
Control of Tumor Progression by Angiocrine Factors


Abstract
Simple Summary
Abstract

1. Introduction
2. Tumor Angiogenesis
3. Beyond Angiogenesis
3.1. Angiocrine Factors
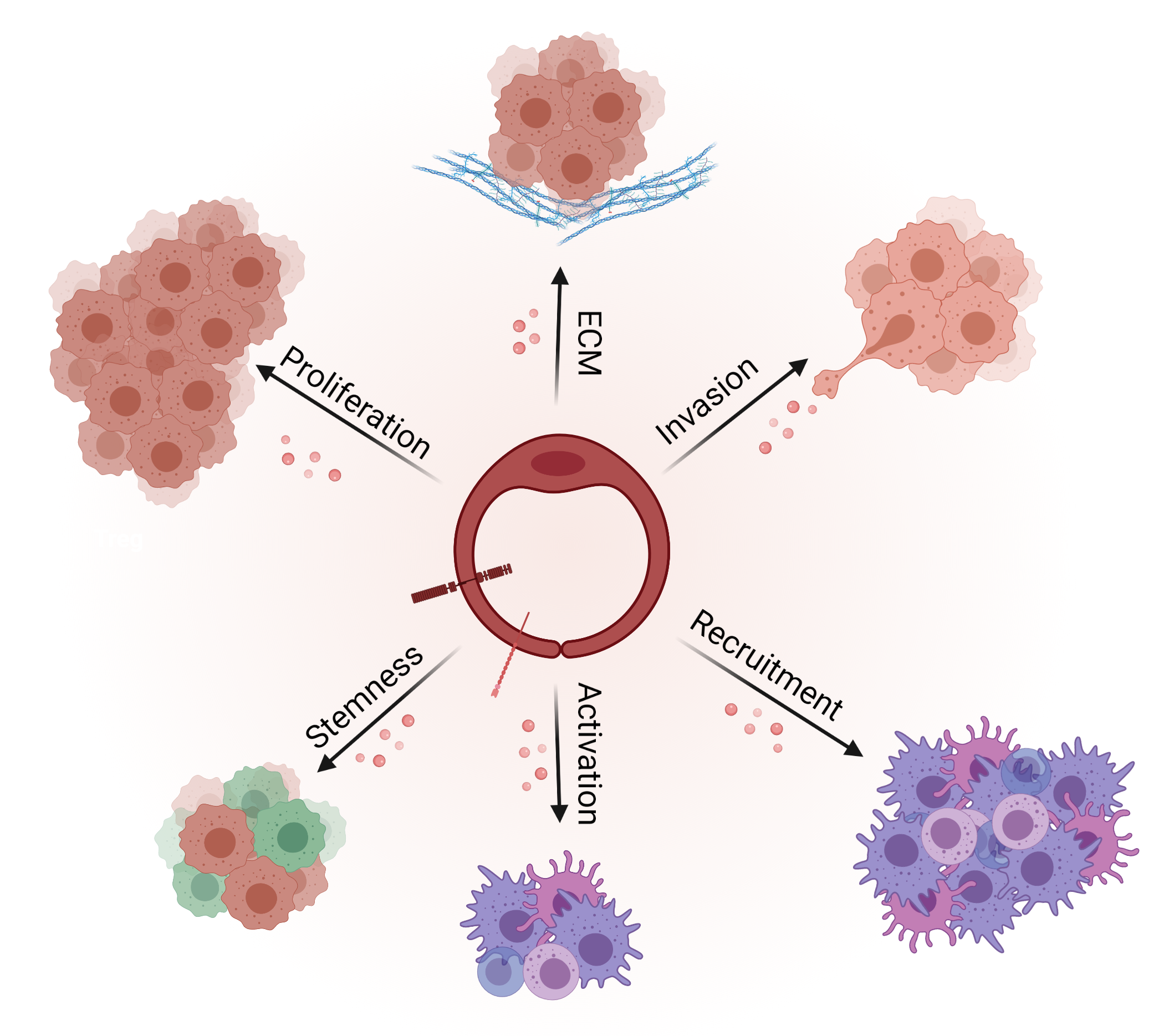
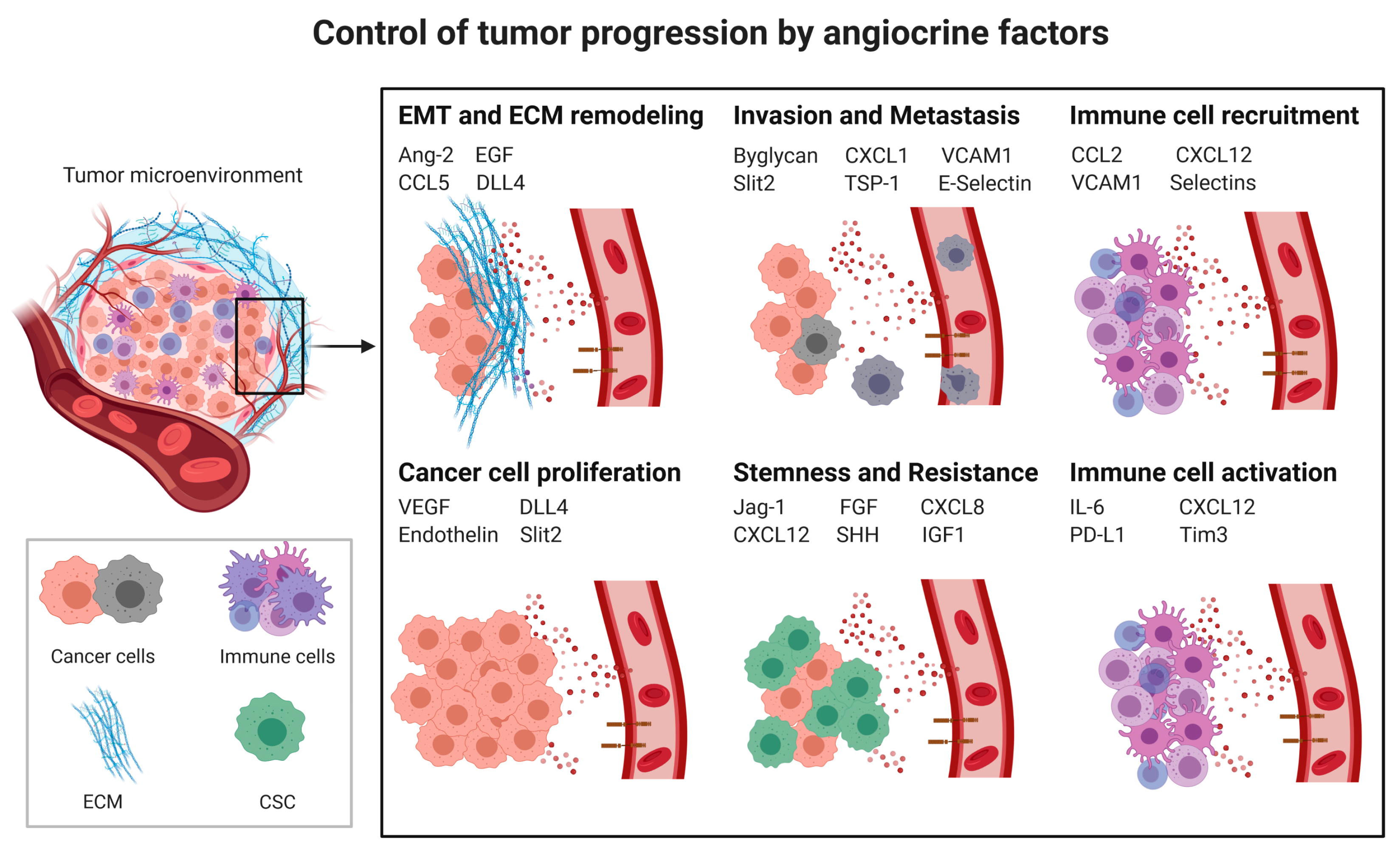
3.2. Angiocrine Control of Cancer Progression
3.2.1. Cancer Cell Proliferation
3.2.2. Cancer Stem Cells (CSCs) and Chemoresistance
3.2.3. Epithelial to Mesenchymal Transition
3.2.4. Invasion and Metastasis
Extracellular Matrix (ECM) Remodeling
Transendothelial Migration
3.2.5. Pre-Metastatic Niche
3.3. Angiocrine Control of the Immune Response
3.3.1. Myeloid-Derived Suppressor Cells (MDSCs) and Tumor-Associated Macrophages (TAMs)
3.3.2. Tumor-Infiltrating Lymphocytes (TILs)
4. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Rafii, S.; Butler, J.M.; Ding, B.S. Angiocrine functions of organ-specific endothelial cells. Nature 2016, 529, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.M.; Kobayashi, H.; Rafii, S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat. Rev. Cancer 2010, 10, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Singhal, M.; Augustin, H.G. Beyond Angiogenesis: Exploiting Angiocrine Factors to Restrict Tumor Progression and Metastasis. Cancer Res. 2020, 80, 659–662. [Google Scholar] [CrossRef]
- Pasquier, J.; Ghiabi, P.; Chouchane, L.; Razzouk, K.; Rafii, S.; Rafii, A. Angiocrine endothelium: From physiology to cancer. J. Transl. Med. 2020, 18, 52. [Google Scholar] [CrossRef]
- Trindade, A.; Duarte, A. Notch Signaling Function in the Angiocrine Regulation of Tumor Development. Cells 2020, 9, 2467. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, S.K.; Kusumbe, A.P.; Adams, R.H. Regulation of tissue morphogenesis by endothelial cell-derived signals. Trends Cell Biol. 2015, 25, 148–157. [Google Scholar] [CrossRef]
- Augustin, H.G.; Koh, G.Y. Organotypic vasculature: From descriptive heterogeneity to functional pathophysiology. Science 2017, 357. [Google Scholar] [CrossRef]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef]
- Khan, K.A.; Kerbel, R.S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 2018, 15, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Larrivée, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Yamamoto, T.; Sakai, Y. Resistance to Anti-Angiogenic Therapy in Cancer-Alterations to Anti-VEGF Pathway. Int. J. Mol. Sci. 2018, 19, 1232. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Gerald, D.; Chintharlapalli, S.; Augustin, H.G.; Benjamin, L.E. Angiopoietin-2: An attractive target for improved antiangiogenic tumor therapy. Cancer Res. 2013, 73, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Gengenbacher, N.; Singhal, M.; Mogler, C.; Hai, L.; Milde, L.; Pari, A.A.A.; Besemfelder, E.; Fricke, C.; Baumann, D.; Gehrs, S.; et al. Timed Ang2-Targeted Therapy Identifies the Angiopoietin-Tie Pathway as Key Regulator of Fatal Lymphogenous Metastasis. Cancer Discov. 2021, 11, 424–445. [Google Scholar] [CrossRef] [PubMed]
- Holopainen, T.; Saharinen, P.; D’Amico, G.; Lampinen, A.; Eklund, L.; Sormunen, R.; Anisimov, A.; Zarkada, G.; Lohela, M.; Helotera, H.; et al. Effects of angiopoietin-2-blocking antibody on endothelial cell-cell junctions and lung metastasis. J. Natl. Cancer Inst. 2012, 104, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, K.; Hu, J.; Korn, C.; Savant, S.; Teichert, M.; Kapel, S.S.; Jugold, M.; Besemfelder, E.; Thomas, M.; Pasparakis, M.; et al. Postsurgical adjuvant tumor therapy by combining anti-angiopoietin-2 and metronomic chemotherapy limits metastatic growth. Cancer Cell 2014, 26, 880–895. [Google Scholar] [CrossRef] [PubMed]
- Maishi, N.; Ohba, Y.; Akiyama, K.; Ohga, N.; Hamada, J.; Nagao-Kitamoto, H.; Alam, M.T.; Yamamoto, K.; Kawamoto, T.; Inoue, N.; et al. Tumour endothelial cells in high metastatic tumours promote metastasis via epigenetic dysregulation of biglycan. Sci. Rep. 2016, 6, 28039. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Dong, Y.Y.; Wang, W.M.; Xie, X.Y.; Wang, Z.M.; Chen, R.X.; Chen, J.; Gao, D.M.; Cui, J.F.; Ren, Z.G. Vascular endothelial cells facilitated HCC invasion and metastasis through the Akt and NF-kappaB pathways induced by paracrine cytokines. J. Exp. Clin. Cancer Res. 2013, 32, 51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xu, J.; Fang, H.; Tang, L.; Chen, W.; Sun, Q.; Zhang, Q.; Yang, F.; Sun, Z.; Cao, L.; et al. Endothelial cells promote triple-negative breast cancer cell metastasis via PAI-1 and CCL5 signaling. FASEB J. 2018, 32, 276–288. [Google Scholar] [CrossRef]
- Zhao, R.; Bei, X.; Yang, B.; Wang, X.; Jiang, C.; Shi, F.; Wang, X.; Zhu, Y.; Jing, Y.; Han, B.; et al. Endothelial cells promote metastasis of prostate cancer by enhancing autophagy. J. Exp. Clin. Cancer Res. 2018, 37, 221. [Google Scholar] [CrossRef]
- Warner, K.A.; Miyazawa, M.; Cordeiro, M.M.; Love, W.J.; Pinsky, M.S.; Neiva, K.G.; Spalding, A.C.; Nor, J.E. Endothelial cells enhance tumor cell invasion through a crosstalk mediated by CXC chemokine signaling. Neoplasia 2008, 10, 131–139. [Google Scholar] [CrossRef]
- Mierke, C.T.; Zitterbart, D.P.; Kollmannsberger, P.; Raupach, C.; Schlotzer-Schrehardt, U.; Goecke, T.W.; Behrens, J.; Fabry, B. Breakdown of the endothelial barrier function in tumor cell transmigration. Biophys. J. 2008, 94, 2832–2846. [Google Scholar] [CrossRef]
- McCoy, M.G.; Nyanyo, D.; Hung, C.K.; Goerger, J.P.; Zipfel, W.R.; Williams, R.M.; Nishimura, N.; Fischbach, C. Endothelial cells promote 3D invasion of GBM by IL-8-dependent induction of cancer stem cell properties. Sci. Rep. 2019, 9, 9069. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Ariyama, H.; Stancikova, J.; Sakitani, K.; Asfaha, S.; Renz, B.W.; Dubeykovskaya, Z.A.; Shibata, W.; Wang, H.; Westphalen, C.B.; et al. Mist1 Expressing Gastric Stem Cells Maintain the Normal and Neoplastic Gastric Epithelium and Are Supported by a Perivascular Stem Cell Niche. Cancer Cell 2015, 28, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Indraccolo, S.; Minuzzo, S.; Masiero, M.; Pusceddu, I.; Persano, L.; Moserle, L.; Reboldi, A.; Favaro, E.; Mecarozzi, M.; Di Mario, G.; et al. Cross-talk between tumor and endothelial cells involving the Notch3-Dll4 interaction marks escape from tumor dormancy. Cancer Res. 2009, 69, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.B.; Ma, X.; Li, H.Z.; Ai, Q.; Liu, S.W.; Zhang, Y.; Gao, Y.; Fan, Y.; Ni, D.; Wang, B.J.; et al. Endothelial Delta-like 4 (DLL4) promotes renal cell carcinoma hematogenous metastasis. Oncotarget 2014, 5, 3066–3075. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, Z.; Dong, Z.; Lauxen, I.S.; Filho, M.S.; Nor, J.E. Endothelial cell-secreted EGF induces epithelial to mesenchymal transition and endows head and neck cancer cells with stem-like phenotype. Cancer Res. 2014, 74, 2869–2881. [Google Scholar] [CrossRef]
- Bagnato, A.; Salani, D.; Di Castro, V.; Wu-Wong, J.R.; Tecce, R.; Nicotra, M.R.; Venuti, A.; Natali, P.G. Expression of endothelin 1 and endothelin A receptor in ovarian carcinoma: Evidence for an autocrine role in tumor growth. Cancer Res. 1999, 59, 720–727. [Google Scholar]
- Grant, K.; Loizidou, M.; Taylor, I. Endothelin-1: A multifunctional molecule in cancer. Br. J. Cancer 2003, 88, 163–166. [Google Scholar] [CrossRef]
- Locard-Paulet, M.; Lim, L.; Veluscek, G.; McMahon, K.; Sinclair, J.; van Weverwijk, A.; Worboys, J.D.; Yuan, Y.; Isacke, C.M.; Jorgensen, C. Phosphoproteomic analysis of interacting tumor and endothelial cells identifies regulatory mechanisms of transendothelial migration. Sci. Signal. 2016, 9, ra15. [Google Scholar] [CrossRef]
- Fessler, E.; Borovski, T.; Medema, J.P. Endothelial cells induce cancer stem cell features in differentiated glioblastoma cells via bFGF. Mol. Cancer 2015, 14, 157. [Google Scholar] [CrossRef]
- Benedicto, A.; Romayor, I.; Arteta, B. Role of liver ICAM-1 in metastasis. Oncol. Lett. 2017, 14, 3883–3892. [Google Scholar] [CrossRef]
- Benedicto, A.; Herrero, A.; Romayor, I.; Marquez, J.; Smedsrod, B.; Olaso, E.; Arteta, B. Liver sinusoidal endothelial cell ICAM-1 mediated tumor/endothelial crosstalk drives the development of liver metastasis by initiating inflammatory and angiogenic responses. Sci. Rep. 2019, 9, 13111. [Google Scholar] [CrossRef]
- Finzel, A.H.; Reininger, A.J.; Bode, P.A.; Wurzinger, L.J. ICAM-1 supports adhesion of human small-cell lung carcinoma to endothelial cells. Clin. Exp. Metastasis 2004, 21, 185–189. [Google Scholar] [CrossRef]
- Cao, Z.; Scandura, J.M.; Inghirami, G.G.; Shido, K.; Ding, B.S.; Rafii, S. Molecular Checkpoint Decisions Made by Subverted Vascular Niche Transform Indolent Tumor Cells into Chemoresistant Cancer Stem Cells. Cancer Cell 2017, 31, 110–126. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.; Warner, K.A.; Dong, Z.; Imai, A.; Nor, C.; Ward, B.B.; Helman, J.I.; Taichman, R.S.; Bellile, E.L.; McCauley, L.K.; et al. Endothelial interleukin-6 defines the tumorigenic potential of primary human cancer stem cells. Stem Cells 2014, 32, 2845–2857. [Google Scholar] [CrossRef]
- Wang, Q.; He, Z.; Huang, M.; Liu, T.; Wang, Y.; Xu, H.; Duan, H.; Ma, P.; Zhang, L.; Zamvil, S.S.; et al. Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2alpha. Nat. Commun. 2018, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Demaria, O.; De Gassart, A.; Coso, S.; Gestermann, N.; Di Domizio, J.; Flatz, L.; Gaide, O.; Michielin, O.; Hwu, P.; Petrova, T.V.; et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 15408–15413. [Google Scholar] [CrossRef] [PubMed]
- Lugassy, C.; Christensen, L.; Le Charpentier, M.; Faure, E.; Escande, J.P. Angio-tumoral laminin in murine tumors derived from human melanoma cell lines. Immunohistochemical and ultrastructural observations. J. Submicrosc. Cytol. Pathol. 1998, 30, 231–237. [Google Scholar] [PubMed]
- Wragg, J.W.; Finnity, J.P.; Anderson, J.A.; Ferguson, H.J.; Porfiri, E.; Bhatt, R.I.; Murray, P.G.; Heath, V.L.; Bicknell, R. MCAM and LAMA4 Are Highly Enriched in Tumor Blood Vessels of Renal Cell Carcinoma and Predict Patient Outcome. Cancer Res. 2016, 76, 2314–2326. [Google Scholar] [CrossRef]
- Foda, H.D.; Zucker, S. Matrix metalloproteinases in cancer invasion, metastasis and angiogenesis. Drug Discov. Today 2001, 6, 478–482. [Google Scholar] [CrossRef]
- Lu, J.; Ye, X.; Fan, F.; Xia, L.; Bhattacharya, R.; Bellister, S.; Tozzi, F.; Sceusi, E.; Zhou, Y.; Tachibana, I.; et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell 2013, 23, 171–185. [Google Scholar] [CrossRef]
- Ghiabi, P.; Jiang, J.; Pasquier, J.; Maleki, M.; Abu-Kaoud, N.; Rafii, S.; Rafii, A. Endothelial cells provide a notch-dependent pro-tumoral niche for enhancing breast cancer survival, stemness and pro-metastatic properties. PLoS ONE 2014, 9, e112424. [Google Scholar] [CrossRef]
- Jiang, H.; Zhou, C.; Zhang, Z.; Wang, Q.; Wei, H.; Shi, W.; Li, J.; Wang, Z.; Ou, Y.; Wang, W.; et al. Jagged1-Notch1-deployed tumor perivascular niche promotes breast cancer stem cell phenotype through Zeb1. Nat. Commun. 2020, 11, 5129. [Google Scholar] [CrossRef] [PubMed]
- Wieland, E.; Rodriguez-Vita, J.; Liebler, S.S.; Mogler, C.; Moll, I.; Herberich, S.E.; Espinet, E.; Herpel, E.; Menuchin, A.; Chang-Claude, J.; et al. Endothelial Notch1 Activity Facilitates Metastasis. Cancer Cell 2017, 31, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- LaGier, A.J.; Pober, J.S. Immune accessory functions of human endothelial cells are modulated by overexpression of B7-H1 (PDL1). Hum. Immunol. 2006, 67, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Laferriere, J.; Houle, F.; Taher, M.M.; Valerie, K.; Huot, J. Transendothelial migration of colon carcinoma cells requires expression of E-selectin by endothelial cells and activation of stress-activated protein kinase-2 (SAPK2/p38) in the tumor cells. J. Biol. Chem. 2001, 276, 33762–33772. [Google Scholar] [CrossRef]
- Woodward, J. Crossing the endothelium: E-selectin regulates tumor cell migration under flow conditions. Cell. Adh. Migr. 2008, 2, 151–152. [Google Scholar] [CrossRef]
- Tremblay, P.L.; Auger, F.A.; Huot, J. Regulation of transendothelial migration of colon cancer cells by E-selectin-mediated activation of p38 and ERK MAP kinases. Oncogene 2006, 25, 6563–6573. [Google Scholar] [CrossRef]
- Brodt, P.; Fallavollita, L.; Bresalier, R.S.; Meterissian, S.; Norton, C.R.; Wolitzky, B.A. Liver endothelial E-selectin mediates carcinoma cell adhesion and promotes liver metastasis. Int. J. Cancer 1997, 71, 612–619. [Google Scholar] [CrossRef]
- Khatib, A.M.; Kontogiannea, M.; Fallavollita, L.; Jamison, B.; Meterissian, S.; Brodt, P. Rapid induction of cytokine and E-selectin expression in the liver in response to metastatic tumor cells. Cancer Res. 1999, 59, 1356–1361. [Google Scholar]
- Kang, S.A.; Blache, C.A.; Bajana, S.; Hasan, N.; Kamal, M.; Morita, Y.; Gupta, V.; Tsolmon, B.; Suh, K.S.; Gorenstein, D.G.; et al. The effect of soluble E-selectin on tumor progression and metastasis. BMC Cancer 2016, 16, 331. [Google Scholar] [CrossRef]
- Borsig, L. Selectins in cancer immunity. Glycobiology 2018, 28, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.N.; Yang, L.; Lv, Y.F.; Shi, Y.; Shen, L.L.; Yao, X.H.; Guo, Q.N.; Zhang, P.; Cui, Y.H.; Zhang, X.; et al. Endothelial cells promote stem-like phenotype of glioma cells through activating the Hedgehog pathway. J. Pathol. 2014, 234, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Brantley-Sieders, D.M.; Dunaway, C.M.; Rao, M.; Short, S.; Hwang, Y.; Gao, Y.; Li, D.; Jiang, A.; Shyr, Y.; Wu, J.Y.; et al. Angiocrine factors modulate tumor proliferation and motility through EphA2 repression of Slit2 tumor suppressor function in endothelium. Cancer Res. 2011, 71, 976–987. [Google Scholar] [CrossRef]
- Tavora, B.; Mederer, T.; Wessel, K.J.; Ruffing, S.; Sadjadi, M.; Missmahl, M.; Ostendorf, B.N.; Liu, X.; Kim, J.Y.; Olsen, O.; et al. Tumoural activation of TLR3-SLIT2 axis in endothelium drives metastasis. Nature 2020, 586, 299–304. [Google Scholar] [CrossRef]
- Wu, F.H.; Yuan, Y.; Li, D.; Lei, Z.; Song, C.W.; Liu, Y.Y.; Li, B.; Huang, B.; Feng, Z.H.; Zhang, G.M. Endothelial cell-expressed Tim-3 facilitates metastasis of melanoma cells by activating the NF-kappaB pathway. Oncol. Rep. 2010, 24, 693–699. [Google Scholar]
- Huang, X.; Bai, X.; Cao, Y.; Wu, J.; Huang, M.; Tang, D.; Tao, S.; Zhu, T.; Liu, Y.; Yang, Y.; et al. Lymphoma endothelium preferentially expresses Tim-3 and facilitates the progression of lymphoma by mediating immune evasion. J. Exp. Med. 2010, 207, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Dias, S.; Choy, M.; Alitalo, K.; Rafii, S. Vascular endothelial growth factor (VEGF)-C signaling through FLT-4 (VEGFR-3) mediates leukemic cell proliferation, survival, and resistance to chemotherapy. Blood 2002, 99, 2179–2184. [Google Scholar] [CrossRef]
- Beck, B.; Driessens, G.; Goossens, S.; Youssef, K.K.; Kuchnio, A.; Caauwe, A.; Sotiropoulou, P.A.; Loges, S.; Lapouge, G.; Candi, A.; et al. A vascular niche and a VEGF-Nrp1 loop regulate the initiation and stemness of skin tumours. Nature 2011, 478, 399–403. [Google Scholar] [CrossRef]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Klemke, M.; Weschenfelder, T.; Konstandin, M.H.; Samstag, Y. High affinity interaction of integrin alpha4beta1 (VLA-4) and vascular cell adhesion molecule 1 (VCAM-1) enhances migration of human melanoma cells across activated endothelial cell layers. J. Cell. Physiol. 2007, 212, 368–374. [Google Scholar] [CrossRef]
- Tamura, R.; Tanaka, T.; Akasaki, Y.; Murayama, Y.; Yoshida, K.; Sasaki, H. The role of vascular endothelial growth factor in the hypoxic and immunosuppressive tumor microenvironment: Perspectives for therapeutic implications. Med. Oncol. 2019, 37, 2. [Google Scholar] [CrossRef]
- Liang, Y.; Brekken, R.A.; Hyder, S.M. Vascular endothelial growth factor induces proliferation of breast cancer cells and inhibits the anti-proliferative activity of anti-hormones. Endocr. Relat. Cancer 2006, 13, 905–919. [Google Scholar] [CrossRef]
- Koistinen, P.; Siitonen, T.; Mantymaa, P.; Saily, M.; Kinnula, V.; Savolainen, E.R.; Soini, Y. Regulation of the acute myeloid leukemia cell line OCI/AML-2 by endothelial nitric oxide synthase under the control of a vascular endothelial growth factor signaling system. Leukemia 2001, 15, 1433–1441. [Google Scholar] [CrossRef]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.M.; Nolan, D.J.; Vertes, E.L.; Varnum-Finney, B.; Kobayashi, H.; Hooper, A.T.; Seandel, M.; Shido, K.; White, I.A.; Kobayashi, M.; et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell 2010, 6, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, P.; Poulos, M.G.; Butler, J.M. Regulation of the hematopoietic stem cell lifecycle by the endothelial niche. Curr. Opin. Hematol. 2017, 24, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Bhattacharya, R.; Ye, X.; Fan, F.; Boulbes, D.R.; Xia, L.; Ellis, L.M. Endothelial cells activate the cancer stem cell-associated NANOGP8 pathway in colorectal cancer cells in a paracrine fashion. Mol. Oncol. 2017, 11, 1023–1034. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef]
- Ohga, N.; Ishikawa, S.; Maishi, N.; Akiyama, K.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Osawa, T.; Yamamoto, K.; Kondoh, M.; et al. Heterogeneity of tumor endothelial cells: Comparison between tumor endothelial cells isolated from high- and low-metastatic tumors. Am. J. Pathol. 2012, 180, 1294–1307. [Google Scholar] [CrossRef]
- Martin-Satue, M.; Marrugat, R.; Cancelas, J.A.; Blanco, J. Enhanced expression of alpha(1,3)-fucosyltransferase genes correlates with E-selectin-mediated adhesion and metastatic potential of human lung adenocarcinoma cells. Cancer Res. 1998, 58, 1544–1550. [Google Scholar]
- Dimitroff, C.J.; Lechpammer, M.; Long-Woodward, D.; Kutok, J.L. Rolling of human bone-metastatic prostate tumor cells on human bone marrow endothelium under shear flow is mediated by E-selectin. Cancer Res. 2004, 64, 5261–5269. [Google Scholar] [CrossRef] [PubMed]
- Carrascal, M.A.; Silva, M.; Ferreira, J.A.; Azevedo, R.; Ferreira, D.; Silva, A.M.N.; Ligeiro, D.; Santos, L.L.; Sackstein, R.; Videira, P.A. A functional glycoproteomics approach identifies CD13 as a novel E-selectin ligand in breast cancer. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2069–2080. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.; Mondal, N.; Greco, T.M.; Wei, Y.; Spadazzi, C.; Lin, S.C.; Zheng, H.; Cheung, C.; Magnani, J.L.; Lin, S.H.; et al. Bone vascular niche E-selectin induces mesenchymal-epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat. Cell Biol. 2019, 21, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, S.; Goel, S.; Kamoun, W.S.; Maru, Y.; Fukumura, D.; Duda, D.G.; Jain, R.K. Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via E-selectin up-regulation. Proc. Natl. Acad. Sci. USA 2011, 108, 3725–3730. [Google Scholar] [CrossRef] [PubMed]
- Barbier, V.; Erbani, J.; Fiveash, C.; Davies, J.M.; Tay, J.; Tallack, M.R.; Lowe, J.; Magnani, J.L.; Pattabiraman, D.R.; Perkins, A.C.; et al. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat. Commun. 2020, 11, 2042. [Google Scholar] [CrossRef] [PubMed]
- Rosette, C.; Roth, R.B.; Oeth, P.; Braun, A.; Kammerer, S.; Ekblom, J.; Denissenko, M.F. Role of ICAM1 in invasion of human breast cancer cells. Carcinogenesis 2005, 26, 943–950. [Google Scholar] [CrossRef]
- Witkowska, A.M.; Borawska, M.H. Soluble intercellular adhesion molecule-1 (sICAM-1): An overview. Eur. Cytokine Netw. 2004, 15, 91–98. [Google Scholar] [PubMed]
- Maruo, Y.; Gochi, A.; Kaihara, A.; Shimamura, H.; Yamada, T.; Tanaka, N.; Orita, K. ICAM-1 expression and the soluble ICAM-1 level for evaluating the metastatic potential of gastric cancer. Int. J. Cancer 2002, 100, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Sherbenou, D.W.; Su, Y.; Behrens, C.R.; Aftab, B.T.; Perez de Acha, O.; Murnane, M.; Bearrows, S.C.; Hann, B.C.; Wolf, J.L.; Martin, T.G.; et al. Potent Activity of an Anti-ICAM1 Antibody-Drug Conjugate against Multiple Myeloma. Clin. Cancer Res. 2020, 26, 6028–6038. [Google Scholar] [CrossRef] [PubMed]
- Wichert, S.; Juliusson, G.; Johansson, A.; Sonesson, E.; Teige, I.; Wickenberg, A.T.; Frendeus, B.; Korsgren, M.; Hansson, M. A single-arm, open-label, phase 2 clinical trial evaluating disease response following treatment with BI-505, a human anti-intercellular adhesion molecule-1 monoclonal antibody, in patients with smoldering multiple myeloma. PLoS ONE 2017, 12, e0171205. [Google Scholar] [CrossRef]
- Lin, Y.C.; Shun, C.T.; Wu, M.S.; Chen, C.C. A novel anticancer effect of thalidomide: Inhibition of intercellular adhesion molecule-1-mediated cell invasion and metastasis through suppression of nuclear factor-kappaB. Clin. Cancer Res. 2006, 12, 7165–7173. [Google Scholar] [CrossRef]
- Guo, P.; Huang, J.; Wang, L.; Jia, D.; Yang, J.; Dillon, D.A.; Zurakowski, D.; Mao, H.; Moses, M.A.; Auguste, D.T. ICAM-1 as a molecular target for triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 14710–14715. [Google Scholar] [CrossRef]
- Wolf, M.J.; Hoos, A.; Bauer, J.; Boettcher, S.; Knust, M.; Weber, A.; Simonavicius, N.; Schneider, C.; Lang, M.; Sturzl, M.; et al. Endothelial CCR2 signaling induced by colon carcinoma cells enables extravasation via the JAK2-Stat5 and p38MAPK pathway. Cancer Cell 2012, 22, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Helfrich, I.; Edler, L.; Sucker, A.; Thomas, M.; Christian, S.; Schadendorf, D.; Augustin, H.G. Angiopoietin-2 levels are associated with disease progression in metastatic malignant melanoma. Clin. Cancer Res. 2009, 15, 1384–1392. [Google Scholar] [CrossRef]
- Daly, C.; Eichten, A.; Castanaro, C.; Pasnikowski, E.; Adler, A.; Lalani, A.S.; Papadopoulos, N.; Kyle, A.H.; Minchinton, A.I.; Yancopoulos, G.D.; et al. Angiopoietin-2 functions as a Tie2 agonist in tumor models, where it limits the effects of VEGF inhibition. Cancer Res. 2013, 73, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Oliner, J.; Min, H.; Leal, J.; Yu, D.; Rao, S.; You, E.; Tang, X.; Kim, H.; Meyer, S.; Han, S.J.; et al. Suppression of angiogenesis and tumor growth by selective inhibition of angiopoietin-2. Cancer Cell 2004, 6, 507–516. [Google Scholar] [CrossRef]
- Coutelle, O.; Schiffmann, L.M.; Liwschitz, M.; Brunold, M.; Goede, V.; Hallek, M.; Kashkar, H.; Hacker, U.T. Dual targeting of Angiopoetin-2 and VEGF potentiates effective vascular normalisation without inducing empty basement membrane sleeves in xenograft tumours. Br. J. Cancer 2015, 112, 495–503. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, I.K.; Han, S.; Park, I.; Kim, C.; Bae, J.; Oh, S.J.; Lee, S.; Kim, J.H.; Woo, D.C.; et al. Normalization of Tumor Vessels by Tie2 Activation and Ang2 Inhibition Enhances Drug Delivery and Produces a Favorable Tumor Microenvironment. Cancer Cell 2017, 31, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Singhal, M.; Gengenbacher, N.; La Porta, S.; Gehrs, S.; Shi, J.; Kamiyama, M.; Bodenmiller, D.M.; Fischl, A.; Schieb, B.; Besemfelder, E.; et al. Preclinical validation of a novel metastasis-inhibiting Tie1 function-blocking antibody. EMBO Mol. Med. 2020, 12, e11164. [Google Scholar] [CrossRef]
- D’Amico, G.; Korhonen, E.A.; Anisimov, A.; Zarkada, G.; Holopainen, T.; Hagerling, R.; Kiefer, F.; Eklund, L.; Sormunen, R.; Elamaa, H.; et al. Tie1 deletion inhibits tumor growth and improves angiopoietin antagonist therapy. J. Clin. Invest. 2014, 124, 824–834. [Google Scholar] [CrossRef]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Barbazan, J.; Alonso-Alconada, L.; Elkhatib, N.; Geraldo, S.; Gurchenkov, V.; Glentis, A.; van Niel, G.; Palmulli, R.; Fernandez, B.; Viano, P.; et al. Liver Metastasis Is Facilitated by the Adherence of Circulating Tumor Cells to Vascular Fibronectin Deposits. Cancer Res. 2017, 77, 3431–3441. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yu, T.; Liang, H.; Wang, J.; Xie, J.; Shao, J.; Gao, Y.; Yu, S.; Chen, S.; Wang, L.; et al. Nitric oxide inhibits hetero-adhesion of cancer cells to endothelial cells: Restraining circulating tumor cells from initiating metastatic cascade. Sci. Rep. 2014, 4, 4344. [Google Scholar] [CrossRef] [PubMed]
- Rejniak, K.A. Circulating Tumor Cells: When a Solid Tumor Meets a Fluid Microenvironment. Adv. Exp. Med. Biol. 2016, 936, 93–106. [Google Scholar] [CrossRef]
- Singh, A.; Veeriah, V.; Xi, P.; Labella, R.; Chen, J.; Romeo, S.G.; Ramasamy, S.K.; Kusumbe, A.P. Angiocrine signals regulate quiescence and therapy resistance in bone metastasis. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Al-Soudi, A.; Kaaij, M.H.; Tas, S.W. Endothelial cells: From innocent bystanders to active participants in immune responses. Autoimmun. Rev. 2017, 16, 951–962. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.A.; Li, M.O. Ontogeny of Tumor-associated Macrophages and Its Implication in Cancer Regulation. Trends Cancer 2016, 2, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and cancer: From mechanisms to therapeutic implications. Trends Immunol. 2015, 36, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Serbina, N.V.; Pamer, E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006, 7, 311–317. [Google Scholar] [CrossRef]
- Häuselmann, I.; Roblek, M.; Protsyuk, D.; Huck, V.; Knopfova, L.; Grässle, S.; Bauer, A.T.; Schneider, S.W.; Borsig, L. Monocyte Induction of E-Selectin-Mediated Endothelial Activation Releases VE-Cadherin Junctions to Promote Tumor Cell Extravasation in the Metastasis Cascade. Cancer Res. 2016, 76, 5302–5312. [Google Scholar] [CrossRef]
- Tormoen, G.W.; Crittenden, M.R.; Gough, M.J. Role of the immunosuppressive microenvironment in immunotherapy. Adv. Radiat. Oncol. 2018, 3, 520–526. [Google Scholar] [CrossRef]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulieres, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodriguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Kloepper, J.; Riedemann, L.; Amoozgar, Z.; Seano, G.; Susek, K.; Yu, V.; Dalvie, N.; Amelung, R.L.; Datta, M.; Song, J.W.; et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl. Acad. Sci. USA 2016, 113, 4476–4481. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.E.; Kirkpatrick, N.D.; Huang, Y.; Farrar, C.T.; Marijt, K.A.; Kloepper, J.; Datta, M.; Amoozgar, Z.; Seano, G.; Jung, K.; et al. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 4470–4475. [Google Scholar] [CrossRef] [PubMed]
- Schmittnaegel, M.; Rigamonti, N.; Kadioglu, E.; Cassara, A.; Wyser Rmili, C.; Kiialainen, A.; Kienast, Y.; Mueller, H.J.; Ooi, C.H.; Laoui, D.; et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Angiocrine Factor 1 | Function | Reference |
|---|---|---|
| Ang-2 | Invasion/metastasis; myeloid cell recruitment | [20,21,22,23] |
| Biglycan | TCs invasion/metastasis | [24] |
| CCL2/MCP-1 | Myeloid cells recruitment; tumor cell extravasation | [25] |
| CCL5 | EMT; invasion | [26,27] |
| CXCL1 | Invasion | [28,29] |
| CXCL8/IL8 | Stemness; invasion; myeloid cell recruitment; lymphocyte phenotype | [25,28,30] |
| CXCL12/SDF-1 | Stemness | [31] |
| DLL4 | Cancer cell proliferation; invasion | [32,33] |
| EGF | EMT | [34] |
| Endothelin | Cancer cell proliferation | [35,36] |
| EphrinA1 | Transendothelial migration | [37] |
| FGF | Stemness | [38] |
| ICAM1 | Myeloid cell recruitment/adhesion; lymphocyte phenotype | [39,40,41] |
| IGF1 | Stemness | [42] |
| IL6 | Myeloid cell recruitment/activation; stemness; lymphocyte phenotype | [43,44] |
| Inf- γ | Lymphocyte phenotype; T cell inhibition | [45] |
| Laminin | ECM; invasion/metastasis | [46,47] |
| MMPs | ECM | [48] |
| Notch signaling/Jag1 | Stemness; CSCs; immune cell recruitment; metastasis | [49,50,51,52] |
| PD-L1 | T cell inhibition | [53,54,55] |
| Selectin | Myeloid cell recruitment; immune cell and tumor cell transmigration/extravasation | [56,57,58,59,60,61,62] |
| Shh | Stemness | [63] |
| Slit2 | Cancer cell proliferation, intravasation, metastasis | [64,65] |
| Tim-3 | T cell inhibition | [66,67] |
| TSP-1 | Pre-metastatic niche | [68] |
| VEGF family | Angiogenesis; cancer cell proliferation; stemness; myeloid cell recruitment; lymphocyte phenotype | [69,70,71] |
| VCAM1 | Myeloid cell recruitment/adhesion; metastasis | [23,52,72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsina-Sanchis, E.; Mülfarth, R.; Fischer, A. Control of Tumor Progression by Angiocrine Factors. Cancers 2021, 13, 2610. https://doi.org/10.3390/cancers13112610
Alsina-Sanchis E, Mülfarth R, Fischer A. Control of Tumor Progression by Angiocrine Factors. Cancers. 2021; 13(11):2610. https://doi.org/10.3390/cancers13112610
Chicago/Turabian StyleAlsina-Sanchis, Elisenda, Ronja Mülfarth, and Andreas Fischer. 2021. "Control of Tumor Progression by Angiocrine Factors" Cancers 13, no. 11: 2610. https://doi.org/10.3390/cancers13112610
APA StyleAlsina-Sanchis, E., Mülfarth, R., & Fischer, A. (2021). Control of Tumor Progression by Angiocrine Factors. Cancers, 13(11), 2610. https://doi.org/10.3390/cancers13112610