
Ubiquitin Specific Protease 29 Functions as an Oncogene Promoting Tumorigenesis in Colorectal Carcinoma

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
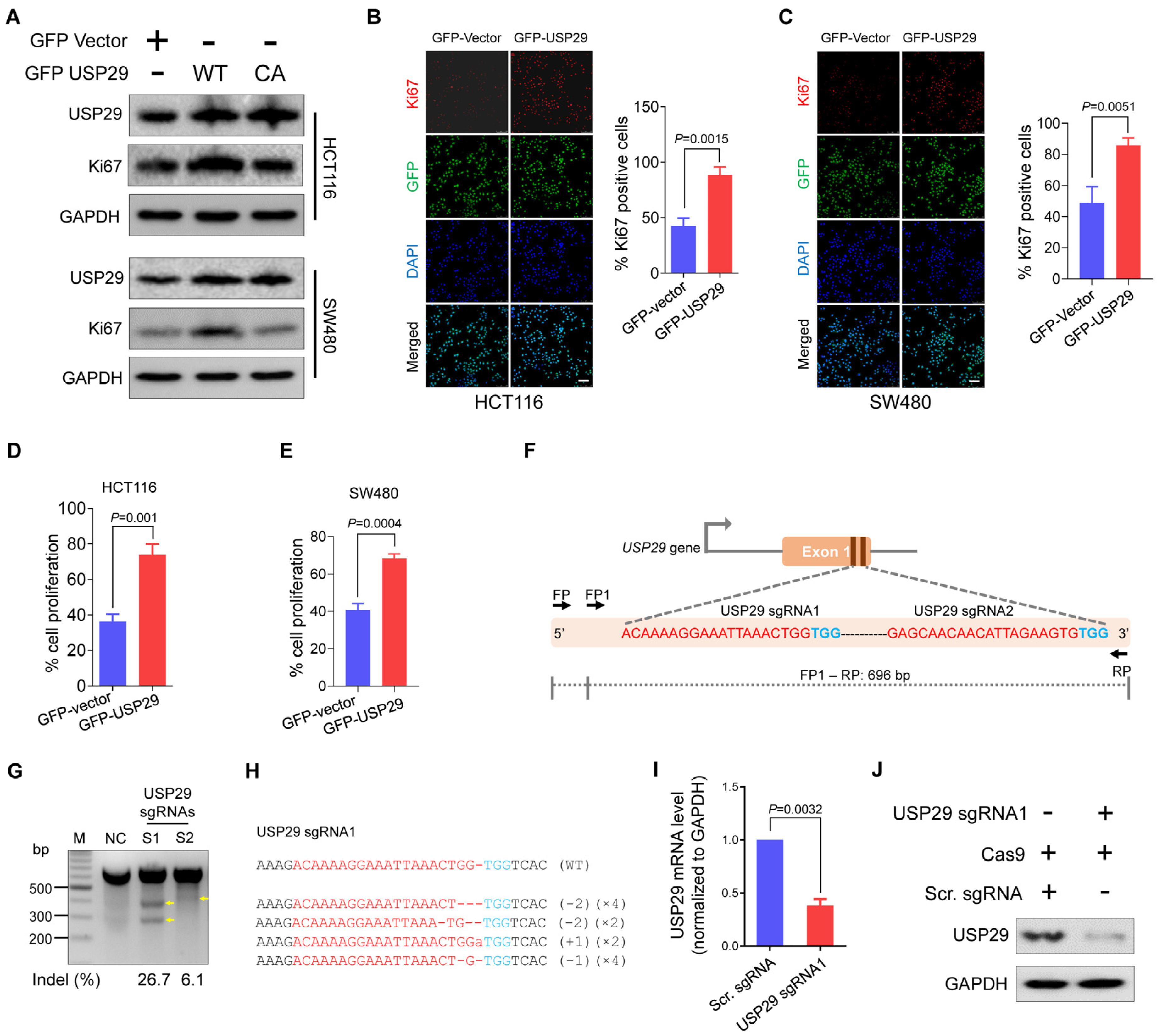
2.1. USP29 Is Highly Upregulated in Clinical Human Colon Cancer Tissues and Promotes Cell Proliferation
2.2. Generation of USP29-Depleted Cells Using CRISPR-Cas9 System
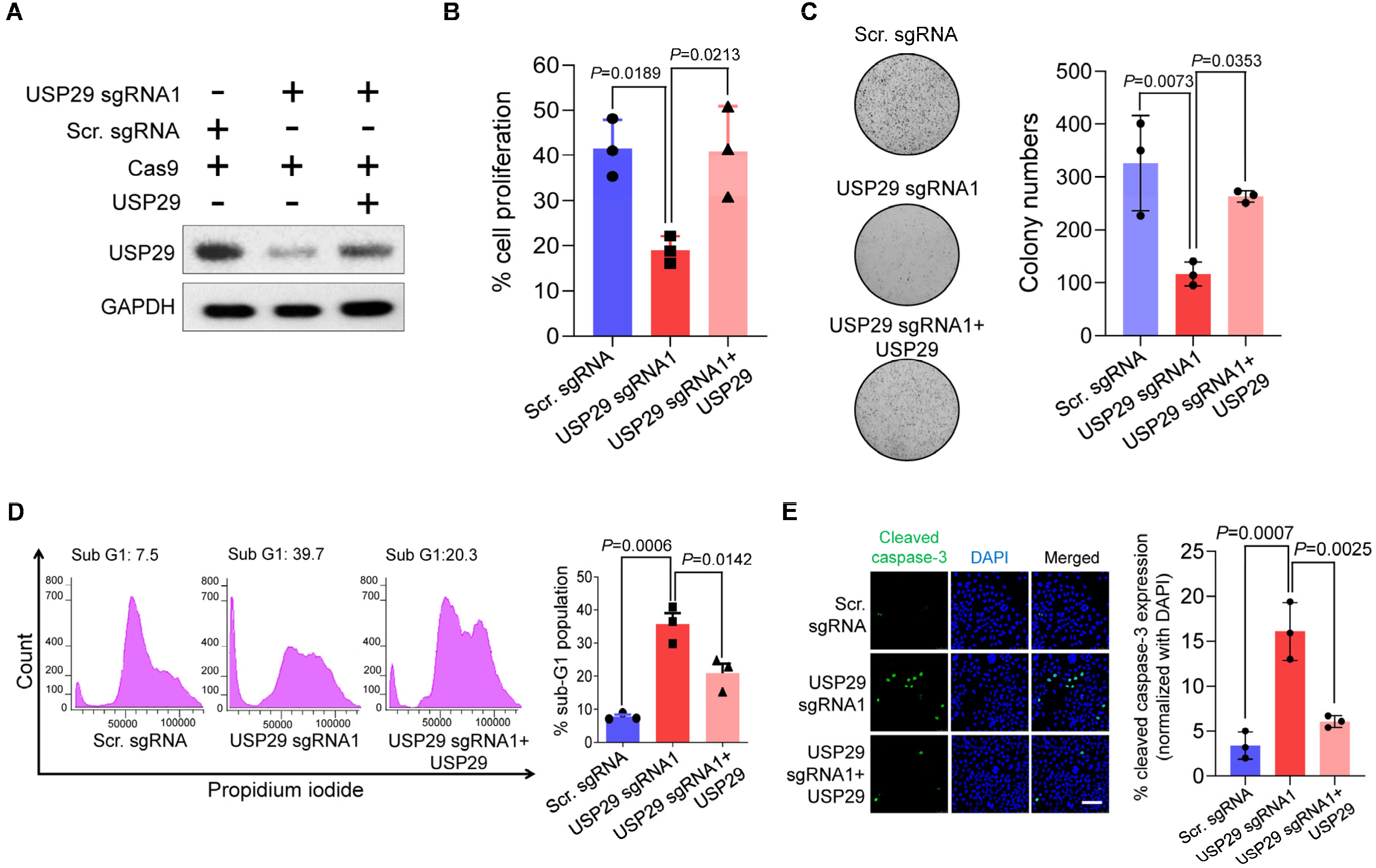
2.3. USP29 Depletion Induces DNA Damage and Delays Cell-Cycle Progression
2.4. USP29-Depleted Cells Undergo Apoptosis
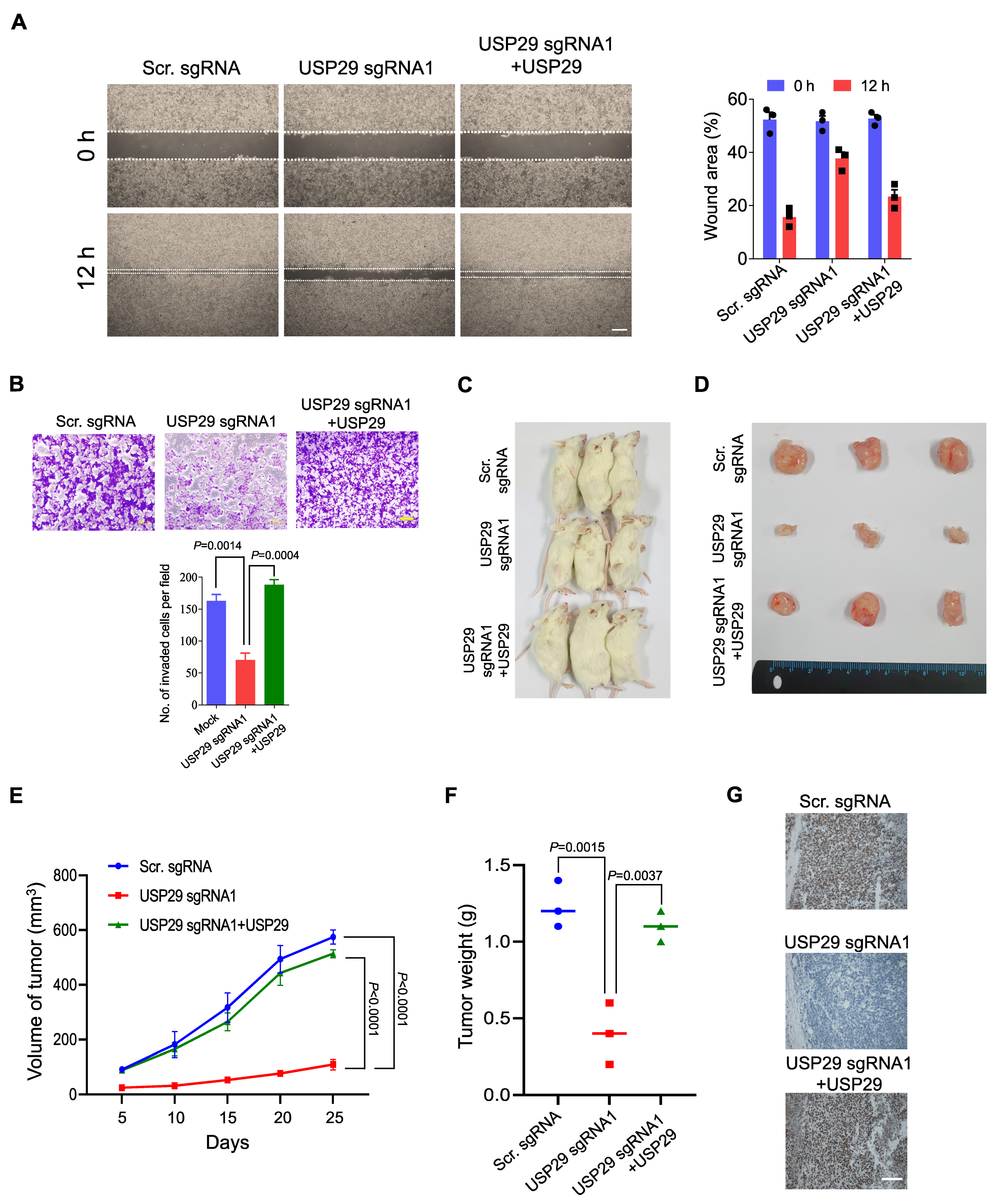
2.5. Loss of USP29 Expression Inhibits Colon Cancer Growth In Vitro and In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Reagents, Plasmids, and Antibodies
4.2. Transfection
4.3. Expression Studies from the TCGA Database
4.4. Immunohistochemistry
4.5. Cell Proliferation Assay
4.6. Immunofluorescence
4.7. Generation of USP29-Depleted HCT116 Cells Using CRISPR-Cas9
4.8. T7E1 Assay
4.9. Western Blot Analysis
4.10. Quantitative PCR Analysis
4.11. Flow Cytometry
4.12. Colony Formation Assay
4.13. In Vitro Scratch Assay
4.14. Matrigel Invasion Assay
4.15. In Vivo Tumor Formation Assay
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA. Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Fakih, M.G. Metastatic Colorectal Cancer: Current State and Future Directions. J. Clin. Oncol. 2015, 33, 1809–1824. [Google Scholar] [CrossRef] [PubMed]
- Amerik, A.Y.; Hochstrasser, M. Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta Mol. Cell Res. 2004, 1695, 189–207. [Google Scholar] [CrossRef]
- Kaushal, K.; Ramakrishna, S. Deubiquitinating Enzyme-Mediated Signaling Networks in Cancer Stem Cells. Cancers (Basel) 2020, 12, 3253. [Google Scholar] [CrossRef]
- Antao, A.M.; Tyagi, A.; Kim, K.-S.; Ramakrishna, S. Advances in Deubiquitinating Enzyme Inhibition and Applications in Cancer Therapeutics. Cancers (Basel) 2020, 12, 1579. [Google Scholar] [CrossRef]
- Tanguturi, P.; Kim, K.-S.; Ramakrishna, S. The role of deubiquitinating enzymes in cancer drug resistance. Cancer Chemother. Pharmacol. 2020, 85, 627–639. [Google Scholar] [CrossRef]
- Poondla, N.; Chandrasekaran, A.P.; Kim, K.-S.; Ramakrishna, S. Deubiquitinating enzymes as cancer biomarkers: New therapeutic opportunities? BMB Rep. 2019, 52, 181–189. [Google Scholar] [CrossRef]
- Sacco, J.J.; Coulson, J.M.; Clague, M.J.; Urbé, S. Emerging roles of deubiquitinases in cancer-associated pathways. IUBMB Life 2010, 62, 140–157. [Google Scholar] [CrossRef]
- Das, S.; Chandrasekaran, A.P.; Suresh, B.; Haq, S.; Kang, J.-H.; Lee, S.-J.; Kim, J.; Kim, J.; Lee, S.; Kim, H.H.; et al. Genome-scale screening of deubiquitinase subfamily identifies USP3 as a stabilizer of Cdc25A regulating cell cycle in cancer. Cell Death Differ. 2020, 27, 3004–3020. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, J.; Romier, C.; Tora, L.; Devys, D. Zinc-finger UBPs: Regulators of deubiquitylation. Trends Biochem. Sci. 2008, 33, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Reverter, D. Molecular Mechanisms of DUBs Regulation in Signaling and Disease. Int. J. Mol. Sci. 2021, 22, 986. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chung, H.-J.; Vogt, M.; Jin, Y.; Malide, D.; He, L.; Dundr, M.; Levens, D. JTV1 co-activates FBP to induce USP29 transcription and stabilize p53 in response to oxidative stress. EMBO J. 2011, 30, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Li, Q.; Wu, X.; Li, W.; Li, Q.; Zhang, J.; Li, M.; Zhang, D.; Zhao, H.; Zou, X.; et al. Deubiquitinase USP29 promotes gastric cancer cell migration by cooperating with phosphatase SCP1 to stabilize Snail protein. Oncogene 2020, 39, 6802–6815. [Google Scholar] [CrossRef] [PubMed]
- Martín, Y.; Cabrera, E.; Amoedo, H.; Hernández-Pérez, S.; Domínguez-Kelly, R.; Freire, R. USP29 controls the stability of checkpoint adaptor Claspin by deubiquitination. Oncogene 2015, 34, 1058–1063. [Google Scholar] [CrossRef]
- Scholzen, T.; Gerdes, J. The Ki-67 protein: From the known and the unknown. J. Cell. Physiol. 2000, 182, 311–322. [Google Scholar] [CrossRef]
- Collins, K.; Jacks, T.; Pavletich, N.P. The cell cycle and cancer. Proc. Natl. Acad. Sci. 1997, 94, 2776–2778. [Google Scholar] [CrossRef]
- Salvesen, G.S. Caspases: Opening the boxes and interpreting the arrows. Cell Death Differ. 2002, 9, 3–5. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Gastroenterol. Rev. 2019, 14, 89–103. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA. Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, J.A.; Jacq, X.; Martin, N.M.; Jackson, S.P. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov. 2018, 17, 57–78. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, P.; Linder, S. Molecular Pathways: Translational Potential of Deubiquitinases as Drug Targets. Clin. Cancer Res. 2014, 20, 3908–3914. [Google Scholar] [CrossRef]
- Fraile, J.M.; Quesada, V.; Rodríguez, D.; Freije, J.M.P.; López-Otín, C. Deubiquitinases in cancer: New functions and therapeutic options. Oncogene 2012, 31, 2373–2388. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.-H.; Baek, K.-H. Deubiquitinating Enzymes as Therapeutic Targets in Cancer. Curr. Pharm. Des. 2013, 19, 4039–4052. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.-I.; Hong, H.K.; Yeo, S.-Y.; Kim, S.-H.; Cho, Y.B.; Kim, K.K. Ubiquitin-Specific Protease 21 Promotes Colorectal Cancer Metastasis by Acting as a Fra-1 Deubiquitinase. Cancers (Basel) 2020, 12, 207. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.; Jung, S.M.; Park, J.S.; Lee, J.; Ha, J.; Kim, M.; Park, S.H. The deubiquitinating enzyme PSMD14 facilitates tumor growth and chemoresistance through stabilizing the ALK2 receptor in the initiation of BMP6 signaling pathway. EBioMedicine 2019, 49, 55–71. [Google Scholar] [CrossRef]
- Singh, S.K.; Mishra, M.K.; Eltoum, I.-E.A.; Bae, S.; Lillard, J.W.; Singh, R. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. Sci. Rep. 2018, 8, 1323. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, H.J.; Kim, H.; Cho, S.W.; Kim, J.-S. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res. 2009, 19, 1279–1288. [Google Scholar] [CrossRef]
- Latifi-Pupovci, H.; Kuçi, Z.; Wehner, S.; Bönig, H.; Lieberz, R.; Klingebiel, T.; Bader, P.; Kuçi, S. In vitro migration and proliferation (“wound healing”) potential of mesenchymal stromal cells generated from human CD271+ bone marrow mononuclear cells. J. Transl. Med. 2015, 13, 315. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandrasekaran, A.P.; Suresh, B.; Sarodaya, N.; Ko, N.-R.; Oh, S.-J.; Kim, K.-S.; Ramakrishna, S. Ubiquitin Specific Protease 29 Functions as an Oncogene Promoting Tumorigenesis in Colorectal Carcinoma. Cancers 2021, 13, 2706. https://doi.org/10.3390/cancers13112706
Chandrasekaran AP, Suresh B, Sarodaya N, Ko N-R, Oh S-J, Kim K-S, Ramakrishna S. Ubiquitin Specific Protease 29 Functions as an Oncogene Promoting Tumorigenesis in Colorectal Carcinoma. Cancers. 2021; 13(11):2706. https://doi.org/10.3390/cancers13112706
Chicago/Turabian StyleChandrasekaran, Arun Pandian, Bharathi Suresh, Neha Sarodaya, Na-Re Ko, Seung-Jun Oh, Kye-Seong Kim, and Suresh Ramakrishna. 2021. "Ubiquitin Specific Protease 29 Functions as an Oncogene Promoting Tumorigenesis in Colorectal Carcinoma" Cancers 13, no. 11: 2706. https://doi.org/10.3390/cancers13112706
APA StyleChandrasekaran, A. P., Suresh, B., Sarodaya, N., Ko, N.-R., Oh, S.-J., Kim, K.-S., & Ramakrishna, S. (2021). Ubiquitin Specific Protease 29 Functions as an Oncogene Promoting Tumorigenesis in Colorectal Carcinoma. Cancers, 13(11), 2706. https://doi.org/10.3390/cancers13112706