
Immune Monitoring in Melanoma and Urothelial Cancer Patients Treated with Anti-PD-1 Immunotherapy and SBRT Discloses Tumor Specific Immune Signatures

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Flow Cytometry
2.3. High Dimensional Data Analysis of Flow Cytometry Data
2.3.1. t-SNE
2.3.2. FlowSOM
2.4. Cytokine Measurement
2.5. UPLC-MS/MS
2.6. Scoring of PD-L1 and Tumor Infiltrating Cells
2.7. Statistics
3. Results
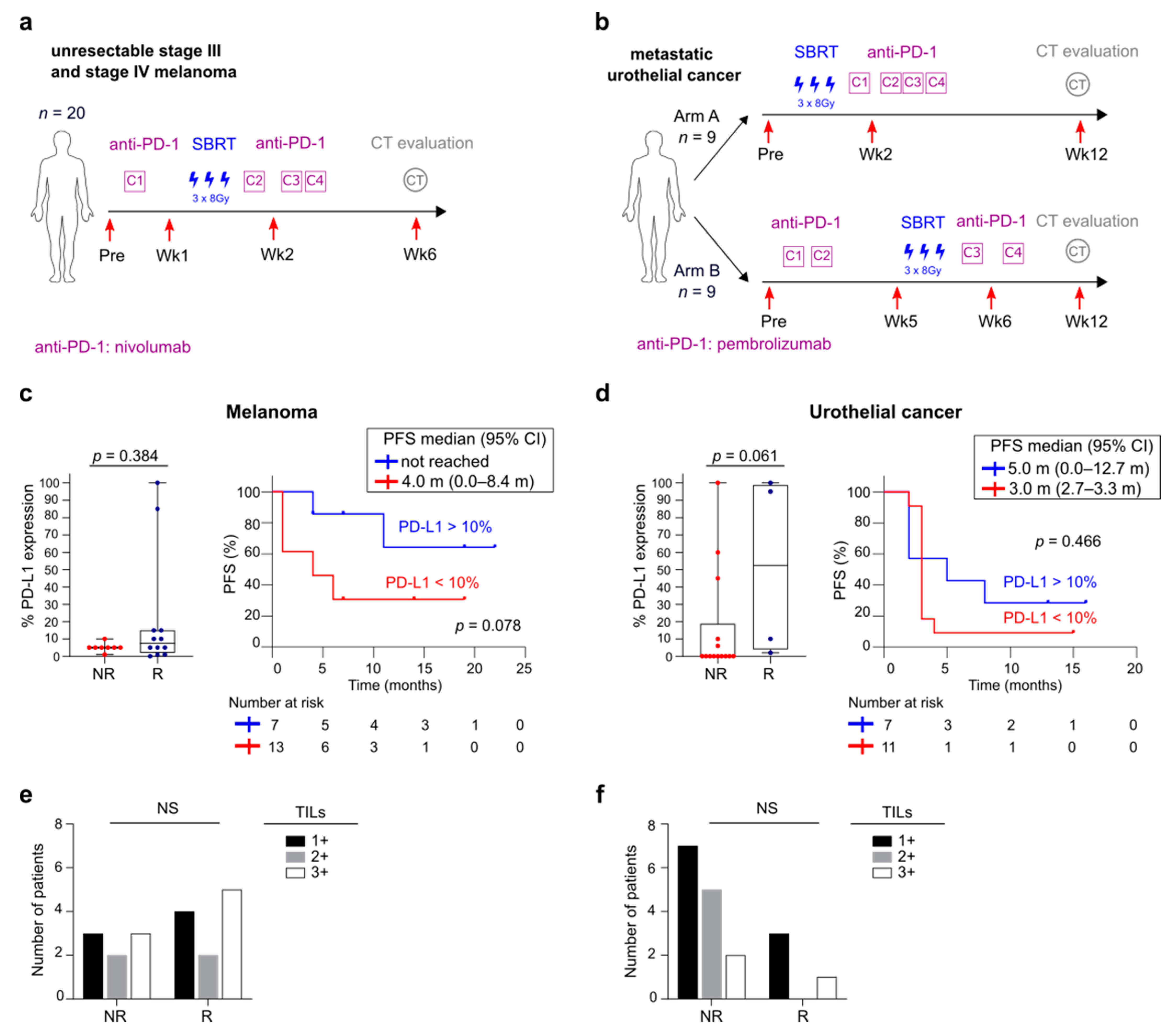
3.1. Overview of Patient Cohorts
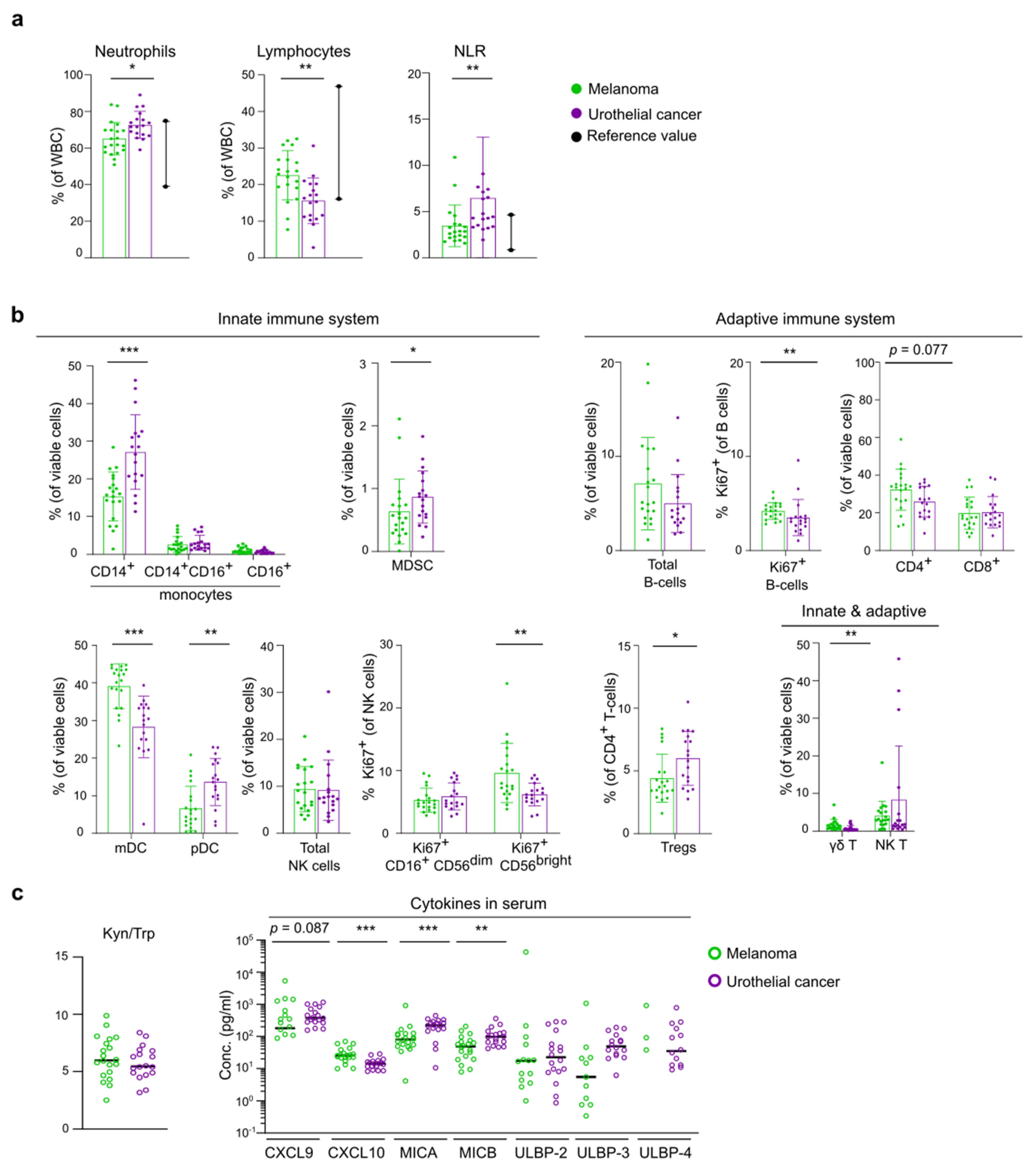
3.2. Differences in Baseline Immunity between Melanoma and UC Cohort
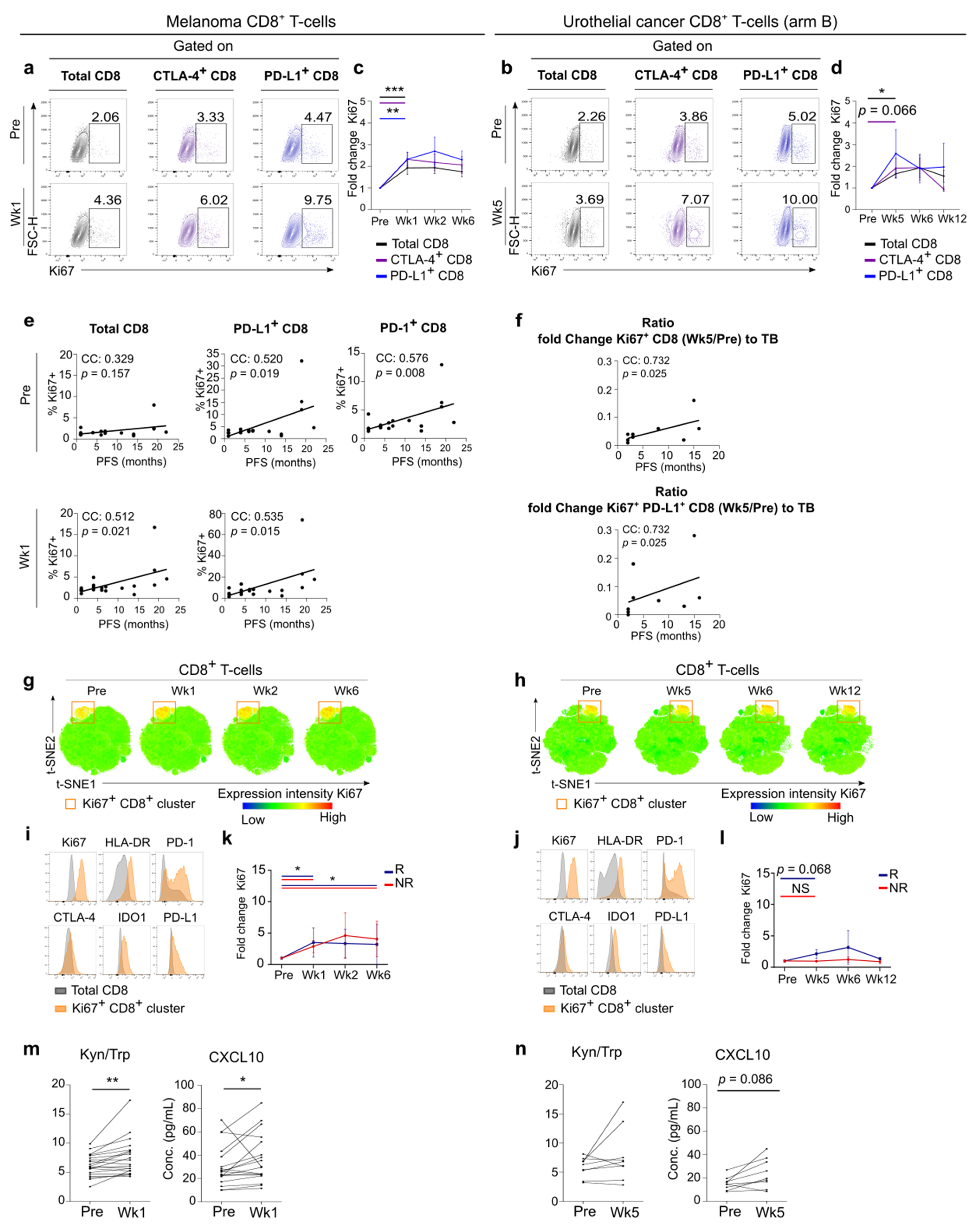
3.3. Early Systemic Immune Changes after Anti-PD-1 Treatment Initiation
3.4. Systemic Immune Changes after SBRT
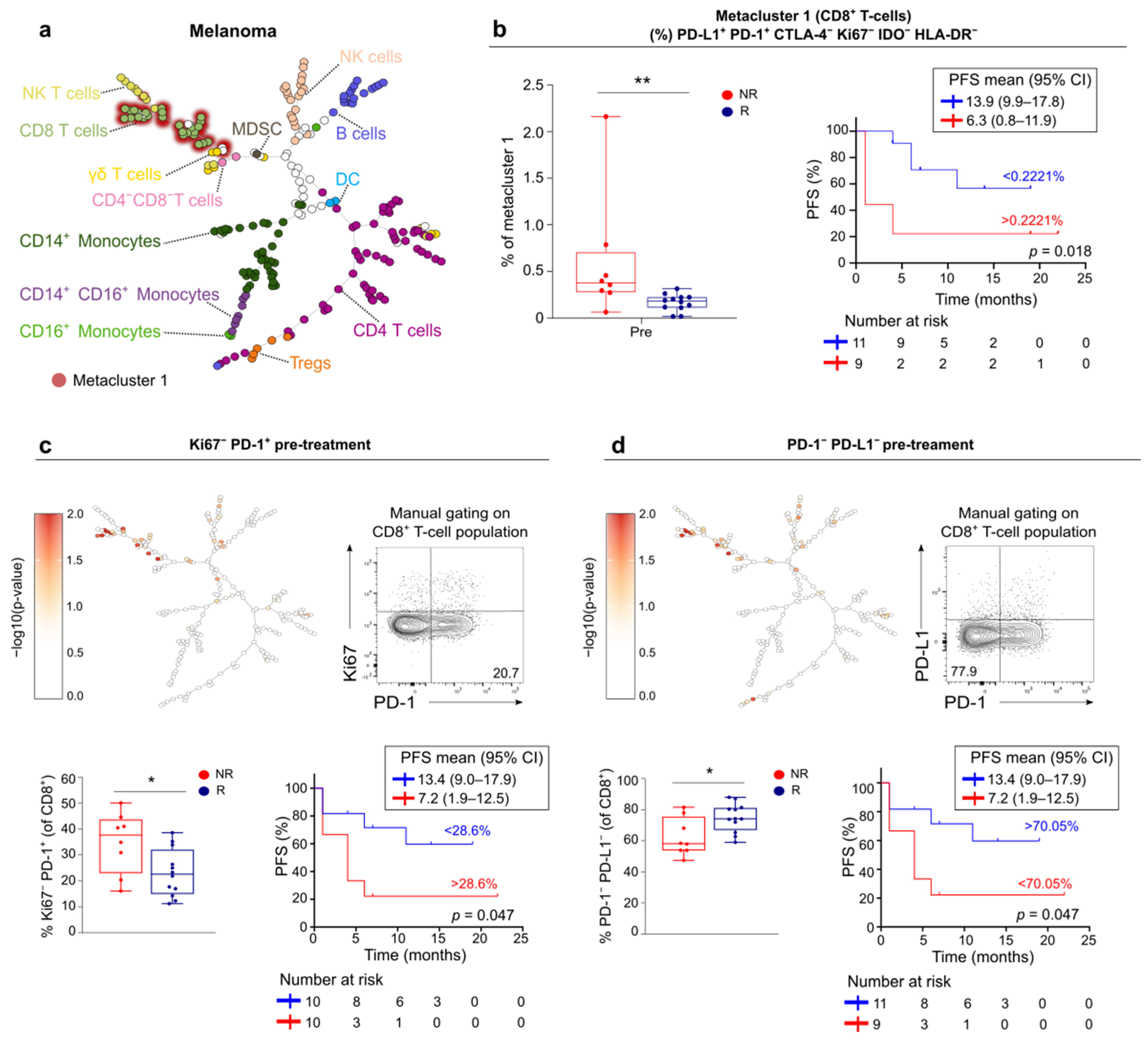
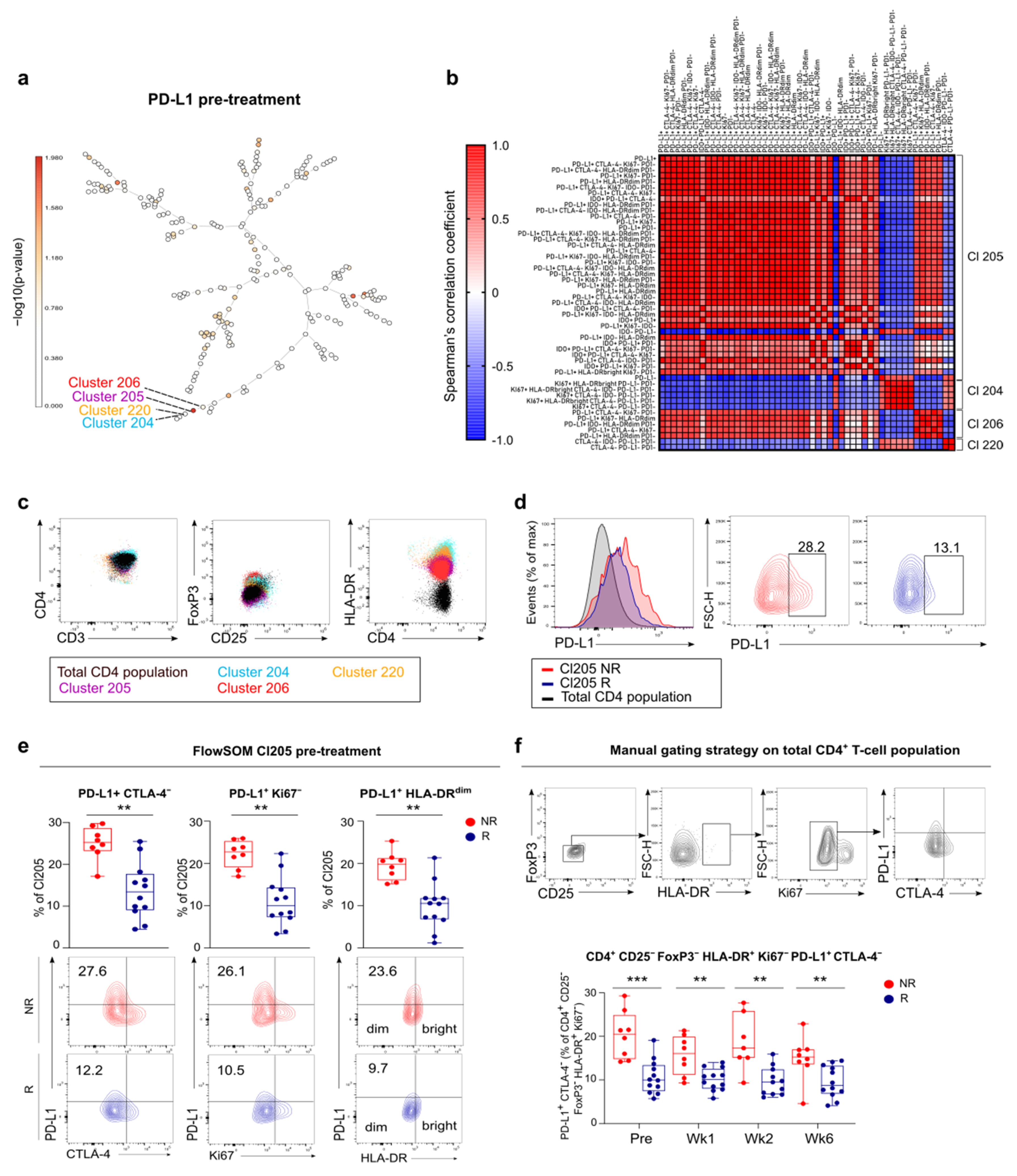
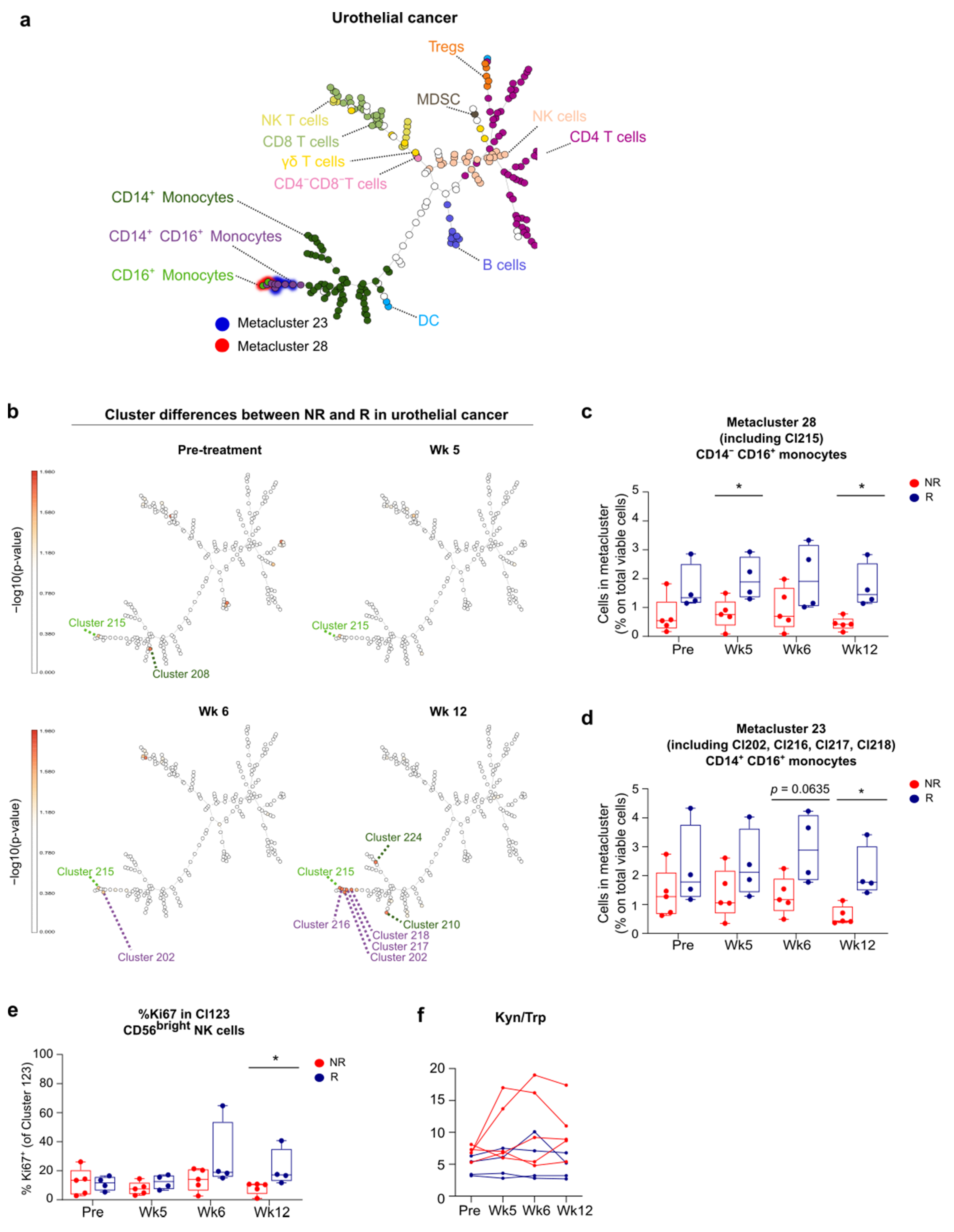
3.5. FlowSOM Analysis to Discover Immune Signatures Correlating with Clinical Outcome
3.6. Link between Blood and Tumor Micro-Environment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haslam, A.; Prasad, V. Estimation of the Percentage of US Patients with Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw. Open 2019, 2, e192535. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Sharma, P.; Siefker-Radtke, A.; de Braud, F.; Basso, U.; Calvo, E.; Bono, P.; Morse, M.A.; Ascierto, P.A.; Lopez-Martin, J.; Brossart, P.; et al. Nivolumab Alone and with Ipilimumab in Previously Treated Metastatic Urothelial Carcinoma: CheckMate 032 Nivolumab 1 mg/kg Plus Ipilimumab 3 mg/kg Expansion Cohort Results. J. Clin. Oncol. 2019, 37, 1608–1616. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.A.; Patel, V.G. The role of PD-L1 expression as a predictive biomarker: An analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef]
- Prasad, V.; Kaestner, V.; Mailankody, S. Cancer Drugs Approved Based on Biomarkers and Not Tumor Type-FDA Approval of Pembrolizumab for Mismatch Repair-Deficient Solid Cancers. JAMA Oncol. 2018, 4, 157–158. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Gide, T.N.; Quek, C.; Menzies, A.M.; Tasker, A.T.; Shang, P.; Holst, J.; Madore, J.; Lim, S.Y.; Velickovic, R.; Wongchenko, M.; et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019, 35, 238–255. [Google Scholar] [CrossRef] [PubMed]
- Sundahl, N.; Seremet, T.; Van Dorpe, J.; Neyns, B.; Ferdinande, L.; Meireson, A.; Brochez, L.; Kruse, V.; Ost, P. Phase 2 Trial of Nivolumab Combined with Stereotactic Body Radiation Therapy in Patients with Metastatic or Locally Advanced Inoperable Melanoma. Int. J. Radiat. Oncol. 2019, 104, 828–835. [Google Scholar] [CrossRef]
- Sundahl, N.; Vandekerkhove, G.; Decaestecker, K.; Meireson, A.; De Visschere, P.; Fonteyne, V.; De Maeseneer, D.; Reynders, D.; Goetghebeur, E.; Van Dorpe, J.; et al. Randomized Phase 1 Trial of Pembrolizumab with Sequential Versus Concomitant Stereotactic Body Radiotherapy in Metastatic Urothelial Carcinoma. Eur. Urol. 2019, 75, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Miyazaki, T.; Shibata, T.; Hara, N.; Tsuchiya, M. Simultaneous measurement of tryptophan and related compounds by liquid chromatography/electrospray ionization tandem mass spectrometry. J. Chromatogr. B 2008, 867, 57–61. [Google Scholar] [CrossRef]
- Schefold, J.C.; Zeden, J.-P.; Fotopoulou, C.; von Haehling, S.; Pschowski, R.; Hasper, D.; Volk, H.-D.; Schuett, C.; Reinke, P. Increased indoleamine 2,3-dioxygenase (IDO) activity and elevated serum levels of tryptophan catabolites in patients with chronic kidney disease: A possible link between chronic inflammation and uraemic symptoms. Nephrol. Dial. Transplant. 2009, 24, 1901–1908. [Google Scholar] [CrossRef]
- Rodríguez-Perea, A.L.; Arcia, E.D.; Rueda, C.M.; Velilla, P.A. Phenotypical characterization of regulatory T cells in humans and rodents. Clin. Exp. Immunol. 2016, 185, 281–291. [Google Scholar] [CrossRef]
- Van Gassen, S.; Callebaut, B.; Van Helden, M.J.; Lambrecht, B.N.; Demeester, P.; Dhaene, T.; Saeys, Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytom. Part A 2015, 87, 636–645. [Google Scholar] [CrossRef]
- Jiang, T.; Qiao, M.; Zhao, C.; Li, X.; Gao, G.; Su, C.; Ren, S.; Zhou, C. Pretreatment neutrophil-to-lymphocyte ratio is associated with outcome of advanced-stage cancer patients treated with immunotherapy: A meta-analysis. Cancer Immunol. Immunother. 2018, 67, 713–727. [Google Scholar] [CrossRef]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef]
- Huang, B.; Han, W.; Sheng, Z.-F.; Shen, G.-L. Identification of immune-related biomarkers associated with tumorigenesis and prognosis in cutaneous melanoma patients. Cancer Cell Int. 2020, 20, 1–15. [Google Scholar] [CrossRef]
- Dhar, P.; Wu, J.D. NKG2D and its ligands in cancer. Curr. Opin. Immunol. 2018, 51, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Callahan, M.K.; Bono, P.; Kim, J.; Spiliopoulou, P.; Calvo, E.; Pillai, R.N.; Ott, P.A.; de Braud, F.; Morse, M.; et al. Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): A multicentre, open-label, two-stage, multi-arm, phase 1/2 trial. Lancet Oncol. 2016, 17, 1590–1598. [Google Scholar] [CrossRef]
- Sharma, P.; Retz, M.; Siefker-Radtke, A.; Baron, A.; Necchi, A.; Bedke, J.; Plimack, E.R.; Vaena, D.; Grimm, M.-O.; Bracarda, S.; et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017, 18, 312–322. [Google Scholar] [CrossRef]
- Bellmunt, J.; de Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.-L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef]
- Vuky, J.; Balar, A.V.; Castellano, D.; O’Donnell, P.H.; Grivas, P.; Bellmunt, J.; Powles, T.; Bajorin, D.; Hahn, N.M.; Savage, M.J.; et al. Long-Term Outcomes in KEYNOTE-052: Phase II Study Investigating First-Line Pembrolizumab in Cisplatin-Ineligible Patients with Locally Advanced or Metastatic Urothelial Cancer. J. Clin. Oncol. 2020, 38, 2658–2666. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Castro, A.; Pyke, R.M.; Zhang, X.; Thompson, W.K.; Day, C.-P.; Alexandrov, L.B.; Zanetti, M.; Carter, H. Strength of immune selection in tumors varies with sex and age. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Conforti, F.; Pala, L.; Bagnardi, V.; Pas, T.D.; Martinetti, M.; Viale, G.; Gelber, R.D.; Goldhirsch, A. Cancer immunotherapy efficacy and patients’ sex: A systematic review and meta-analysis. Lancet Oncol. 2018, 19, 737–746. [Google Scholar] [CrossRef]
- Gupta, S.; Artomov, M.; Goggins, W.; Daly, M.; Tsao, H. Gender Disparity and Mutation Burden in Metastatic Melanoma. JNCI J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Pan, H.; Li, F.; Wu, K.; Zhang, X.; He, J. Analysis of ultra-deep targeted sequencing reveals mutation burden is associated with gender and clinical outcome in lung adenocarcinoma. Oncotarget 2016, 7, 22857–22864. [Google Scholar] [CrossRef]
- Allen, E.M.V.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef]
- Rozeman, E.A.; Hoefsmit, E.P.; Reijers, I.L.M.; Saw, R.P.M.; Versluis, J.M.; Krijgsman, O.; Dimitriadis, P.; Sikorska, K.; van de Wiel, B.A.; Eriksson, H.; et al. Survival and biomarker analyses from the OpACIN-neo and OpACIN neoadjuvant immunotherapy trials in stage III melanoma. Nat. Med. 2021, 27, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Renkema, K.R.; Li, G.; Wu, A.; Smithey, M.J.; Nikolich-Žugich, J. Two separate defects affecting true naive or virtual memory T cell precursors combine to reduce naive T cell responses with aging. J. Immunol. 1950 2014, 192, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Briceño, O.; Lissina, A.; Wanke, K.; Afonso, G.; von Braun, A.; Ragon, K.; Miquel, T.; Gostick, E.; Papagno, L.; Stiasny, K.; et al. Reduced naïve CD8+ T-cell priming efficacy in elderly adults. Aging Cell 2016, 15, 14–21. [Google Scholar] [CrossRef]
- Agrawal, A.; Agrawal, S.; Gupta, S. Dendritic cells in human aging. Exp. Gerontol. 2007, 42, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Kugel, C.H.; Douglass, S.M.; Webster, M.R.; Kaur, A.; Liu, Q.; Yin, X.; Weiss, S.A.; Darvishian, F.; Al-Rohil, R.N.; Ndoye, A.; et al. Age Correlates with Response to Anti-PD1, Reflecting Age-Related Differences in Intratumoral Effector and Regulatory T-Cell Populations. Clin. Cancer Res. 2018, 24, 5347–5356. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, E.K.; Flynn, J.R.; Panageas, K.S.; Ferraro, R.A.; Sta. Cruz, J.M.; Postow, M.A.; Coit, D.G.; Ariyan, C.E. High neutrophil-to-lymphocyte ratio (NLR) is associated with treatment failure and death in patients who have melanoma treated with PD-1 inhibitor monotherapy. Cancer 2020, 126, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Matsukane, R.; Watanabe, H.; Minami, H.; Hata, K.; Suetsugu, K.; Tsuji, T.; Masuda, S.; Okamoto, I.; Nakagawa, T.; Ito, T.; et al. Continuous monitoring of neutrophils to lymphocytes ratio for estimating the onset, severity, and subsequent prognosis of immune related adverse events. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cornen, S.; Vivier, E. Chemotherapy and tumor immunity. Science 2018, 362, 1355–1356. [Google Scholar] [CrossRef]
- Ruscetti, M.; Leibold, J.; Bott, M.J.; Fennell, M.; Kulick, A.; Salgado, N.R.; Chen, C.-C.; Ho, Y.-J.; Sanchez-Rivera, F.J.; Feucht, J.; et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 2018, 362, 1416–1422. [Google Scholar] [CrossRef]
- Zhang, P.; Su, D.-M.; Liang, M.; Fu, J. Chemopreventive agents induce programmed death-1-ligand 1 (PD-L1) surface expression in breast cancer cells and promote PD-L1-mediated T cell apoptosis. Mol. Immunol. 2008, 45, 1470–1476. [Google Scholar] [CrossRef]
- Qin, X.; Liu, C.; Zhou, Y.; Wang, G. Cisplatin induces programmed death-1-ligand 1(PD-L1) over-expression in hepatoma H22 cells via Erk /MAPK signaling pathway. Cell. Mol. Biol. 2010, 56, 1366–1372. [Google Scholar]
- Brochez, L.; Chevolet, I.; Kruse, V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur. J. Cancer 2017, 76, 167–182. [Google Scholar] [CrossRef]
- Huang, A.C.; Orlowski, R.J.; Xu, X.; Mick, R.; George, S.M.; Yan, P.K.; Manne, S.; Kraya, A.A.; Wubbenhorst, B.; Dorfman, L.; et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat. Med. 2019, 25, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, A.O.; Pillai, R.N.; Yang, S.; Nasti, T.H.; Akondy, R.S.; Wieland, A.; Sica, G.L.; Yu, K.; Koenig, L.; Patel, N.T.; et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc. Natl. Acad. Sci. USA 2017, 114, 4993–4998. [Google Scholar] [CrossRef] [PubMed]
- Jacquelot, N.; Roberti, M.P.; Enot, D.P.; Rusakiewicz, S.; Ternès, N.; Jegou, S.; Woods, D.M.; Sodré, A.L.; Hansen, M.; Meirow, Y.; et al. Predictors of responses to immune checkpoint blockade in advanced melanoma. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Brochez, L.; Meireson, A.; Chevolet, I.; Sundahl, N.; Ost, P.; Kruse, V. Challenging PD-L1 expressing cytotoxic T cells as a predictor for response to immunotherapy in melanoma. Nat. Commun. 2018, 9, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Harrison, D.L.; Song, Y.; Ji, J.; Huang, J.; Hui, E. Antigen-Presenting Cell-Intrinsic PD-1 Neutralizes PD-L1 in cis to Attenuate PD-1 Signaling in T Cells. Cell Rep. 2018, 24, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, M.; Lenehan, J.G.; Maleki Vareki, S. Neoadjuvant Immunotherapy for High-Risk, Resectable Malignancies: Scientific Rationale and Clinical Challenges. J. Natl. Cancer Inst. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kinter, A.L.; Godbout, E.J.; McNally, J.P.; Sereti, I.; Roby, G.A.; O’Shea, M.A.; Fauci, A.S. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J. Immunol. 2008, 181, 6738–6746. [Google Scholar] [CrossRef]
- Dammeijer, F.; van Gulijk, M.; Mulder, E.E.; Lukkes, M.; Klaase, L.; van den Bosch, T.; van Nimwegen, M.; Lau, S.P.; Latupeirissa, K.; Schetters, S.; et al. The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell 2020, 38, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Qiu, X.; Zhang, Z.; Zhang, S.; Zhang, Y.; Liang, Y.; Guo, J.; Peng, H.; Chen, M.; Fu, Y.-X.; et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Lin, H.; Wei, S.; Hurt, E.M.; Green, M.D.; Zhao, L.; Vatan, L.; Szeliga, W.; Herbst, R.; Harms, P.W.; Fecher, L.A.; et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J. Clin. Investig. 2018, 128, 805–815. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Sunshine, J.; Taube, J.M. PD-1/PD-L1 inhibitors. Curr. Opin. Pharmacol. 2015, 23, 32–38. [Google Scholar] [CrossRef]
- Mukherjee, N.; Ji, N.; Hurez, V.; Curiel, T.J.; Montgomery, M.O.; Braun, A.J.; Nicolas, M.; Aguilera, M.; Kaushik, D.; Liu, Q.; et al. Intratumoral CD56bright natural killer cells are associated with improved survival in bladder cancer. Oncotarget 2018, 9, 36492–36502. [Google Scholar] [CrossRef]
- Meireson, A.; Devos, M.; Brochez, L. IDO Expression in Cancer: Different Compartment, Different Functionality? Front. Immunol. 2020, 11, 2340. [Google Scholar] [CrossRef]
- Hayashi, H.; Nakagawa, K. Combination therapy with PD-1 or PD-L1 inhibitors for cancer. Int. J. Clin. Oncol. 2020, 25, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Rozeman, E.A.; Menzies, A.M.; van Akkooi, A.C.J.; Adhikari, C.; Bierman, C.; van de Wiel, B.A.; Scolyer, R.A.; Krijgsman, O.; Sikorska, K.; Eriksson, H.; et al. Identification of the optimal combination dosing schedule of neoadjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma (OpACIN-neo): A multicentre, phase 2, randomised, controlled trial. Lancet Oncol. 2019, 20, 948–960. [Google Scholar] [CrossRef]
- van Dijk, N.; Gil-Jimenez, A.; Silina, K.; Hendricksen, K.; Smit, L.A.; de Feijter, J.M.; van Montfoort, M.L.; van Rooijen, C.; Peters, D.; Broeks, A.; et al. Preoperative ipilimumab plus nivolumab in locoregionally advanced urothelial cancer: The NABUCCO trial. Nat. Med. 2020, 26, 1839–1844. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Anang, N.-A.A.S.; Sharma, R.; Andrews, M.C.; Reuben, A.; Levine, J.H.; Cogdill, A.P.; Mancuso, J.J.; Wargo, J.A.; Pe’er, D.; et al. Combination anti–CTLA-4 plus anti–PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc. Natl. Acad. Sci. USA 2019, 116, 22699–22709. [Google Scholar] [CrossRef] [PubMed]
- Woods, D.M.; Laino, A.S.; Winters, A.; Alexandre, J.; Freeman, D.; Rao, V.; Adavani, S.S.; Weber, J.S.; Chattopadhyay, P.K. Nivolumab and ipilimumab are associated with distinct immune landscape changes and response-associated immunophenotypes. JCI Insight 2020, 5, e137066. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meireson, A.; Tavernier, S.J.; Van Gassen, S.; Sundahl, N.; Demeyer, A.; Spaas, M.; Kruse, V.; Ferdinande, L.; Van Dorpe, J.; Hennart, B.; et al. Immune Monitoring in Melanoma and Urothelial Cancer Patients Treated with Anti-PD-1 Immunotherapy and SBRT Discloses Tumor Specific Immune Signatures. Cancers 2021, 13, 2630. https://doi.org/10.3390/cancers13112630
Meireson A, Tavernier SJ, Van Gassen S, Sundahl N, Demeyer A, Spaas M, Kruse V, Ferdinande L, Van Dorpe J, Hennart B, et al. Immune Monitoring in Melanoma and Urothelial Cancer Patients Treated with Anti-PD-1 Immunotherapy and SBRT Discloses Tumor Specific Immune Signatures. Cancers. 2021; 13(11):2630. https://doi.org/10.3390/cancers13112630
Chicago/Turabian StyleMeireson, Annabel, Simon J. Tavernier, Sofie Van Gassen, Nora Sundahl, Annelies Demeyer, Mathieu Spaas, Vibeke Kruse, Liesbeth Ferdinande, Jo Van Dorpe, Benjamin Hennart, and et al. 2021. "Immune Monitoring in Melanoma and Urothelial Cancer Patients Treated with Anti-PD-1 Immunotherapy and SBRT Discloses Tumor Specific Immune Signatures" Cancers 13, no. 11: 2630. https://doi.org/10.3390/cancers13112630
APA StyleMeireson, A., Tavernier, S. J., Van Gassen, S., Sundahl, N., Demeyer, A., Spaas, M., Kruse, V., Ferdinande, L., Van Dorpe, J., Hennart, B., Allorge, D., Haerynck, F., Decaestecker, K., Rottey, S., Saeys, Y., Ost, P., & Brochez, L. (2021). Immune Monitoring in Melanoma and Urothelial Cancer Patients Treated with Anti-PD-1 Immunotherapy and SBRT Discloses Tumor Specific Immune Signatures. Cancers, 13(11), 2630. https://doi.org/10.3390/cancers13112630