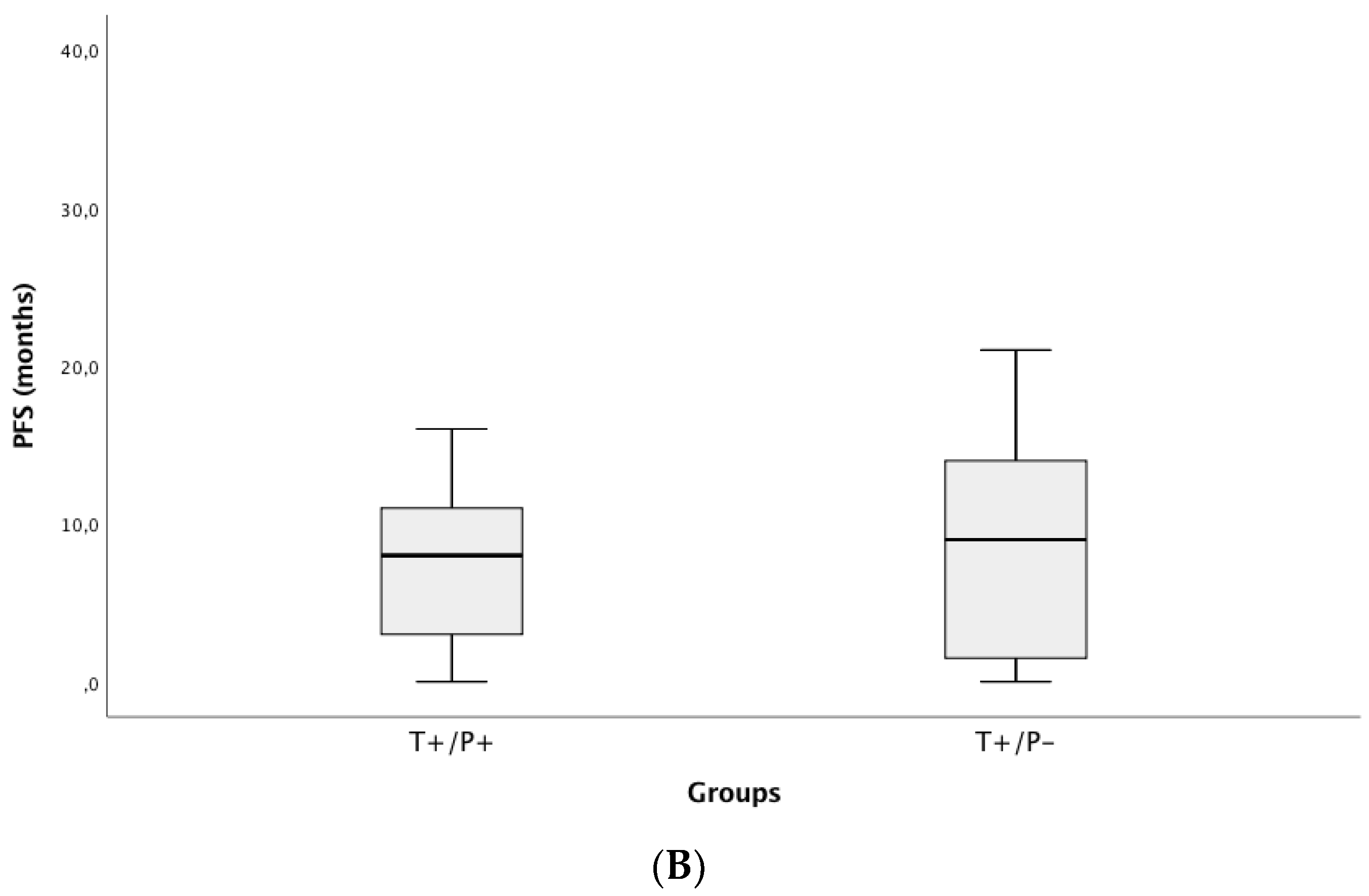
3.4. Correlation of ctDNA Positivity with Clinical Outcomes (PFS and OS)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalemkerian, G.P.; Narula, N.; Kennedy, E.B.; Biermann, W.A.; Donington, J.; Leighl, N.B.; Lew, M.; Pantelas, J.; Ramalingam, S.S.; Reck, M.; et al. Molecular Testing Guideline for the Selection of Patients With Lung Cancer for Treatment With Targeted Tyrosine Kinase Inhibitors: American Society of Clinical Oncology Endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology Clinical Practice Guideline Update. J. Clin. Oncol. 2018, 36, 911–919. [Google Scholar]
- Lindeman, N.I.; Cagle, P.T.; Aisner, D.L.; Arcila, M.E.; Beasley, M.B.; Bernicker, E.H.; Colasacco, C.; Dacic, S.; Hirsch, F.R.; Kerr, K.; et al. Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment With Targeted Tyrosine Kinase Inhibitors: Guideline From the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch. Pathol. Lab. Med. 2018, 142, 321–346. [Google Scholar]
- Riely, G.L. What, When, and How of Biomarker Testing in Non–Small Cell Lung Cancer. J. Natl. Compr. Cancer Netw. 2017, 15, 686–688. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef] [PubMed]
- Ryška, A.; Berzinec, P.; Brcic, L.; Cufer, T.; Dziadziuszko, R.; Gottfried, M.; Kovalszky, I.; Olszewski, W.; Öz, B.; Plank, L.; et al. NSCLC molecular testing in Central and Eastern European countries. BMC Cancer 2018, 18, 269. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.E.; Choi, K.; Lanman, R.B.; Licitra, E.J.; Skrzypczak, S.M.; Benito, R.P.; Wu, T.; Arunajadai, S.; Kaur, S.; Harper, H.; et al. Genomic Profiling of Advanced Non–Small Cell Lung Cancer in Community Settings: Gaps and Opportunities. Clin. Lung Cancer 2017, 18, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.G.O.; Jacob, M.; Martins, N.; Moura, C.S.; Guimarães, S.; Reis, J.P.; Justino, A.; Pina, M.J.; Cirnes, L.; Sousa, C.; et al. Targeted Gene Next-Generation Sequencing Panel in Patients with Advanced Lung Adenocarcinoma: Paving the Way for Clinical Implementation. Cancers 2019, 11, 1229. [Google Scholar] [CrossRef]
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.-F.; et al. Using Multiplexed Assays of Oncogenic Drivers in Lung Cancers to Select Targeted Drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Kimura, H.; Suminoe, M.; Kasahara, K.; Sone, T.; Araya, T.; Tamori, S.; Koizumi, F.; Nishio, K.; Miyamoto, K.; Fujimura, M.; et al. Evaluation of epidermal growth factor receptor mutation status in serum DNA as a predictor of response to gefitinib (IRESSA). Br. J. Cancer 2007, 97, 778–784. [Google Scholar] [CrossRef]
- Sacher, A.G.; Paweletz, C.; Dahlberg, S.E.; Alden, R.S.; O’Connell, A.; Feeney, N.; Mach, S.L.; Jänne, P.A.; Oxnard, G.R. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol. 2016, 2, 1014–1022. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Thress, K.S.; Alden, R.S.; Lawrance, R.; Paweletz, C.P.; Cantarini, M.; Yang, J.C.-H.; Barrett, J.C.; Jänne, P.A. Association Between Plasma Genotyping and Outcomes of Treatment With Osimertinib (AZD9291) in Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 3375–3382. [Google Scholar] [CrossRef] [PubMed]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Lindeman, N.; Lockwood, C.M.; Rai, A.J.; Schilsky, R.L.; et al. Circulating Tumor DNA Analysis in Patients With Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J. Clin. Oncol. 2018, 36, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Rolfo, C.; Mack, P.C.; Scagliotti, G.V.; Baas, P.; Barlesi, F.; Bivona, T.G.; Herbst, R.S.; Mok, T.S.; Peled, N.; Pirker, R.; et al. Liquid Biopsy for Advanced Non-Small Cell Lung Cancer (NSCLC): A Statement Paper from the IASLC. J. Thorac. Oncol. 2018, 13, 1248–1268. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Cheng, D.T.; Mitchell, T.N.; Zehir, A.; Shah, R.H.; Benayed, R.; Syed, A.; Chandramohan, R.; Liu, Z.Y.; Won, H.H.; Scott, S.N.; et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridisation Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J. Mol. Diagn. 2015, 17, 251–264. [Google Scholar] [CrossRef]
- D’Haene, N.; Le Mercier, M.; De Nève, N.; Blanchard, O.; Delaunoy, M.; El Housni, H.; Dessars, B.; Heimann, P.; Remmelink, M.; Demetter, P.; et al. Clinical Validation of Targeted Next Generation Sequencing for Colon and Lung Cancers. PLoS ONE 2015, 10, e0138245. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Wang, L.; Arcila, M.E.; Balasubramanian, S.; Greenbowe, J.R.; Ross, J.S.; Stephens, P.J.; Lipson, D.; Miller, V.A.; Kris, M.G.; et al. Broad, Hybrid Capture–Based Next-Generation Sequencing Identifies Actionable Genomic Alterations in Lung Adenocarcinomas Otherwise Negative for Such Alterations by Other Genomic Testing Approaches. Clin. Cancer Res. 2015, 21, 3631–3639. [Google Scholar] [CrossRef]
- Lim, S.M.; Kim, E.Y.; Kim, H.R.; Ali, S.M.; Greenbowe, J.R.; Shim, H.S.; Chang, H.; Lim, S.; Paik, S.; Cho, B.C. Genomic profiling of lung adenocarcinoma patients reveals therapeutic targets and confers clinical benefit when standard molecular testing is negative. Oncotarget 2016, 7, 24172–24178. [Google Scholar] [CrossRef]
- McCourt, C.M.; McArt, D.G.; Mills, K.; Catherwood, M.A.; Maxwell, P.; Waugh, D.J.; Hamilton, P.; O’Sullivan, J.M.; Salto-Tellez, M. Validation of Next Generation Sequencing Technologies in Comparison to Current Diagnostic Gold Standards for BRAF, EGFR and KRAS Mutational Analysis. PLoS ONE 2013, 8, e69604. [Google Scholar] [CrossRef]
- Tops, B.B.J.; Normanno, N.; Kurth, H.; Amato, E.; Mafficini, A.; Rieber, N.; Le Corre, D.; Rachiglio, A.M.; Reiman, A.; Sheils, O.; et al. Development of a semi-conductor sequencing-based panel for genotyping of colon and lung cancer by the Onconetwork consortium. BMC Cancer 2015, 15, 26. [Google Scholar] [CrossRef]
- Vendrell, J.A.; Grand, D.; Rouquette, I.; Costes, V.; Icher, S.; Selves, J.; Larrieux, M.; Barbe, A.; Brousset, P.; Solassol, J. High-throughput detection of clinically targetable alterations using next-generation sequencing. Oncotarget 2017, 8, 40345–40358. [Google Scholar] [CrossRef]
- Tuononen, K.; Maki-Nevala, S.; Sarhadi, V.K.; Wirtanen, A.; Rönty, M.; Salmenkivi, K.; Andrews, J.M.; Telaranta-Keerie, A.I.; Hannula, S.; Lagström, S.; et al. Comparison of targeted next-generation sequencing (NGS) and real-time PCR in the detection of EGFR, KRAS, and BRAF mutations on formalin-fixed, paraffin-embedded tumor material of non-small cell lung carcinoma-superiority of NGS. Genes Chromosomes Cancer 2013, 52, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.L.; Walsh, K.; Diamond, A.; Oniscu, A.; Deans, Z.C. Validation of the Oncomine™ focus panel for next-generation sequencing of clinical tumour samples. Virchows Archiv 2018, 473, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.-Y.; Ostoros, G.; Cobo, M.; Ciuleanu, T.; Cole, R.; McWalter, G.; Walker, J.; Dearden, S.; Webster, A.; Milenkova, T.; et al. Gefitinib Treatment in EGFR Mutated Caucasian NSCLC: Circulating-Free Tumor DNA as a Surrogate for Determination of EGFR Status. J. Thorac. Oncol. 2014, 9, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, B.S.; Wu, L.; Wei, W.; Tsai, J.; Weber, B.; Nexo, E.; Meldgaard, P. Monitoring of epidermal growth factor receptor tyrosine kinase inhibitor-ssensitising and resistance mutations in the plasma DNA of patients with advanced non-small cell lung cancer during treatment with erlotinib. Cancer 2014, 120, 3896–3901. [Google Scholar] [CrossRef]
- Luo, J.; Shen, L.; Zheng, D. Diagnostic value of circulating free DNA for the detection of EGFR mutation status in NSCLC: A systematic review and meta-analysis. Sci. Rep. 2015, 4, srep06269. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Wang, J.; Xu, Y.; Ding, X.; Li, M.; Jiang, F.; Xu, L.; Yin, R. Circulating Tumor DNA Is Effective for the Detection of EGFR Mutation in Non–Small Cell Lung Cancer: A Meta-analysis. Cancer Epidemiol. Biomark. Prev. 2015, 24, 206–212. [Google Scholar] [CrossRef]
- Couraud, S.; Vaca-Paniagua, F.; Villar, S.; Oliver, J.; Schuster, T.; Blanché, H.; Girard, N.; Trédaniel, J.; Guilleminault, L.; Gervais, R.; et al. Noninvasive Diagnosis of Actionable Mutations by Deep Sequencing of Circulating Free DNA in Lung Cancer from Never-Smokers: A Proof-of-Concept Study from BioCAST/IFCT-1002. Clin. Cancer Res. 2014, 20, 4613–4624. [Google Scholar] [CrossRef]
- Paweletz, C.P.; Sacher, A.G.; Raymond, C.K.; Alden, R.S.; O’Connell, A.; Mach, S.L.; Kuang, Y.; Gandhi, L.; Kirschmeier, P.; English, J.M.; et al. Bias-Corrected Targeted Next-Generation Sequencing for Rapid, Multiplexed Detection of Actionable Alterations in Cell-Free DNA from Advanced Lung Cancer Patients. Clin. Cancer Res. 2016, 22, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.C.; Yee, S.S.; Troxel, A.B.; Savitch, S.L.; Fan, R.; Balli, D.; Lieberman, D.B.; Morrissette, J.D.; Evans, T.L.; Bauml, J.M.; et al. Detection of Therapeutically Targetable Driver and Resistance Mutations in Lung Cancer Patients by Next-Generation Sequencing of Cell-Free Circulating Tumor DNA. Clin. Cancer Res. 2016, 22, 5772–5782. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Tsao, M.S.; Le, L.W.; Shepherd, F.A.; Feld, R.; Burkes, R.L.; Liu, G.; Kamel-Reid, S.; Hwang, D.; Tanguay, J.; et al. Biomarker testing and time to treatment decision in patients with advanced nonsmall-cell lung cancer. Ann. Oncol. 2015, 26, 1415–1421. [Google Scholar] [CrossRef]
- Leighl, N.B.; Page, R.D.; Raymond, V.M.; Daniel, D.B.; Divers, S.G.; Reckamp, K.L.; Villalona-Calero, M.A.; Dix, D.; Odegaard, J.I.; Lanman, R.B.; et al. Clinical Utility of Comprehensive Cell-free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non–small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 4691–4700. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, C.; Thompson, J.C.; Black, T.A.; Katz, S.I.; Fan, R.; Yee, S.S.; Chien, A.L.; Evans, T.L.; Bauml, J.M.; Alley, E.W.; et al. Clinical Implications of Plasma-Based Genotyping With the Delivery of Personalised Therapy in Metastatic Non–Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 173–180. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Lee, V.; Liam, C.-K.; Lu, S.; Park, K.; Srimuninnimit, V.; Wang, J.; Zhou, C.; Appius, A.; Button, P.; et al. Clinical utility of a blood-based EGFR mutation test in patients receiving first-line erlotinib therapy in the ENSURE, FASTACT-2, and ASPIRATION studies. Lung Cancer 2018, 126, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Remon, J.; Caramella, C.; Jovelet, C.; Lacroix, L.; Lawson, A.; Smalley, S.; Howarth, K.; Gale, D.; Green, E.; Plagnol, V.; et al. Osimertinib benefit inEGFR-mutant NSCLC patients withT790M-mutation detected by circulating tumour DNA. Ann. Oncol. 2017, 28, 784–790. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Value (n, %) | |
|---|---|---|
| Age (median, range) | 66 (38,92) | |
| Gender, n (%) | Males | 71 (61.7) |
| Females | 44 (38.3) | |
| Performance Status, n (%) | 0 | 42 (36.5) |
| 1 | 53 (46.1) | |
| 2 | 15 (13.0) | |
| 3 | 5 (4.4) | |
| Smoking Status, n (%) | Smoker or Former smoker | 73 (63.5) |
| Never smoker | 42 (36.5) | |
| Histology, n (%) | Adenocarcinoma | 114 (99.1) |
| Adenosquamous | 1 (0.9) | |
| Tumor specimen type | Histologic | 93 (83.9) |
| Cytologic | 22 (19.1) | |
| Disease stage, n (%) | III: (IIIA/B/C) | 21 (18.3): (7/10/4) |
| IV | 94 (81.7) | |
| TNM discriminator, n (%) | ||
| T | Tx | 7 (6.1) |
| T1 | 19 (16.5) | |
| T2 | 21 (18.3) | |
| T3 | 17 (14.8) | |
| T4 | 51 (44.3) | |
| N | N0 | 43 (37.4) |
| N1 | 10 (8.7) | |
| N2 | 36 (31.3) | |
| N3 | 26 (22.6) | |
| M | M0 | 20 (17.4) |
| M1a | 22 (19.1) | |
| M1b | 21 (18.3) | |
| M1c | 52 (45.2) | |
| First-Line treatment | BSC | 9 (7.8) |
| Multimodal (ChT+RT) ChT TKIs | 13 (11.3) 69 (60.0) 24 (20.9) | |
| # | TNM | DNA Concentration ng/µL | Gene | Codon | Amino Acid | Variant Allelic Fraction % | ||
|---|---|---|---|---|---|---|---|---|
| Tissue | cfDNA | Tissue DNA | ctDNA | |||||
| Discordant Results (n = 15) | ||||||||
| 86 | IIIB | 36.80 | 0.41 | KRAS | c.35G > T | p.(G12V) | 44.70 | 0 |
| 3 | IV | 0.06 | 31.10 | EGFR | c.2238_2252del15 | p.(L747_T751del) | 27.80 | 0 |
| 40 | IV | 1.55 | 0.44 | EGFR | c.2235_2249del15 | p.(E746_A750del) | 56.50 | 0 |
| 29 | IV | 12.1 | 0.18 | KRAS | c.35G > T | p.(G12V) | 6.50 | 0 |
| 140 | IV | 2.83 | 2.44 | EGFR | c.2239_2248del10insC | p.(L747_A750 > P) | 14.00 | 0 |
| 54 | IV | 7.27 | 0.19 | EGFR | c.2248_2276del29ins5 | p.(A750_L760del) | 84.50 | 0 |
| 23 | IV | 1.62 | 1.63 | EGFR | c.2235_2249del15 | p.(E746_A750del) | 74.10 | 0 |
| 35 | IV | 1.17 | 0.25 | KRAS | c.35G > T | p.(G12V) | 3.30 | 0 |
| 37 | IV | 1.29 | 0.26 | KRAS | c.34G > T | p.(G12C) | 15.00 | 0 |
| 104 | IV | 0.11 | 1.00 | KRAS | c.35G > T | p.(G12V) | 50.60 | 0 |
| 118 | IV | 10.5 | 1.18 | EGFR | c.2235_2249del15 | p.(E746_A750del) | 20.40 | 0 |
| 154 | IV | 30.8 | 0.13 | KRAS | c.35G > T | p.(G12V) | 46.30 | 0 |
| 39 | IV | n.a. | 3.25 | KRAS | c.34G > T | p.(G12C) | 0 | 0.45 |
| 43 | IV | 1.20 | 1.05 | EGFR | c.2240_2257del18 | p.(L747_P753 > S) | 0 | 0.9 |
| 93 * | IV | 5.70 | 0.76 | EGFR | c.2235_2249del15 | p.(E746_A750del) | 0 | 0.11 |
| Concordant positive results (n = 51) | ||||||||
| 87 | IIIB | 4.14 | 0.27 | KRAS | c.34G > T | p.(G12C) | 5.90 | 0.21 |
| 163 | IV | 2.38 | 0.67 | KRAS | c.35G > T | p.(G12V) | 28.60 | 0.03 |
| 22 | IV | 0.22 | 0.44 | EGFR | c.2294T > G | p.(V765G) | 50.90 | 0.10 |
| 32 | IV | 2.58 | 0.27 | KRAS | c.35G > T | p.(G12V) | 19.90 | 0.12 |
| 62 | IV | 3.63 | 3.93 | BRAF | c.1799T > A | p.(V600E) | 36.30 | 0.53 |
| 81 * | IV | 0.26 | 1.62 | EGFR | c.2236_2250del15 | p.(E746_A750del) | 36.70 | 0.18 |
| 81 * | EGFR | c.2369C > T | p.(T790M) | 15.00 | 0.14 | |||
| 93 * | IV | 5.70 | 0.76 | EGFR | c.2573T > G | p.(L785R) | 31.30 | 0.18 |
| 146 | IV | 6.38 | 0.33 | EGFR | c.2235_2249del | p.(E746_A750del) | 35.80 | 1.39 |
| 100 | IV | 0.69 | 3.59 | KRAS | c.35G > T | p.(G12V) | 55.20 | 0.01 |
| 44 * | IV | n.a. | 3.65 | KRAS | c.38_39delGCinsAA | p.(G13G) | 14.20 | 3.62 |
| 44 * | ERBB4 | c.1033G > T | p.(A345S) | 14.90 | 1.90 | |||
| 80 * | IV | 4.17 | 1.55 | BRAF | c.1799T > A | p.(V600E) | 50.40 | 9.90 |
| 80 * | TP53 | c.476C > G | p.(A159V) | 39.20 | 4.90 | |||
| 119 | IV | 0.38 | 0.27 | KRAS | c.34G > T | p.(G12C) | 72.50 | 0.60 |
| 142 | IV | 22.0 | n.a. | EGFR | c.2573T > G | p.(L785R) | 15.30 | 0.12 |
| 165 * | IV | 4.94 | 0.72 | ERBB2 | c.2310_2311insGCATAC | p.(A775_Gl776insT) | 20.00 | 23.3 |
| 165 * | TP53 | c.1024C > T | p.(R342 *) | 21.00 | 1.00 | |||
| 187 | IV | n.a. | 0.10 | EGFR | c.2235_2249del15 | p.(E746_A750del) | 31.70 | 9.05 |
| 1 | IV | 2.53 | 1.31 | KRAS | c.35G > A | p.(G12A) | 6.10 | 1.37 |
| 2 | IV | 0.34 | 3.36 | EGFR | c.2240_2257del18 | p.(L747_P753 > S) | 40.00 | 0.72 |
| 12 | IV | 1.35 | 6.38 | TP53 | c.527G > T | p.(C176F) | 41.10 | 1.27 |
| 14 | IV | 2.45 | 55.00 | KRAS | c.35G > T | p.(G12V) | 11.70 | 0.09 |
| 15 * | IV | 0.06 | 2.27 | KRAS | c.182A > G | p.(Q61R) | 8.80 | 0.43 |
| 15 * | TP53 | c.461G > T | p.(Gl154V) | 24.70 | 0.71 | |||
| 15 * | STK11 | c.597G > C | p.(E199D) | 12.80 | 0.53 | |||
| 16 | IV | 0.18 | 1.01 | KRAS | c.34G > T | p.(G12C) | 3.80 | 0.72 |
| 38 | IV | 0.95 | 0.19 | EGFR | c.2573T > G | p.(L785R) | 6.30 | 0.91 |
| 52 | IV | 2.95 | 0.82 | EGFR | c.2236_2250del15 | p.(E746_A750del) | 0.17 | 4.10 |
| 51 | IV | n.a. | 3.91 | KRAS | c.34G > T | p.(G12C) | 5.90 | 1.50 |
| 57 | IV | 3.39 | 2.67 | EGFR | c.2573T > G | p.(L785R) | 29.20 | 0.03 |
| 60 | IV | 4.46 | 1.26 | KRAS | c.35G > T | p.(G12V) | 20.40 | 7.72 |
| 65 | IV | 0.28 | 0.28 | EGFR | c.2235_2249del15 | p.(E746_A750del) | 7.60 | 2.01 |
| 74 * | IV | 11.70 | 17.7 | EGFR | c.2236_2250del15 | p.(E746_A750del) | 11.10 | 0.27 |
| 74 * | KRAS | c.182A > G | p.(G61R) | 0.38 | 0.07 | |||
| 98 * | IV | 0.96 | 1.26 | EGFR | c.2239_2248del | p.(L747_A750 > P) | 66.00 | 0.22 |
| 98 * | ALK | c.3512T > A | p.(I1171N) | 0.08 | 0.08 | |||
| 95 | IV | n.a. | 0.55 | EGFR | c.2240_2254del15 | p.(L747_T751del) | 96.50 | 0.65 |
| 101 | IV | 0.39 | 1.29 | KRAS | c.34G > T | p.(G12C) | 17.40 | 0.06 |
| 107 * | IV | 22.10 | 1.03 | EGFR | c.2240_2257del18 | p.(L747_P753 > S) | 67.20 | 5.50 |
| 107 * | EGFR | c.2369C > T | p.(T790M) | 0.60 | 1.25 | |||
| 117 | IV | 48.80 | 0.39 | KRAS | c.35G > A | p.(G12A) | 22.30 | 47.0 |
| 130 | IV | n.a. | 0.56 | EGFR | c.2235_2249del15 | p.(E746_A750del) | 13.20 | 1.06 |
| 131 | IV | 0.93 | n.a. | BRAF | c.1799T > A | p.(V600E) | 29.10 | 0.61 |
| 144 * | IV | 0.51 | 0.37 | EGFR | c.2235_2249del15 | p.(E746_A750del) | 34.60 | 0.62 |
| 144 * | PIK3CA | c.1633G > A | p.E545K) | 27.80 | 0.75 | |||
| 143 | IV | 0.55 | 2.10 | BRAF | c.1799T > A | p.V600E) | 10.90 | 1.89 |
| 136 * | IV | 8.85 | 1.14 | KRAS | c.35G > T | p.(G12V) | 26.70 | 10.30 |
| 136 * | TP53 | c.839G > A | p.(R280K) | 13.00 | 0.06 | |||
| 161 | IV | 13.90 | 0.58 | KRAS | c.35G > T | p.(G12V) | 27.40 | 2.22 |
| 78 | IV | 2.15 | 2.53 | EGFR | c.2573T > G | p.(L785R) | 18.60 | 0.09 |
| Concordant negative results (n = 61) | ||||||||
| Cases Compared (N) | Concordant Cases [tDNA vs. ctDNA (N)] | Discordant Cases [tDNA vs. ctDNA (N)] | Concordant Cases (%) | Kappa | ||
|---|---|---|---|---|---|---|
| Negative/Negative | Positive/Positive | Negative/Positive | Positive/Negative | |||
| (a) 115 patients | 61 | 40 | 2 | 12 | 87.8 | 0.75 |
| (b) 127 hotspot | 61 | 51 | 3 | 12 | 88.2 | 0.76 |
| TNM Discriminator Value, n (%) | All | Plasma-Negative | Plasma-Positive | p Value | |
|---|---|---|---|---|---|
| Age (median, IQR) | 66 (14) | 66 (12) | 64 (18) | 0.716 | |
| Gender | Male | 71 (61.7) | 46 (63.0) | 25 (59.5) | 0.711 |
| Female | 44 (38.3) | 27 (37.0) | 17 (40.5) | ||
| Performance Status | 0 | 42 (36.5) | 35 (47.9) | 7 (16.7) | 0.009 |
| 1 | 53 (46.1) | 29 (39.7) | 25 (59.5) | ||
| 2 | 15 (13.0) | 7 (9.6) | 8 (19.0) | ||
| 3 | 5 (4.4) | 2 (2.8) | 2 (4.8) | ||
| TNM discriminator | |||||
| T, n (%) | Tx | 7 (6.1) | 2 (2.7) | 5 (11.9) | 0.328 |
| T1 | 13 (11.3) | 7 (9.6) | 6 (14.3) | ||
| T2 | 15 (13.0) | 10 (13.7) | 5 (11.9) | ||
| T3 | 12 (10.4) | 7 (9.6) | 5 (11.9) | ||
| T4 | 68 (59.1) | 47 (69.1) | 21 (50.0) | ||
| N, n (%) | N0 | 4 (38.3) | 26 (35.6) | 18 (42.9) | 0.623 |
| N1 | 10 (8.7) | 7 (9.6) | 3 (7.1) | ||
| N2 | 35 (30.4) | 21 (28.8) | 14 (33.3) | ||
| N3 | 26 (22.6) | 19 (26.0) | 7 (16.7) | ||
| M, n (%) | M0 | 20 (17.4) | 19 (26.0) | 1 (2.4) | 0.013 |
| M1a | 22 (19.1) | 13 (17.8) | 9 (21.4) | ||
| M1b | 21 (18.3) | 13 (17.8) | 8 (19.0) | ||
| M1c | 52 (45.2) | 28 (38.4) | 24 (57.1) | ||
| Number of organs involved Median (min-max) | 1 (0–6) | 1 (0–6) | 2 (0–5) | 0.004 | |
| Disease stage | IIIA | 7 (6.1) | 7 (9.6) | 0 | 0.010 |
| IIIB | 10 (8.7) | 9 (12.3) | 1 (2.4) | ||
| IIIC | 4 (3.5) | 4 (3.5) | 0 | ||
| IV | 94 (81.7) | 53 (72.6) | 41 (97.6) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, M.G.O.; Cruz-Martins, N.; Souto Moura, C.; Guimarães, S.; Pereira Reis, J.; Justino, A.; Pina, M.J.; Magalhães, A.; Queiroga, H.; Machado, J.C.; et al. Clinical Application of Next-Generation Sequencing of Plasma Cell-Free DNA for Genotyping Untreated Advanced Non-Small Cell Lung Cancer. Cancers 2021, 13, 2707. https://doi.org/10.3390/cancers13112707
Fernandes MGO, Cruz-Martins N, Souto Moura C, Guimarães S, Pereira Reis J, Justino A, Pina MJ, Magalhães A, Queiroga H, Machado JC, et al. Clinical Application of Next-Generation Sequencing of Plasma Cell-Free DNA for Genotyping Untreated Advanced Non-Small Cell Lung Cancer. Cancers. 2021; 13(11):2707. https://doi.org/10.3390/cancers13112707
Chicago/Turabian StyleFernandes, Maria Gabriela O., Natália Cruz-Martins, Conceição Souto Moura, Susana Guimarães, Joana Pereira Reis, Ana Justino, Maria João Pina, Adriana Magalhães, Henrique Queiroga, José Carlos Machado, and et al. 2021. "Clinical Application of Next-Generation Sequencing of Plasma Cell-Free DNA for Genotyping Untreated Advanced Non-Small Cell Lung Cancer" Cancers 13, no. 11: 2707. https://doi.org/10.3390/cancers13112707
APA StyleFernandes, M. G. O., Cruz-Martins, N., Souto Moura, C., Guimarães, S., Pereira Reis, J., Justino, A., Pina, M. J., Magalhães, A., Queiroga, H., Machado, J. C., Hespanhol, V., & Costa, J. L. (2021). Clinical Application of Next-Generation Sequencing of Plasma Cell-Free DNA for Genotyping Untreated Advanced Non-Small Cell Lung Cancer. Cancers, 13(11), 2707. https://doi.org/10.3390/cancers13112707