
Focus on the Complex Interconnection between Cancer, Narcolepsy and Other Neurodegenerative Diseases: A Possible Case of Orexin-Dependent Inverse Comorbidity

 ,
,  , and
, and
Abstract
Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Cancer and Neurodegenerative Diseases
1.1. The Connection between Cancer and Neurodegenerative Diseases
1.2. The Biological Bases of the Inverse Comorbidity between Cancer and Neurodegeneration
1.3. The Role of Mitochondria
1.4. Other Factors
2. Cancer, Narcolepsy, and Other Neurodegenerative Diseases
2.1. Narcolepsy and Cancer
2.2. Narcolepsy and Neurodegenerative Diseases
3. Orexin, Cancer, and Neurodegenerative Diseases
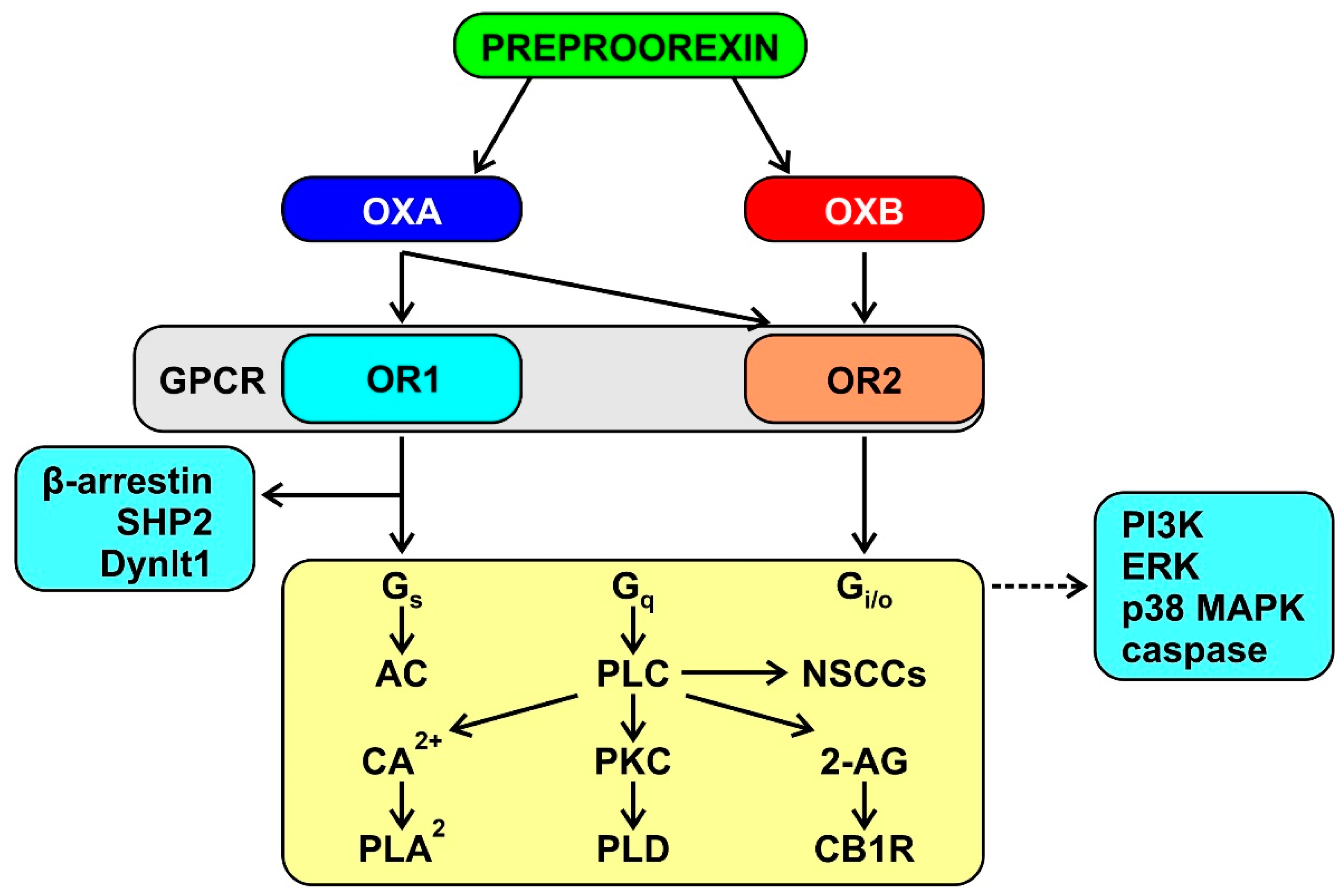
3.1. The Orexin System
3.2. Orexin and Mitochondrial Function
3.3. Microbiota
3.4. Genetic Factors
3.5. Orexin in Cancer
3.6. Orexin in Neurodegenerative Diseases
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021. [Google Scholar] [CrossRef] [PubMed]
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef]
- Trapp, B.D.; Nave, K.-A. Multiple Sclerosis: An Immune or Neurodegenerative Disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef]
- Giovannoni, G. The neurodegenerative prodrome in multiple sclerosis. Lancet Neurol. 2017, 16, 413–414. [Google Scholar] [CrossRef]
- Chaudhuri, A. Multiple sclerosis is primarily a neurodegenerative disease. J. Neural Transm. 2013, 120, 1463–1466. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Pugliatti, M.; Rosati, G.; Carton, H.; Riise, T.; Drulovic, J.; Vecsei, L.; Milanov, I. The epidemiology of multiple sclerosis in Europe. Eur. J. Neurol. 2006, 13, 700–722. [Google Scholar] [CrossRef]
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; Van den Berg, L.H. Amyotrophic Lateral Sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Palmer, S.; Albergante, L.; Blackburn, C.C.; Newman, T.J. Thymic involution and rising disease incidence with age. Proc. Natl. Acad. Sci. USA 2018, 115, 1883–1888. [Google Scholar] [CrossRef]
- de Pedro-Cuesta, J.; Rabano, A.; Martinez-Martin, P.; Ruiz-Tovar, M.; Alcalde-Cabero, E.; Almazan-Isla, J.; Avellanal, F.; Calero, M. Comparative Incidence of Conformational, Neurodegenerative Disorders. PLoS ONE 2015, 10, e0137342. [Google Scholar]
- Catalá-López, F.; Suárez-Pinilla, M.; Suárez-Pinilla, P.; Valderas, J.M.; Gómez-Beneyto, M.; Martinez, S.; Balanzá-Martínez, V.; Climent, J.; Valencia, A.; McGrath, J.; et al. Inverse and Direct Cancer Comorbidity in People with Central Nervous System Disorders: A Meta-Analysis of Cancer Incidence in 577,013 Participants of 50 Observational Studies. Psychother. Psychosom. 2014, 83, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Driver, J.A.; Beiser, A.; Au, R.; Kreger, B.E.; Splansky, G.L.; Kurth, T.; Kiel, D.P.; Lu, K.P.; Seshadri, S.; Wolf, P.A. Inverse Association between Cancer and Alzheimer’s Disease: Results from the Framingham Heart Study. BMJ 2012, 344, e1442. [Google Scholar] [CrossRef]
- Fratiglioni, L.; Wang, H.-X. Smoking and Parkinson’s and Alzheimer’s disease: Review of the epidemiological studies. Behav. Brain Res. 2000, 113, 117–120. [Google Scholar] [CrossRef]
- Ospina-Romero, M.; Glymour, M.M.; Hayes-Larson, E.; Mayeda, E.R.; Graff, R.E.; Brenowitz, W.D.; Ackley, S.F.; Witte, J.S.; Kobayashi, L.C. Association between Alzheimer Disease and Cancer with Evaluation of Study Biases: A Systematic Review and Meta-Analysis. JAMA Netw. Open 2020, 3, e2025515. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Valle, J.; Tejero, H.; Ibáñez, K.; Portero, J.L.; Krallinger, M.; Al-Shahrour, F.; Tabares-Seisdedos, R.; Baudot, A.; Valencia, A. A molecular hypothesis to explain direct and inverse co-morbidities between Alzheimer’s Disease, Glioblastoma and Lung cancer. Sci. Rep. 2017, 7, 4474. [Google Scholar] [CrossRef]
- Cui, X.; Liew, Z.; Hansen, J.; Lee, P.-C.; Arah, O.A.; Ritz, B. Cancers Preceding Parkinson’s Disease after Adjustment for Bias in a Danish Population-Based Case-Control Study. Neuroepidemiology 2019, 52, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Ghajarzadeh, M.; Mohammadi, A.; Sahraian, M.A. Risk of cancer in multiple sclerosis (MS): A systematic review and meta-analysis. Autoimmun. Rev. 2020, 19, 102650. [Google Scholar] [CrossRef] [PubMed]
- Thormann, A.; Koch-Henriksen, N.; Laursen, B.; Sørensen, P.S.; Magyari, M. Inverse comorbidity in multiple sclerosis: Findings in a complete nationwide cohort. Mult. Scler. Relat. Disord. 2016, 10, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Houck, A.L.; Seddighi, S.; Driver, J.A. At the Crossroads between Neurodegeneration and Cancer: A Review of Overlapping Biology and Its Implications. Curr. Aging Sci. 2018, 11, 77–89. [Google Scholar] [CrossRef]
- Kesler, S.R.; Watson, C.L.; Blayney, D.W. Brain network alterations and vulnerability to simulated neurodegeneration in breast cancer. Neurobiol. Aging 2015, 36, 2429–2442. [Google Scholar] [CrossRef] [PubMed]
- Frain, L.; Swanson, D.; Cho, K.; Gagnon, D.; Lu, K.P.; Betensky, R.A.; Driver, J. Association of Cancer and Alzheimer’s Disease Risk in a National Cohort of Veterans. Alzheimer’s Dement. 2017, 13, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Tabarés-Seisdedos, R.; Rubenstein, J.L. Inverse cancer comorbidity: A serendipitous opportunity to gain insight into CNS disorders. Nat. Rev. Neurosci. 2013, 14, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Klus, P.; Cirillo, D.; Orfila, T.B.; Tartaglia, G.G. Neurodegeneration and Cancer: Where the Disorder Prevails. Sci. Rep. 2015, 5, 15390. [Google Scholar] [CrossRef] [PubMed]
- Tallaksen, C.M.; Muller, U. Cancer and Neurodegeneration: Time to Move Beyond Janus? Neurology 2017, 88, 1106–1107. [Google Scholar] [CrossRef] [PubMed]
- Rojas, N.G.; Cesarini, M.; Etcheverry, J.L.; Da Prat, G.A.; Arciuch, V.A.; Gatto, E.M. Neurodegenerative diseases and cancer: Sharing common mechanisms in complex interactions. J. Integr. Neurosci. 2020, 19, 187–199. [Google Scholar] [PubMed]
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: Linking Aging to Chronic Disease. Cell 2014, 159, 709–713. [Google Scholar] [CrossRef]
- Garcia-Ratés, S.; Greenfield, S. Cancer and neurodegeneration: Two sides, same coin? Oncotarget 2017, 8, 22307–22308. [Google Scholar] [CrossRef]
- Duffner, P.K. Risk factors for cognitive decline in children treated for brain tumors. Eur. J. Paediatr. Neurol. 2010, 14, 106–115. [Google Scholar] [CrossRef]
- Mogavero, M.P.; Bruni, O.; DelRosso, L.M.; Ferri, R. Neurodevelopmental Consequences of Pediatric Cancer and Its Treatment: The Role of Sleep. Brain Sci. 2020, 10, 411. [Google Scholar] [CrossRef]
- Marusak, H.A.; Iadipaolo, A.S.; Harper, F.W.; Elrahal, F.; Taub, J.W.; Goldberg, E.; Rabinak, C.A. Neurodevelopmental consequences of pediatric cancer and its treatment: Applying an early adversity framework to understanding cognitive, behavioral, and emotional outcomes. Neuropsychol. Rev. 2018, 28, 123–175. [Google Scholar] [CrossRef]
- Catalan-Figueroa, J.; Palma-Florez, S.; Álvarez, G.; Fritz, H.F.; O Jara, M.; O Morales, J. Nanomedicine and nanotoxicology: The pros and cons for neurodegeneration and brain cancer. Nanomedicine 2016, 11, 171–187. [Google Scholar] [CrossRef]
- Cheung, Y.T.; Brinkman, T.M.; Mulrooney, D.A.; Mzayek, Y.; Liu, W.; Banerjee, P.; Panoskaltsis-Mortari, A.; Srivastava, D.; Pui, C.-H.; Robison, L.L.; et al. Impact of sleep, fatigue, and systemic inflammation on neurocognitive and behavioral outcomes in long-term survivors of childhood acute lymphoblastic leukemia. Cancer 2017, 123, 3410–3419. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Y.T.; Khan, R.B.; Liu, W.; Brinkman, T.M.; Edelmann, M.N.; Reddick, W.E.; Pei, D.; Panoskaltsis-Mortari, A.; Srivastava, D.; Cheng, C.; et al. Association of Cerebrospinal Fluid Biomarkers of Central Nervous System Injury with Neurocognitive and Brain Imaging Outcomes in Children Receiving Chemotherapy for Acute Lymphoblastic Leukemia. JAMA Oncol. 2018, 4, e180089. [Google Scholar] [CrossRef] [PubMed]
- Tabarés-Seisdedos, R.; Dumont, N.; Baudot, A.; Valderas, J.M.; Climent, J.; Valencia, A.; Crespo-Facorro, B.; Vieta, E.; Gómez-Beneyto, M.; Martinez, S.; et al. No paradox, no progress: Inverse cancer comorbidity in people with other complex diseases. Lancet Oncol. 2011, 12, 604–608. [Google Scholar] [CrossRef]
- Devine, M.J.; Plun-Favreau, H.; Wood, N.W. Parkinson’s Disease and Cancer: Two Wars, One Front. Nat. Rev. Cancer 2011, 11, 812–823. [Google Scholar] [CrossRef]
- West, A.B.; Dawson, V.L.; Dawson, T.M. To Die or Grow: Parkinson’s Disease and Cancer. Trends Neurosci. 2005, 28, 348–352. [Google Scholar] [CrossRef]
- Inzelberg, R.; Samuels, Y.; Azizi, E.; Qutob, N.; Inzelberg, L.; Domany, E.; Schechtman, E.; Friedman, E. Parkinson disease (PARK) genes are somatically mutated in cutaneous melanoma. Neurol. Genet. 2016, 2, e70. [Google Scholar] [CrossRef]
- Matsushima-Nishiu, M.; Unoki, M.; Ono, K.; Tsunoda, T.; Minaguchi, T.; Kuramoto, H.; Nishida, M.; Satoh, T.; Tanaka, T.; Nakamura, Y. Growth and gene expression profile analyses of endometrial cancer cells expressing exogenous PTEN. Cancer Res. 2001, 61, 3741–3749. [Google Scholar]
- Salemi, M.; Mazzetti, S.; de Leonardis, M.; Giampietro, F.; Medici, V.; Poloni, T.E.; Cannarella, R.; Giaccone, G.; Pezzoli, G.; Cappelletti, G.; et al. Poly (Adp-Ribose) Polymerase 1 and Parkinson’s Disease: A Study in Post-Mortem Human Brain. Neurochem. Int. 2021, 144, 104978. [Google Scholar] [CrossRef]
- Salemi, M.; Galia, A.; Fraggetta, F.; La Corte, C.; Pepe, P.; La Vignera, S.; Improta, G.; Bosco, P.; Calogero, A. Poly (ADP-ribose) polymerase 1 protein expression in normal and neoplastic prostatic tissue. Eur. J. Histochem. 2013, 57, e13. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.-C.A.; Cho, K.; Lindstrom, S.; Kraft, P.; Cormack, J.; Liang, L.; Driver, J.A. Investigating the genetic relationship between Alzheimer’s disease and cancer using GWAS summary statistics. Hum. Genet. 2017, 136, 1341–1351. [Google Scholar] [CrossRef]
- Checler, F.; da Costa, C.A. P53 in Neurodegenerative Diseases and Brain Cancers. Pharmacol. Ther. 2014, 142, 99–113. [Google Scholar] [CrossRef]
- Seo, J.; Park, M. Molecular crosstalk between cancer and neurodegenerative diseases. Cell. Mol. Life Sci. 2020, 77, 2659–2680. [Google Scholar] [CrossRef] [PubMed]
- Ariga, H. Common Mechanisms of Onset of Cancer and Neurodegenerative Diseases. Biol. Pharm. Bull. 2015, 38, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Modulation of mitochondrial apoptosis by PI3K inhibitors. Mitochondrion 2013, 13, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Boveris, A.; Cadenas, E. Mitochondrial Energy Metabolism and Redox Signaling in Brain Aging and Neurodegeneration. Antioxid. Redox Signal. 2014, 20, 353–371. [Google Scholar] [CrossRef]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef]
- Nagy, A.; Eder, K.; Selak, M.A.; Kalman, B. Mitochondrial energy metabolism and apoptosis regulation in glioblastoma. Brain Res. 2015, 1595, 127–142. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M. Gut Dysbiosis Dysregulates Central and Systemic Homeostasis Via Suboptimal Mitochondrial Function: Assessment, Treatment and Classification Implications. Curr. Top Med. Chem. 2020, 20, 524–539. [Google Scholar] [CrossRef]
- Cai, Z.; Yan, L.-J.; Ratka, A. Telomere Shortening and Alzheimer’s Disease. Neuromol. Med. 2013, 15, 25–48. [Google Scholar] [CrossRef] [PubMed]
- Panossian, L.A.; Porter, V.R.; Valenzuela, H.F.; Zhu, X.; Reback, E.; Masterman, D.; Cummings, J.L.; Effros, R.B. Telomere Shortening in T Cells Correlates with Alzheimer’s Disease Status. Neurobiol. Aging 2003, 24, 77–84. [Google Scholar] [CrossRef]
- Depinho, R.A. The age of cancer. Nat. Cell Biol. 2000, 408, 248–254. [Google Scholar] [CrossRef]
- Kim, N.; Piatyszek, M.; Prowse, K.; Harley, C.; West, M.; Ho, P.; Coviello, G.; Wright, W.; Weinrich, S.; Shay, J. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/Beta-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Sun, L.; Lian, J.; Zhang, M.; Liu, D. Correlation of Alzheimer’s Disease with Wnt Signaling Pathway and Neural Stem Cells. In Proceedings of the 4th International Conference on Mechatronics, Materials, Chemistry and Computer Engineering, Xi’an, China, 12–13 December 2015; pp. 119–122. [Google Scholar]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nat. Cell Biol. 2005, 434, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Serafino, A.; Sferrazza, G.; Baldeschi, A.C.; Nicotera, G.; Andreola, F.; Pittaluga, E.; Pierimarchi, P. Developing drugs that target the Wnt pathway: Recent approaches in cancer and neurodegenerative diseases. Expert Opin. Drug Discov. 2017, 12, 169–186. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-Synuclein in Lewy Bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Houck, A.L.; Hernández, F.; Ávila, J. A Simple Model to Study Tau Pathology. J. Exp. Neurosci. 2016, 10, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L. Heat shock protein 90: The cancer chaperone. J. Biosci. 2007, 32, 517–530. [Google Scholar] [CrossRef]
- Frezza, M.; Schmitt, S.; Dou, Q.P. Targeting the ubiquitin-proteasome pathway: An emerging concept in cancer therapy. Curr. Top. Med. Chem. 2011, 11, 2888–2905. [Google Scholar] [CrossRef] [PubMed]
- Chouliaras, L.; Mastroeni, D.; Delvaux, E.; Grover, A.; Kenis, G.; Hof, P.R.; Steinbusch, H.W.; Coleman, P.D.; Rutten, B.P.; van den Hove, D.L. Consistent Decrease in Global DNA Methylation and Hydroxymethylation in the Hippocampus of Alzheimer’s Disease Patients. Neurobiol. Aging 2013, 34, 2091–2099. [Google Scholar] [CrossRef]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; Laurent, G.S., 3rd; Kenny, P.J.; Wahlestedt, C. Expression of a Noncoding Rna Is Elevated in Alzheimer’s Disease and Drives Rapid Feed-Forward Regulation of Beta-Secretase. Nat. Med. 2008, 14, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jaber, V.; Alexandrov, P.N.; Vergallo, A.; Lista, S.; Hampel, H.; Lukiw, W.J. Microrna-Based Biomarkers in Alzheimer’s Disease (Ad). Front. Neurosci. 2020, 14, 585432. [Google Scholar] [CrossRef]
- Vergallo, A.; Lista, S.; Zhao, Y.; Lemercier, P.; Teipel, S.J.; Potier, M.C.; Habert, M.O.; Dubois, B.; Lukiw, W.J.; Hampel, H.; et al. Mirna-15b and Mirna-125b Are Associated with Regional Abeta-Pet and Fdg-Pet Uptake in Cognitively Normal Individuals with Subjective Memory Complaints. Transl. Psychiatry 2021, 11, 78. [Google Scholar] [CrossRef]
- Du, L.; Pertsemlidis, A. Cancer and neurodegenerative disorders: Pathogenic convergence through microRNA regulation. J. Mol. Cell Biol. 2011, 3, 176–180. [Google Scholar] [CrossRef]
- Vishnoi, A.; Rani, S. MiRNA Biogenesis and Regulation of Diseases: An Overview. Methods Mol. Biol. 2017, 1509, 1–10. [Google Scholar]
- Bassetti, C.L.A.; Adamantidis, A.; Burdakov, D.; Han, F.; Gay, S.; Kallweit, U.; Khatami, R.; Koning, F.; Kornum, B.R.; Lammers, G.J.; et al. Narcolepsy—Clinical spectrum, aetiopathophysiology, diagnosis and treatment. Nat. Rev. Neurol. 2019, 15, 519–539. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, S.; Mayeli, M.; Saghazadeh, A.; Rezaei, N. Cytokines in narcolepsy: A systematic review and meta-analysis. Cytokine 2020, 131, 155103. [Google Scholar] [CrossRef] [PubMed]
- Jennum, P.; Ibsen, R.; Knudsen, S.; Kjellberg, J. Comorbidity and Mortality of Narcolepsy: A Controlled Retro- and Prospective National Study. Sleep 2013, 36, 835–840. [Google Scholar] [CrossRef]
- Black, J.; Reaven, N.; Funk, S.; McGaughey, K.; Ohayon, M.; Guilleminault, C.; Ruoff, C. Medical comorbidity in narcolepsy: Findings from the Burden of Narcolepsy Disease (BOND) study. Sleep Med. 2017, 33, 13–18. [Google Scholar] [CrossRef]
- Jennum, P.; Pickering, L.; Thorstensen, E.W.; Ibsen, R.; Kjellberg, J. Morbidity of childhood onset narcolepsy: A controlled national study. Sleep Med. 2017, 29, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-M.; Chen, Y.-T.; Tao, C.-W.; Ou, S.-M.; Hsiao, Y.-H.; Li, S.-Y.; Chen, T.-J.; Perng, D.-W.; Chou, K.-T. Adult narcoleptic patients have increased risk of cancer: A nationwide population-based study. Cancer Epidemiol. 2015, 39, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Kukkonen, J.P. Physiology of the orexinergic/hypocretinergic system: A revisit in 2012. Am. J. Physiol. Physiol. 2013, 304, C2–C32. [Google Scholar] [CrossRef]
- Leonard, C.S.; Kukkonen, J.P. Orexin/hypocretin receptor signalling: A functional perspective. Br. J. Pharmacol. 2014, 171, 294–313. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, C.S.; Kiss, M.G.; Rattik, S.; He, S.; Vassalli, A.; Valet, C.; Anzai, A.; Chan, C.T.; Mindur, J.E.; Kahles, F.; et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nat. Cell Biol. 2019, 566, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Economou, N.-T.; Manconi, M.; Ghika, J.; Raimondi, M.; Bassetti, C.L. Development of Parkinson and Alzheimer Diseases in Two Cases of Narcolepsy-Cataplexy. Eur. Neurol. 2012, 67, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Kallweit, U.; Bassetti, C.L.A.; Oberholzer, M.; Fronczek, R.; Beguin, M.; Strub, M.; Lammers, G.J. Coexisting Narcolepsy (with and without Cataplexy) and Multiple Sclerosis: Six New Cases and a Literature Review. J. Neurol. 2018, 265, 2071–2078. [Google Scholar] [CrossRef] [PubMed]
- Scammell, T.E.; Matheson, J.K.; Honda, M.; Thannickal, T.C.; Siegel, J.M. Coexistence of Narcolepsy and Alzheimer’s Disease. Neurobiol. Aging 2012, 33, 1318–1319. [Google Scholar] [CrossRef]
- Long, J.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Liguori, C.; Placidi, F.; Izzi, F.; Nuccetelli, M.; Bernardini, S.; Sarpa, M.G.; Cum, F.; Marciani, M.G.; Mercuri, N.B.; Romigi, A. Beta-amyloid and phosphorylated tau metabolism changes in narcolepsy over time. Sleep Breath. 2016, 20, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Liguori, C.; Placidi, F.; Albanese, M.; Nuccetelli, M.; Izzi, F.; Marciani, M.G.; Mercuri, N.B.; Bernardini, S.; Romigi, A. CSF beta-amyloid levels are altered in narcolepsy: A link with the inflammatory hypothesis? J. Sleep Res. 2014, 23, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Jennum, P.J.; Østergaard Pedersen, L.; Bahl, J.M.C.; Modvig, S.; Fog, K.; Holm, A.; Kornum, B.R.; Gammeltoft, S. Cerebrospinal Fluid Biomarkers of Neurodegeneration Are Decreased or Normal in Narcolepsy. Sleep 2017, 40. [Google Scholar] [CrossRef]
- Baiardi, S.; Pizza, F.; Polischi, B.; Moresco, M.; Abu-Rumeileh, S.; Plazzi, G.; Parchi, P. Cerebrospinal Fluid Biomarkers of Neurodegeneration in Narcolepsy Type 1. Sleep 2020, 43. [Google Scholar] [CrossRef] [PubMed]
- Gabelle, A.; Jaussent, I.; Ben Bouallègue, F.; Lehmann, S.; Lopez, R.; Barateau, L.; Grasselli, C.; Pesenti, C.; De Verbizier, D.; Béziat, S.; et al. Reduced brain amyloid burden in elderly patients with narcolepsy type. Ann. Neurol. 2019, 85, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.E.; Lim, M.M.; Bateman, R.J.; Lee, J.J.; Smyth, L.P.; Cirrito, J.R.; Fujiki, N.; Nishino, S.; Holtzman, D.M. Amyloid-Beta Dynamics Are Regulated by Orexin and the Sleep-Wake Cycle. Science 2009, 326, 1005–1007. [Google Scholar] [CrossRef]
- Sakurai, T.; Amemiya, A.; Ishii, M.; Matsuzaki, I.; Chemelli, R.M.; Tanaka, H.; Williams, S.; A Richardson, J.; Kozlowski, G.P.; Wilson, S.; et al. Orexins and Orexin Receptors: A Family of Hypothalamic Neuropeptides and G Protein-Coupled Receptors that Regulate Feeding Behavior. Cell 1998, 92, 573–585. [Google Scholar] [CrossRef]
- de Lecea, L.; Kilduff, T.S.; Peyron, C.; Gao, X.; Foye, P.E.; Danielson, P.E.; Fukuhara, C.; Battenberg, E.L.; Gautvik, V.T.; Bartlett, F.S., 2nd; et al. The Hypocretins: Hypothalamus-Specific Peptides with Neuroexcitatory Activity. Proc. Natl. Acad. Sci. USA 1998, 95, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Mileykovskiy, B.Y.; Kiyashchenko, L.I.; Siegel, J.M. Behavioral Correlates of Activity in Identified Hypocretin/Orexin Neurons. Neuron 2005, 46, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Peyron, C.; Tighe, D.K.; Pol, A.N.V.D.; De Lecea, L.; Heller, H.C.; Sutcliffe, J.G.; Kilduff, T. Neurons Containing Hypocretin (Orexin) Project to Multiple Neuronal Systems. J. Neurosci. 1998, 18, 9996–10015. [Google Scholar] [CrossRef] [PubMed]
- Kukkonen, J.P.; Leonard, C.S. Orexin/hypocretin receptor signalling cascades. Br. J. Pharmacol. 2014, 171, 314–331. [Google Scholar] [CrossRef]
- Kukkonen, J.P. OX2 orexin/hypocretin receptor signal transduction in recombinant Chinese hamster ovary cells. Cell. Signal. 2016, 28, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Kukkonen, J.P. G-Protein-Dependency of Orexin/Hypocretin Receptor Signalling in Recombinant Chinese Hamster Ovary Cells. Biochem. Biophys. Res. Commun. 2016, 476, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T. The neural circuit of orexin (hypocretin): Maintaining sleep and wakefulness. Nat. Rev. Neurosci. 2007, 8, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, D.; Silvani, A.; Benarroch, E.E.; Cortelli, P. Orexin/hypocretin system and autonomic control: New insights and clinical correlations. Neurology 2014, 82, 271–278. [Google Scholar] [CrossRef]
- Bastianini, S.; Silvani, A. Clinical Implications of Basic Research: The Role of Hypocretin/Orexin Neurons in the Central Autonomic Network. Clin. Transl. Neurosci. 2018, 2. [Google Scholar] [CrossRef]
- Jennum, P.J.; Plazzi, G.; Silvani, A.; Surkin, L.A.; Dauvilliers, Y. Cardiovascular disorders in narcolepsy: Review of associations and determinants. Sleep Med. Rev. 2021, 58, 101440. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.V.; Pavlovic, A.; Shang, C.; Wheeler, M.T.; Miller, C.L.; Liu, J.; Dewey, F.E.; Pan, S.; Thanaporn, P.K.; Absher, D.; et al. Systems Genomics Identifies a Key Role for Hypocretin/Orexin Receptor-2 in Human Heart Failure. J. Am. Coll. Cardiol. 2015, 66, 2522–2533. [Google Scholar] [CrossRef]
- Kastin, A.J.; Akerstrom, V. Orexin A but not orexin B rapidly enters brain from blood by simple diffusion. J. Pharmacol. Exp. Ther. 1999, 289, 219–223. [Google Scholar]
- Yoshida, Y.; Fujiki, N.; Maki, R.A.; Schwarz, D.; Nishino, S. Differential kinetics of hypocretins in the cerebrospinal fluid after intracerebroventricular administration in rats. Neurosci. Lett. 2003, 346, 182–186. [Google Scholar] [CrossRef]
- Bingham, S.; Davey, P.T.; Babbs, A.J.; Irving, E.A.; Sammons, M.J.; Wyles, M.; Jeffrey, P.; Cutler, L.; Riba, I.; Johns, A.; et al. Orexin-A, an hypothalamic peptide with analgesic properties. Pain 2001, 92, 81–90. [Google Scholar] [CrossRef]
- Zhu, Z.; Xu, L.; Cao, D.; Song, C.; Wang, Y.; Li, M.; Yan, J.; Xie, Z. Effect of Orexin-a on Mitochondrial Biogenesis, Mitophagy and Structure in Hek293-Appswe Cell Model of Alzheimer’s Disease. Clin. Exp. Pharmacol. Physiol. 2021, 48, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Meng, Y.; Chu, B.; Shen, Y.; Xue, X.; Song, C.; Liu, X.; Ding, M.; Cao, X.; Wang, P.; et al. Orexin-a Exacerbates Alzheimer’s Disease by Inducing Mitochondrial Impairment. Neurosci. Lett. 2020, 718, 134741. [Google Scholar] [CrossRef] [PubMed]
- Lorenzoni, P.J.; Werneck, L.C.; Crippa, A.C.D.S.; Zanatta, A.; Kay, C.S.K.; Silvado, C.E.S.; Scola, R.H. Is there a relationship between narcolepsy, multiple sclerosis and HLA-DQB1*06:02? Arq. Neuro-Psiquiatr. 2017, 75, 345–348. [Google Scholar] [CrossRef] [PubMed]
- West, A.P. Mitochondrial dysfunction as a trigger of innate immune responses and inflammation. Toxicology 2017, 391, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Diakos, C.; A Charles, K.; McMillan, D.C.; Clarke, S.J. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 2014, 15, e493–e503. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- LeComte, A.; Barateau, L.; Pereira, P.; Paulin, L.; Auvinen, P.; Scheperjans, F.; Dauvilliers, Y. Gut microbiota composition is associated with narcolepsy type. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e896. [Google Scholar] [CrossRef]
- Foster, J.A.; Lyte, M.; Meyer, E.; Cryan, J.F. Gut Microbiota and Brain Function: An Evolving Field in Neuroscience. Int. J. Neuropsychopharmacol. 2016, 19, pyv114. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef]
- Spielman, L.J.; Gibson, D.L.; Klegeris, A. Unhealthy gut, unhealthy brain: The role of the intestinal microbiota in neurodegenerative diseases. Neurochem. Int. 2018, 120, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, M.; Miyagawa, T.; Toyoda, H.; Khor, S.-S.; Koike, A.; Nitta, A.; Akiyama, K.; Sasaki, T.; Honda, Y.; Honda, M.; et al. Genome-wide analysis of CNV (copy number variation) and their associations with narcolepsy in a Japanese population. J. Hum. Genet. 2014, 59, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Sripada, L.; Tomar, D.; Singh, R. Mitochondria: One of the destinations of miRNAs. Mitochondrion 2012, 12, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yang, L.; Zhao, Y.; Tang, M.; Wang, F.; Wang, X.; Li, G.; Du, Y. Reproducibility of quantitative real-time PCR assay in microRNA expression profiling and comparison with microarray analysis in narcolepsy. SpringerPlus 2015, 4, 812. [Google Scholar] [CrossRef]
- Holm, A.; Bang-Berthelsen, C.H.; Knudsen, S.; Kornum, B.R.; Modvig, S.; Jennum, P.; Gammeltoft, S. miRNA Profiles in Plasma from Patients with Sleep Disorders Reveal Dysregulation of miRNAs in Narcolepsy and Other Central Hypersomnias. Sleep 2014, 37, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Possovre, M.-L.; Tafti, M. Conditional Deletion of Mirna in Hypocretin Neurons: A New Mouse Model of Narcolepsy. J. Sleep Res. 2020, 29, e13181. [Google Scholar]
- Mogavero, M.P.; DelRosso, L.M.; Fanfulla, F.; Bruni, O.; Ferri, R. Sleep disorders and cancer: State of the art and future perspectives. Sleep Med. Rev. 2020, 56, 101409. [Google Scholar] [CrossRef]
- Graybill, N.L.; Weissig, V. A review of orexin’s unprecedented potential as a novel, highly-specific treatment for various localized and metastatic cancers. SAGE Open Med. 2017, 5. [Google Scholar] [CrossRef]
- Kim, M.-K.; Park, H.-J.; Kim, S.-R.; Choi, Y.K.; Bae, S.-K.; Bae, M.-K. Involvement of Heme Oxygenase-1 in Orexin-A-induced Angiogenesis in Vascular Endothelial Cells. Korean J. Physiol. Pharmacol. 2015, 19, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D. The Heme Oxygenase System: A Regulator of Second Messenger Gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef]
- Mediavilla, C. Bidirectional gut-brain communication: A role for orexin-A. Neurochem. Int. 2020, 141, 104882. [Google Scholar] [CrossRef] [PubMed]
- Rouet-Benzineb, P.; Rouyer-Fessard, C.; Jarry, A.; Avondo, V.; Pouzet, C.; Yanagisawa, M.; Laboisse, C.; Laburthe, M.; Voisin, T. Orexins Acting at Native OX1 Receptor in Colon Cancer and Neuroblastoma Cells or at Recombinant OX1 Receptor Suppress Cell Growth by Inducing Apoptosis. J. Biol. Chem. 2004, 279, 45875–45886. [Google Scholar] [CrossRef] [PubMed]
- Voisin, T.; El Firar, A.; Fasseu, M.; Rouyer-Fessard, C.; Descatoire, V.; Walker, F.; Paradis, V.; Bedossa, P.; Henin, D.; Lehy, T.; et al. Aberrant Expression of OX1 Receptors for Orexins in Colon Cancers and Liver Metastases: An Openable Gate to Apoptosis. Cancer Res. 2011, 71, 3341–3351. [Google Scholar] [CrossRef]
- Wen, J.; Zhao, Y.; Guo, L. Orexin A induces autophagy in HCT-116 human colon cancer cells through the ERK signaling pathway. Int. J. Mol. Med. 2016, 37, 126–132. [Google Scholar] [CrossRef]
- Bai, B.; Chen, X.; Zhang, R.; Wang, X.; Jiang, Y.; Li, D.; Wang, Z.; Chen, J. Dual-agonist occupancy of orexin receptor 1 and cholecystokinin A receptor heterodimers decreases G-protein–dependent signaling and migration in the human colon cancer cell line HT-29. Biochim. Biophys. Acta (BBA)-Mol. Bioenerg. 2017, 1864, 1153–1164. [Google Scholar] [CrossRef]
- Messal, N.; Fernandez, N.; Dayot, S.; Gratio, V.; Nicole, P.; Prochasson, C.; Chantret, I.; Leguilloux, G.; Jarry, A.; Couvelard, A.; et al. Ectopic expression of OX1R in ulcerative colitis mediates anti-inflammatory effect of orexin-A. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 3618–3628. [Google Scholar] [CrossRef]
- Biegańska, K.; Sokołowska, P.; Joehren, O.; Zawilska, J.B. Orexin A Suppresses the Growth of Rat C6 Glioma Cells via a Caspase-Dependent Mechanism. J. Mol. Neurosci. 2012, 48, 706–712. [Google Scholar] [CrossRef][Green Version]
- Wen, J.; Zhao, Y.; Shen, Y.; Guo, L. Effect of orexin A on apoptosis in BGC-823 gastric cancer cells via OX1R through the AKT signaling pathway. Mol. Med. Rep. 2015, 11, 3439–3444. [Google Scholar] [CrossRef][Green Version]
- Liu, Y.; Zhao, Y.; Ju, S.; Guo, L. Orexin A upregulates the protein expression of OX1R and enhances the proliferation of SGC-7901 gastric cancer cells through the ERK signaling pathway. Int. J. Mol. Med. 2015, 35, 539–545. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Suo, L.; Chang, X.; Zhao, Y. The Orexin-A-Regulated Akt/mTOR Pathway Promotes Cell Proliferation through Inhibiting Apoptosis in Pancreatic Cancer Cells. Front. Endocrinol. 2018, 9, 647. [Google Scholar] [CrossRef] [PubMed]
- Dayot, S.; Speisky, D.; Couvelard, A.; Bourgoin, P.; Gratio, V.; Cros, J.; Rebours, V.; Sauvanet, A.; Bedossa, P.; Paradis, V.; et al. In vitro, in vivo and ex vivo demonstration of the antitumoral role of hypocretin-1/orexin-A and almorexant in pancreatic ductal adenocarcinoma. Oncotarget 2018, 9, 6952–6967. [Google Scholar] [CrossRef]
- Alexandre, D.; Hautot, C.; Mehio, M.; Jeandel, L.; Courel, M.; Voisin, T.; Couvineau, A.; Gobet, F.; Leprince, J.; Pfister, C.; et al. The orexin type 1 receptor is overexpressed in advanced prostate cancer with a neuroendocrine differentiation, and mediates apoptosis. Eur. J. Cancer 2014, 50, 2126–2133. [Google Scholar] [CrossRef] [PubMed]
- Valiante, S.; Liguori, G.; Tafuri, S.; Pavone, L.M.; Campese, R.; Monaco, R.; Iachetta, G.; Assisi, L.; Mirabella, N.; Forte, M.; et al. Expression and potential role of the peptide orexin-A in prostate cancer. Biochem. Biophys. Res. Commun. 2015, 464, 1290–1296. [Google Scholar] [CrossRef]
- Szyszka, M.; Paschke, L.; Tyczewska, M.; Rucinski, M.; Grabowska, P.; Malendowicz, L.K. Lack of expression of preproorexin and orexin receptors genes in human normal and prostate cancer cell lines. Folia Histochem. Cytobiol. 2015, 53, 333–341. [Google Scholar] [CrossRef][Green Version]
- Fronczek, R.; van Geest, S.; Frolich, M.; Overeem, S.; Roelandse, F.W.; Lammers, G.J.; Swaab, D.F. Hypocretin (Orexin) Loss in Alzheimer’s Disease. Neurobiol. Aging 2012, 33, 1642–1650. [Google Scholar] [CrossRef]
- Oh, J.; Eser, R.A.; Ehrenberg, A.J.; Morales, D.; Petersen, C.; Kudlacek, J.; Dunlop, S.R.; Theofilas, P.; Resende, E.; Cosme, C.; et al. Profound Degeneration of Wake-Promoting Neurons in Alzheimer’s Disease. Alzheimer’s Dement. 2019, 15, 1253–1263. [Google Scholar] [CrossRef]
- Shimizu, S.; Takenoshita, N.; Inagawa, Y.; Tsugawa, A.; Hirose, D.; Kaneko, Y.; Ogawa, Y.; Serisawa, S.; Sakurai, S.; Hirao, K.; et al. Positive Association Between Cognitive Function and Cerebrospinal Fluid Orexin A Levels in Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 73, 117–123. [Google Scholar] [CrossRef]
- Gabelle, A.; Jaussent, I.; Hirtz, C.; Vialaret, J.; Navucet, S.; Grasselli, C.; Robert, P.; Lehmann, S.; Dauvilliers, Y. Cerebrospinal fluid levels of orexin-A and histamine, and sleep profile within the Alzheimer process. Neurobiol. Aging 2017, 53, 59–66. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. J. Park. Dis. 2017, 7, S51–S69. [Google Scholar] [CrossRef]
- Thannickal, T.C.; Lai, Y.Y.; Siegel, J.M. Hypocretin (Orexin) Cell Loss in Parkinson’s Disease. Brain 2007, 130, 1586–1595. [Google Scholar] [CrossRef]
- Kasanuki, K.; Iseki, E.; Kondo, D.; Fujishiro, H.; Minegishi, M.; Sato, K.; Katsuse, O.; Hino, H.; Kosaka, K.; Arai, H. Neuropathological investigation of hypocretin expression in brains of dementia with Lewy bodies. Neurosci. Lett. 2014, 569, 68–73. [Google Scholar] [CrossRef]
- Coon, E.A.; Cutsforth-Gregory, J.K.; Benarroch, E.E. Neuropathology of autonomic dysfunction in synucleinopathies. Mov. Disord. 2018, 33, 349–358. [Google Scholar] [CrossRef]
- Wienecke, M.; Werth, E.; Poryazova, R.; Baumann-Vogel, H.; Bassetti, C.L.; Weller, M.; Waldvogel, D.; Storch, A.; Baumann, C.R. Progressive dopamine and hypocretin deficiencies in Parkinson’s disease: Is there an impact on sleep and wakefulness? J. Sleep Res. 2012, 21, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Baumann, C.R.; Dauvilliers, Y.; Mignot, E.; Bassetti, C.L. Normal CSF Hypocretin-1 (Orexin A) Levels in Dementia with Lewy Bodies Associated with Excessive Daytime Sleepiness. Eur. Neurol. 2004, 52, 73–76. [Google Scholar] [CrossRef]
- Abdo, W.; Bloem, B.; Kremer, H.; Lammers, G.; Verbeek, M.; Overeem, S. CSF hypocretin-1 levels are normal in multiple-system atrophy. Park. Relat. Disord. 2008, 14, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Niepel, G.; Patterson, M.; Judd, A.; Braitch, M.; Fahey, A.J.; Harikrishnan, S.; Edwards, L.J.; Tench, C.R.; Bennett, G.W.; et al. Orexin A (hypocretin-1) levels are not reduced while cocaine/amphetamine regulated transcript levels are increased in the cerebrospinal fluid of patients with multiple sclerosis: No correlation with fatigue and sleepiness. J. Neurol. Sci. 2011, 307, 127–131. [Google Scholar] [CrossRef]
- Oka, Y.; Kanbayashi, T.; Mezaki, T.; Iseki, K.; Matsubayashi, J.; Murakami, G.; Matsui, M.; Shimizu, T.; Shibasaki, H. Low CSF hypocretin-1/orexin-A associated with hypersomnia secondary to hypothalamic lesion in a case of multiple sclerosis. J. Neurol. 2004, 251, 885–886. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, P.; Sturek, M.; Neyra, J.A.; Basile, D.P. Calcium channel Orai1 promotes lymphocyte IL-17 expression and progressive kidney injury. J. Clin. Investig. 2019, 129, 4951–4961. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Ogawa, Y.; Irukayama-Tomobe, Y.; Murakoshi, N.; Kiyama, M.; Ishikawa, Y.; Hosokawa, N.; Tominaga, H.; Uchida, S.; Kimura, S.; Kanuka, M.; et al. Peripherally administered orexin improves survival of mice with endotoxin shock. Elife 2016, 5, e21055. [Google Scholar] [CrossRef]
- Li, T.; Xu, W.; Ouyang, J.; Lu, X.; Sherchan, P.; Lenahan, C.; Irio, G.; Zhang, J.H.; Zhao, J.; Zhang, Y.; et al. Orexin a Alleviates Neuroinflammation Via Oxr2/Camkkbeta/Ampk Signaling Pathway after Ich in Mice. J. Neuroinflamm. 2020, 17, 187. [Google Scholar] [CrossRef]
- Becquet, L.; Abad, C.; Leclercq, M.; Miel, C.; Jean, L.; Riou, G.; Couvineau, A.; Boyer, O.; Tan, Y.-V. Systemic administration of orexin A ameliorates established experimental autoimmune encephalomyelitis by diminishing neuroinflammation. J. Neuroinflamm. 2019, 16, 64. [Google Scholar] [CrossRef]
- Tunisi, L.; Forte, N.; Fernández-Rilo, A.C.; Mavaro, I.; Capasso, R.; D’Angelo, L.; Milić, N.; Cristino, L.; Di Marzo, V.; Palomba, L. Orexin-A Prevents Lipopolysaccharide-Induced Neuroinflammation at the Level of the Intestinal Barrier. Front. Endocrinol. 2019, 10, 219. [Google Scholar] [CrossRef] [PubMed]
- Couvineau, A.; Voisin, T.; Nicole, P.; Gratio, V.; Abad, C.; Tan, Y.-V. Orexins as Novel Therapeutic Targets in Inflammatory and Neurodegenerative Diseases. Front. Endocrinol. 2019, 10, 709. [Google Scholar] [CrossRef]
- Zhou, F.; Yan, X.-D.; Wang, C.; He, Y.-X.; Li, Y.-Y.; Zhang, J.; Wang, Z.-J.; Cai, H.-Y.; Qi, J.-S.; Wu, M.-N. Suvorexant ameliorates cognitive impairments and pathology in APP/PS1 transgenic mice. Neurobiol. Aging 2020, 91, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Kylkilahti, T.M.; Berends, E.; Ramos, M.; Shanbhag, N.C.; Töger, J.; Bloch, K.M.; Lundgaard, I. Achieving brain clearance and preventing neurodegenerative diseases—A glymphatic perspective. Br. J. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Bishir, M.; Bhat, A.; Essa, M.M.; Ekpo, O.; Ihunwo, A.O.; Veeraraghavan, V.P.; Mohan, S.K.; Mahalakshmi, A.M.; Ray, B.; Tuladhar, S.; et al. Sleep Deprivation and Neurological Disorders. BioMed Res. Int. 2020, 2020, 5764017. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, A.Q.; Xue, Y.; Liu, M.F.; Liu, C.; Liu, Y.H.; Pan, Y.P.; Diao, H.L.; Chen, L. Orexins Alleviate Motor Deficits via Increasing Firing Activity of Pallidal Neurons in a Mouse Model of Parkinson’s Disease. Am. J. Physiol. Cell. Physiol. 2019, 317, C800–C812. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mogavero, M.P.; Silvani, A.; DelRosso, L.M.; Salemi, M.; Ferri, R. Focus on the Complex Interconnection between Cancer, Narcolepsy and Other Neurodegenerative Diseases: A Possible Case of Orexin-Dependent Inverse Comorbidity. Cancers 2021, 13, 2612. https://doi.org/10.3390/cancers13112612
Mogavero MP, Silvani A, DelRosso LM, Salemi M, Ferri R. Focus on the Complex Interconnection between Cancer, Narcolepsy and Other Neurodegenerative Diseases: A Possible Case of Orexin-Dependent Inverse Comorbidity. Cancers. 2021; 13(11):2612. https://doi.org/10.3390/cancers13112612
Chicago/Turabian StyleMogavero, Maria P., Alessandro Silvani, Lourdes M. DelRosso, Michele Salemi, and Raffaele Ferri. 2021. "Focus on the Complex Interconnection between Cancer, Narcolepsy and Other Neurodegenerative Diseases: A Possible Case of Orexin-Dependent Inverse Comorbidity" Cancers 13, no. 11: 2612. https://doi.org/10.3390/cancers13112612
APA StyleMogavero, M. P., Silvani, A., DelRosso, L. M., Salemi, M., & Ferri, R. (2021). Focus on the Complex Interconnection between Cancer, Narcolepsy and Other Neurodegenerative Diseases: A Possible Case of Orexin-Dependent Inverse Comorbidity. Cancers, 13(11), 2612. https://doi.org/10.3390/cancers13112612