
TNFα Enhances Tamoxifen Sensitivity through Dissociation of ERα-p53-NCOR1 Complexes in ERα-Positive Breast Cancer

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
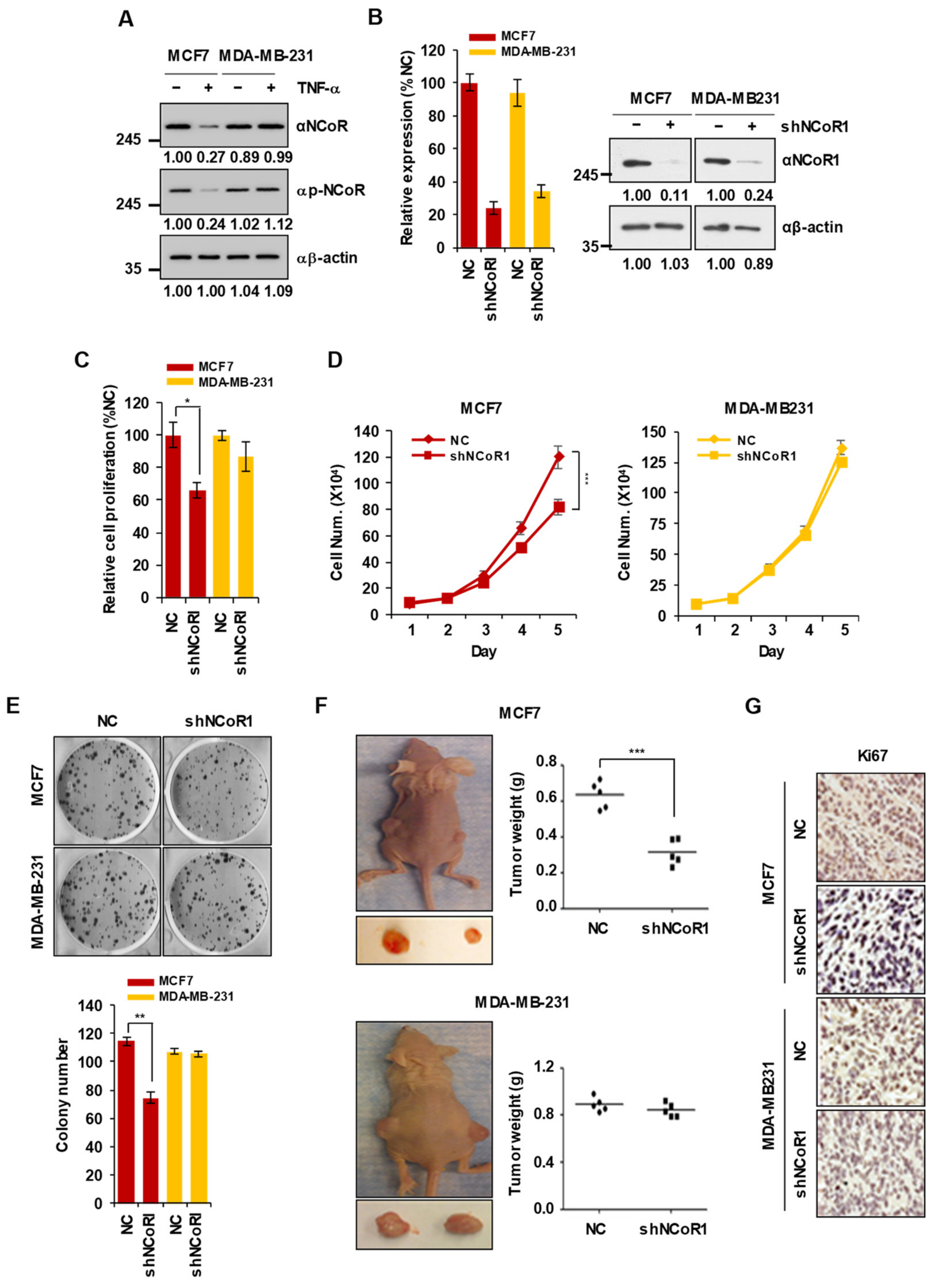
2.1. NCOR1 Knockdown Suppresses the Growth of MCF7 Cells but Not of MDA-MB-231 Cells In Vitro and in Xenograft Mouse Models
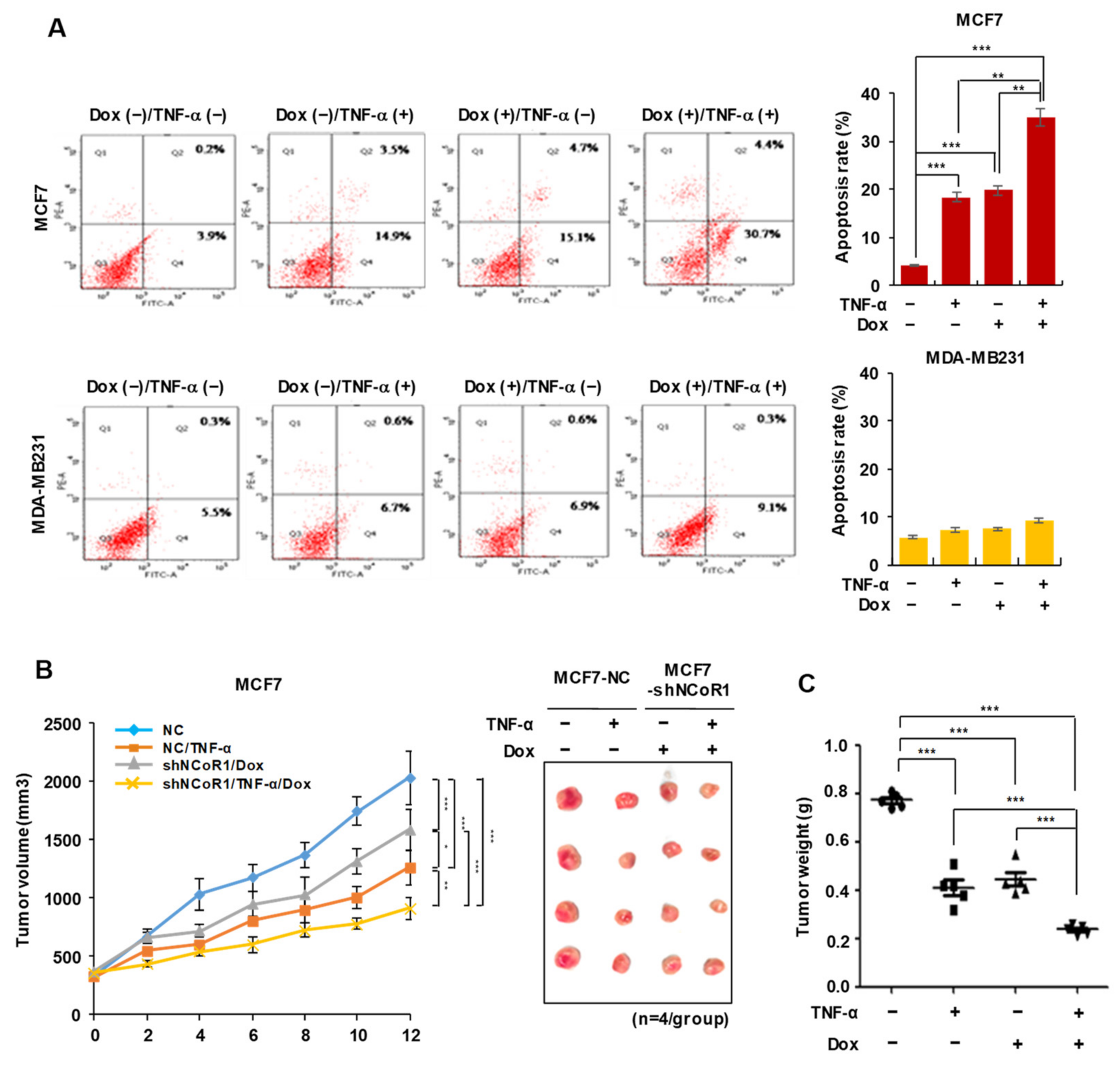
2.2. NCOR1 Knockdown and TNFα Treatment Exert Similar Apoptosis-Inducing Effects in MCF7 Cells or Tumor-Suppressing Effects in MCF7 Xenograft Model
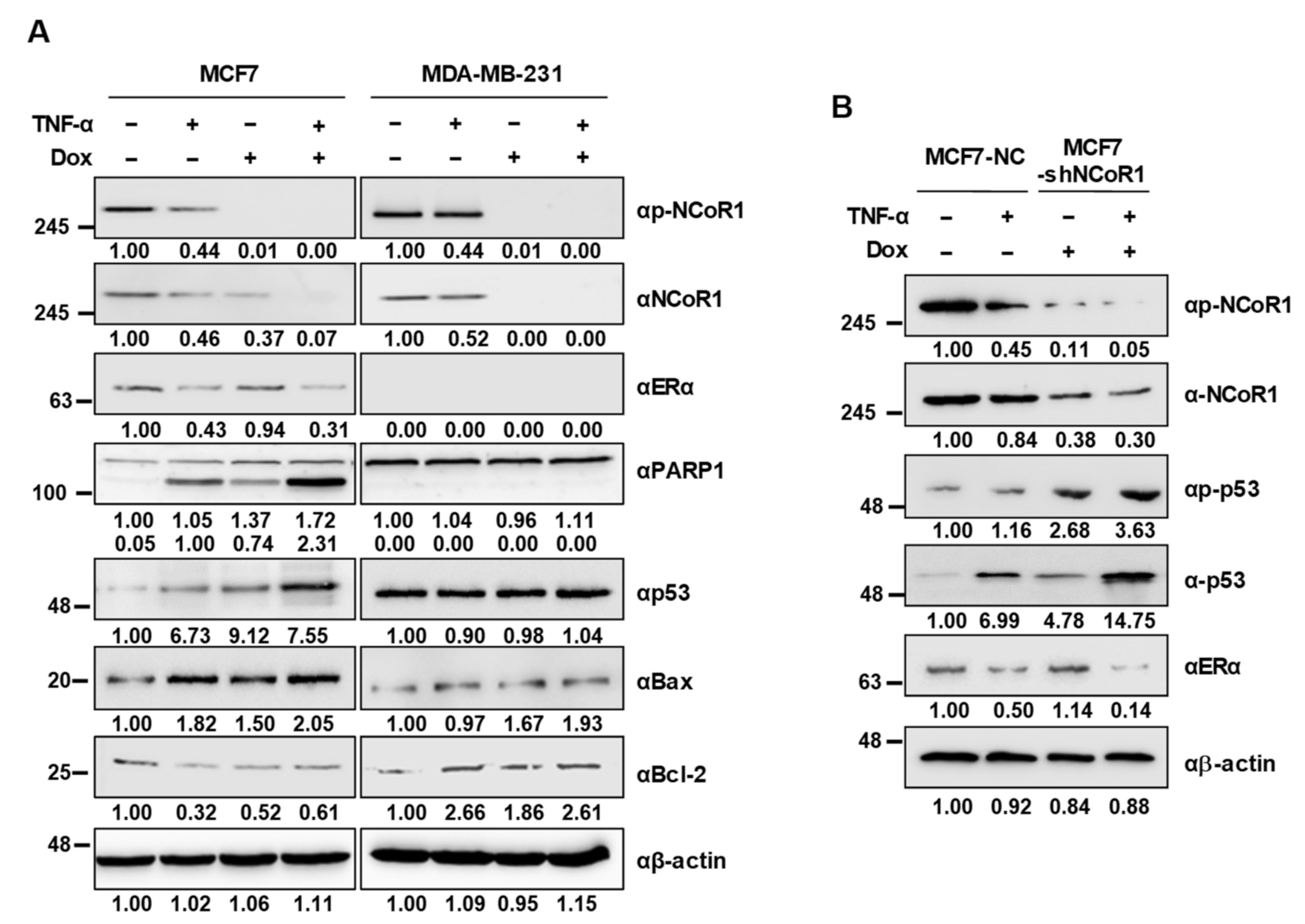
2.3. NCOR1 Knockdown Induces p53 Stabilization upon TNFα Treatment in MCF7 Cells and MCF7 Xenograft Mice
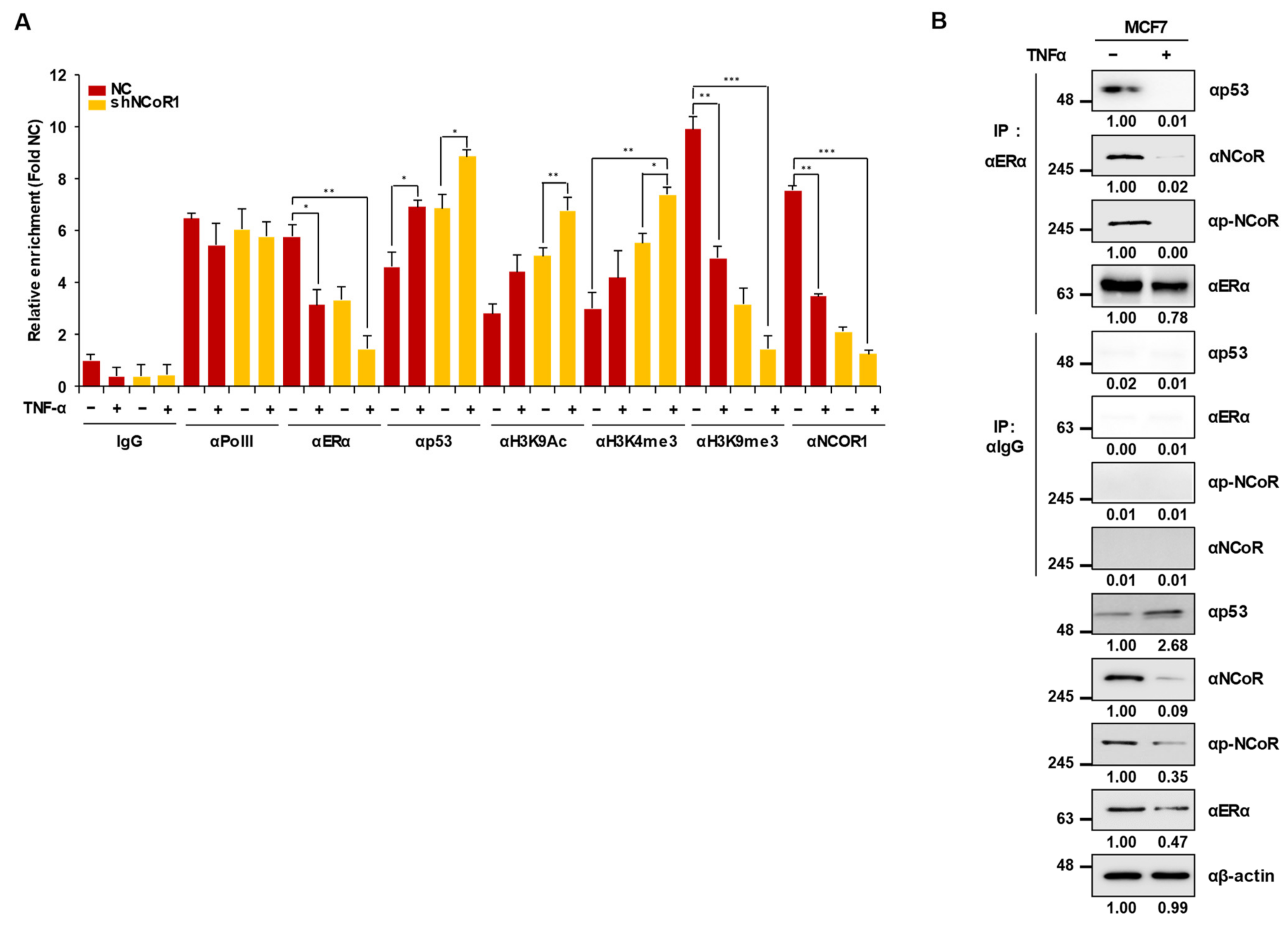
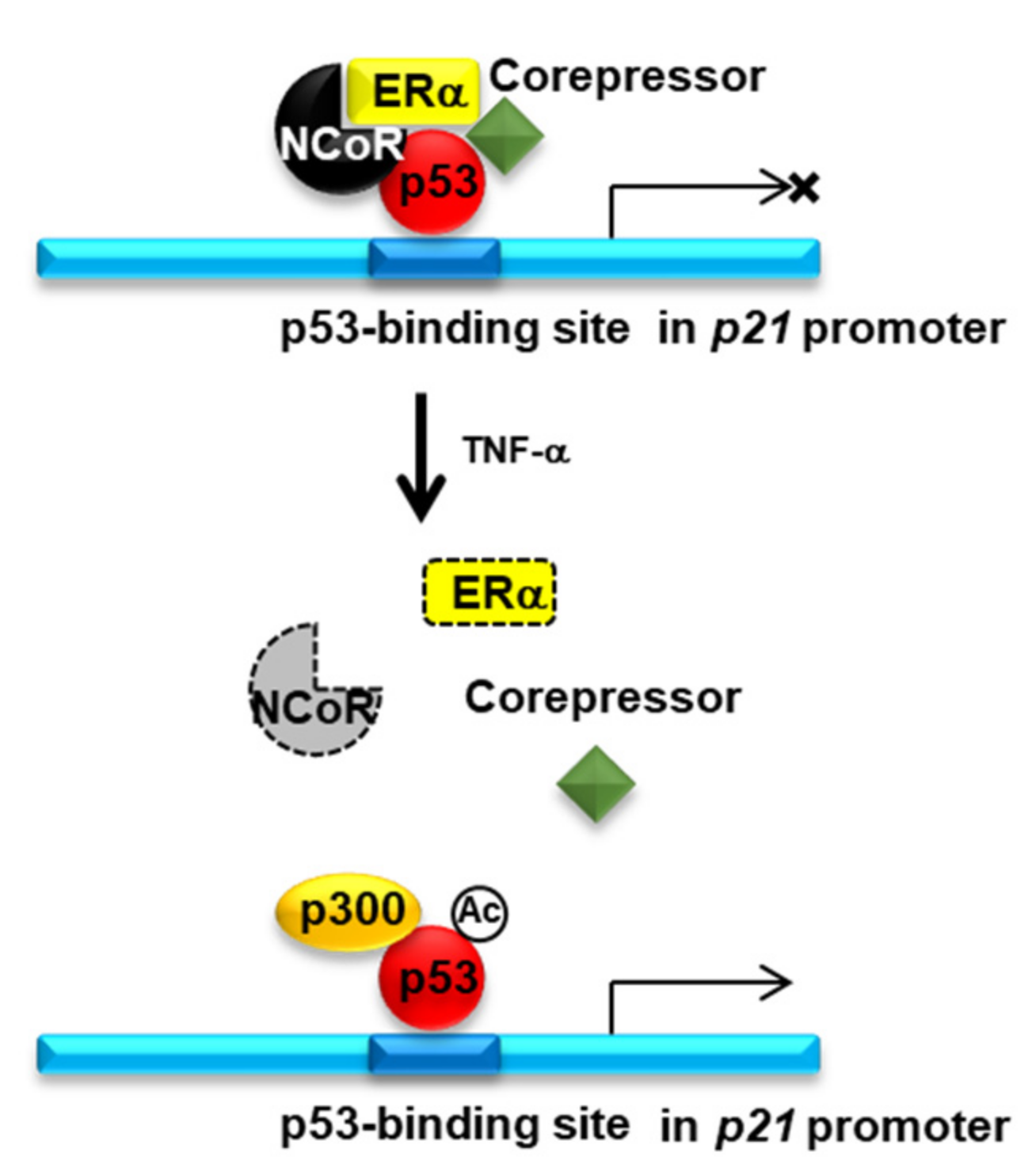
2.4. Knockdown of NCOR1 Activates the p21 Promoter via Recruitment of p53 Instead of ERα
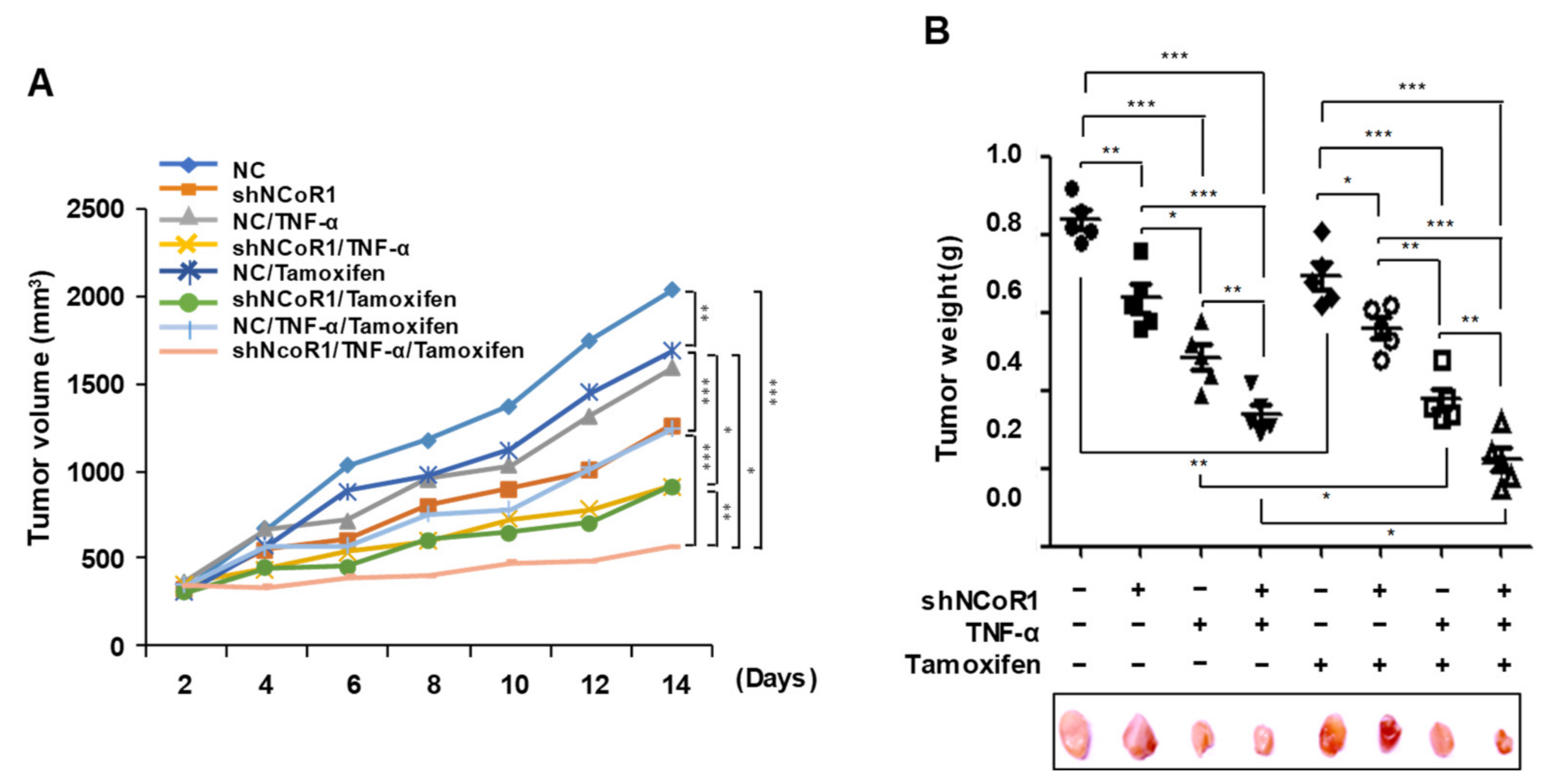
2.5. Tamoxifen Sensitivity Is Enhanced by TNFα or NCOR1 Knockdown in the MCF7 Xenograft Mouse Model
3. Discussion
4. Materials and Methods
4.1. Cell Culture Conditions, Reagents, and Antibodies
4.2. Western Blot
4.3. Quantification of Apoptotic Cells by Flow Cytometry
4.4. Cell Proliferation Measurement by MTT Assay
4.5. Lentiviral Short Hairpin RNAs
4.6. Chromatin Immunoprecipitation Assays
4.7. Colony Formation Assay
4.8. Generation of Dox-Inducible Stable Cell Lines
4.9. Xenograft Mouse Model
4.10. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Libson, S.; Lippman, M. A review of clinical aspects of breast cancer. Int. Rev. Psychiatry 2014, 26, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Jia, T.; Zhang, L.; Duan, Y.; Zhang, M.; Wang, G.; Zhang, J.; Zhao, Z. The differential susceptibilities of MCF-7 and MDA-MB-231 cells to the cytotoxic effects of curcumin are associated with the PI3K/Akt-SKP2-Cip/Kips pathway. Cancer Cell Int. 2014, 14, 126. [Google Scholar] [CrossRef] [PubMed]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.R.; Kim, H.J.; Kim, Y.A.; Chang, M.S.; Hwang, K.T.; Park, S.Y. Diversity index as a novel prognostic factor in breast cancer. Oncotarget 2017, 8, 97114–97126. [Google Scholar] [CrossRef] [PubMed]
- Melchor, L.; Honrado, E.; Huang, J.; Alvarez, S.; Naylor, T.L.; Garcia, M.J.; Osorio, A.; Blesa, D.; Stratton, M.R.; Weber, B.L.; et al. Estrogen receptor status could modulate the genomic pattern in familial and sporadic breast cancer. Clin. Cancer Res. 2007, 13, 7305–7313. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.D.; Darimont, B.; Zhou, W.; Arrazate, A.; Young, A.; Ingalla, E.; Walter, K.; Blake, R.A.; Nonomiya, J.; Guan, Z.; et al. The selective estrogen receptor downregulator GDC-0810 is efficacious in diverse models of ER+ breast cancer. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Omoto, Y.; Iwase, H.; Yamashita, H.; Toyama, T.; Coombes, R.C.; Filipovic, A.; Warner, M.; Gustafsson, J.A. Differential expression of estrogen receptor alpha, beta1, and beta2 in lobular and ductal breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1933–1938. [Google Scholar] [CrossRef]
- Rugo, H.S.; Rumble, R.B.; Macrae, E.; Barton, D.L.; Connolly, H.K.; Dickler, M.N.; Fallowfield, L.; Fowble, B.; Ingle, J.N.; Jahanzeb, M.; et al. Endocrine Therapy for Hormone Receptor-Positive Metastatic Breast Cancer: American Society of Clinical Oncology Guideline. J. Clin. Oncol. 2016, 34, 3069–3103. [Google Scholar] [CrossRef]
- Reinert, T.; Barrios, C.H. Optimal management of hormone receptor positive metastatic breast cancer in 2016. Ther. Adv. Med. Oncol. 2015, 7, 304–320. [Google Scholar] [CrossRef]
- Ring, A.; Dowsett, M. Mechanisms of tamoxifen resistance. Endocr. Relat. Cancer 2004, 11, 643–658. [Google Scholar] [CrossRef]
- Hultsch, S.; Kankainen, M.; Paavolainen, L.; Kovanen, R.-M.; Ikonen, E.; Kangaspeska, S.; Pietiäinen, V.; Kallioniemi, O. Association of tamoxifen resistance and lipid reprogramming in breast cancer. BMC Cancer 2018, 18, 850. [Google Scholar] [CrossRef]
- Hosford, S.R.; Miller, T.W. Clinical potential of novel therapeutic targets in breast cancer: CDK4/6, Src, JAK/STAT, PARP, HDAC, and PI3K/AKT/mTOR pathways. Pharm. Pers. Med. 2014, 7, 203. [Google Scholar]
- Hurtado, A.; Holmes, K.A.; Geistlinger, T.R.; Hutcheson, I.R.; Nicholson, R.I.; Brown, M.; Jiang, J.; Howat, W.J.; Ali, S.; Carroll, J.S. Regulation of ERBB2 by oestrogen receptor–PAX2 determines response to tamoxifen. Nature 2008, 456, 663–666. [Google Scholar] [CrossRef]
- Park, S.-H.; Kim, H.; Kwak, S.; Jeong, J.-H.; Lee, J.; Hwang, J.-T.; Choi, H.-K.; Choi, K.-C. HDAC3–ERα Selectively Regulates TNF-α-Induced Apoptotic Cell Death in MCF-7 Human Breast Cancer Cells via the p53 Signaling Pathway. Cells 2020, 9, 1280. [Google Scholar] [CrossRef]
- McKenna, N.J.; Lanz, R.B.; O’Malley, B.W. Nuclear receptor coregulators: Cellular and molecular biology. Endocr. Rev. 1999, 20, 321–344. [Google Scholar] [CrossRef]
- Perissi, V.; Jepsen, K.; Glass, C.K.; Rosenfeld, M.G. Deconstructing repression: Evolving models of co-repressor action. Nat. Rev. Genet. 2010, 11, 109–123. [Google Scholar] [CrossRef]
- Girault, I.; Lerebours, F.; Amarir, S.; Tozlu, S.; Tubiana-Hulin, M.; Lidereau, R.; Bieche, I. Expression analysis of estrogen receptor alpha coregulators in breast carcinoma: Evidence that NCOR1 expression is predictive of the response to tamoxifen. Clin. Cancer Res. 2003, 9, 1259–1266. [Google Scholar]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hara, Y.; Kobayashi, S.; Iwase, H. NCOR1 mRNA is an independent prognostic factor for breast cancer. Cancer Lett. 2006, 237, 123–129. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Yamashita, H.; Toyama, T.; Yamamoto, Y.; Kawasoe, T.; Ibusuki, M.; Tomita, S.; Sugiura, H.; Kobayashi, S.; Fujii, Y.; et al. Nuclear corepressor 1 expression predicts response to first-line endocrine therapy for breast cancer patients on relapse. Chin. Med. J. 2009, 122, 1764–1768. [Google Scholar]
- Elledge, R.M.; Allred, D.C. The p53 tumor suppressor gene in breast cancer. Breast Cancer Res. Treat. 1994, 32, 39–47. [Google Scholar] [CrossRef]
- Elledge, R.M.; Fuqua, S.A.; Clark, G.M.; Pujol, P.; Allred, D.C. The role and prognostic significance of p53 gene alterations in breast cancer. Breast Cancer Res. Treat. 1993, 27, 95–102. [Google Scholar] [CrossRef]
- Criscitiello, C.; Fumagalli, D.; Saini, K.S.; Loi, S. Tamoxifen in early-stage estrogen receptor-positive breast cancer: Overview of clinical use and molecular biomarkers for patient selection. OncoTargets Ther. 2011, 4, 1. [Google Scholar]
- Fan, W.; Chang, J.; Fu, P. Endocrine therapy resistance in breast cancer: Current status, possible mechanisms and overcoming strategies. Future Med. Chem. 2015, 7, 1511–1519. [Google Scholar] [CrossRef]
- Osborne, C.K.; Shou, J.; Massarweh, S.; Schiff, R. Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin. Cancer Res. 2005, 11, 865s–870s. [Google Scholar]
- Clemons, M.; Danson, S.; Howell, A. Tamoxifen (“Nolvadex”): A review. Cancer Treat. Rev. 2002, 28, 165–180. [Google Scholar] [CrossRef]
- Deschenes, L. Droloxifene, a new antiestrogen, in advanced breast cancer. A double-blind dose-finding study. The Droloxifene 002 International Study Group. Am. J. Clin. Oncol 1991, 14 (Suppl. 2), S52–S55. [Google Scholar] [CrossRef]
- Ingle, J.N.; Ahmann, D.L.; Green, S.J.; Edmonson, J.H.; Creagan, E.T.; Hahn, R.G.; Rubin, J. Randomized clinical trial of megestrol acetate versus tamoxifen in paramenopausal or castrated women with advanced breast cancer. Am. J. Clin. Oncol 1982, 5, 155–160. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Buzdar, A.U.; Frye, D.; Yap, H.Y.; Hug, V.; Pinnamaneni, K.; Fraschini, G.; Halvorson, H.C.; Blumenschein, G.R. Oral medroxyprogesterone acetate in the treatment of metastatic breast cancer. Breast Cancer Res. Treat. 1985, 5, 321–326. [Google Scholar] [CrossRef]
- Gale, K.E.; Andersen, J.W.; Tormey, D.C.; Mansour, E.G.; Davis, T.E.; Horton, J.; Wolter, J.M.; Smith, T.J.; Cummings, F.J. Hormonal treatment for metastatic breast cancer. An Eastern Cooperative Oncology Group Phase III trial comparing aminoglutethimide to tamoxifen. Cancer 1994, 73, 354–361. [Google Scholar] [CrossRef]
- Falkson, C.I.; Falkson, H.C. A randomised study of CGS 16949A (fadrozole) versus tamoxifen in previously untreated postmenopausal patients with metastatic breast cancer. Ann. Oncol 1996, 7, 465–469. [Google Scholar] [CrossRef]
- Munster, P.; Thurn, K.; Thomas, S.; Raha, P.; Lacevic, M.; Miller, A.; Melisko, M.; Ismail-Khan, R.; Rugo, H.; Moasser, M. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br. J. Cancer 2011, 104, 1828–1835. [Google Scholar] [CrossRef]
- Bachelot, T.; Bourgier, C.; Cropet, C.; Ray-Coquard, I.; Ferrero, J.-M.; Freyer, G.; Abadie-Lacourtoisie, S.; Eymard, J.-C.; Debled, M.; Spaëth, D. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor–positive, human epidermal growth factor receptor 2–negative metastatic breast cancer with prior exposure to aromatase inhibitors: A GINECO study. J. Clin. Oncol. 2012, 30, 2718–2724. [Google Scholar] [CrossRef]
- Ciardiello, F.; Troiani, T.; Caputo, F.; De Laurentiis, M.; Tortora, G.; Palmieri, G.; De Vita, F.; Diadema, M.; Orditura, M.; Colantuoni, G. Phase II study of gefitinib in combination with docetaxel as first-line therapy in metastatic breast cancer. Br. J. Cancer 2006, 94, 1604–1609. [Google Scholar] [CrossRef]
- Mo, Z.; Liu, M.; Yang, F.; Luo, H.; Li, Z.; Tu, G.; Yang, G. GPR30 as an initiator of tamoxifen resistance in hormone-dependent breast cancer. Breast Cancer Res. 2013, 15, 1–15. [Google Scholar] [CrossRef]
- van Horssen, R.; Ten Hagen, T.L.; Eggermont, A.M. TNF-alpha in cancer treatment: Molecular insights, antitumor effects, and clinical utility. Oncologist 2006, 11, 397–408. [Google Scholar] [CrossRef]
- Wu, X.; Wu, M.Y.; Jiang, M.; Zhi, Q.; Bian, X.; Xu, M.D.; Gong, F.R.; Hou, J.; Tao, M.; Shou, L.M.; et al. TNF-alpha sensitizes chemotherapy and radiotherapy against breast cancer cells. Cancer Cell Int. 2017, 17, 13. [Google Scholar] [CrossRef]
- Lazar, M.A. Nuclear receptor corepressors. Nucl. Recept. Signal. 2003, 1, nrs-01001. [Google Scholar] [CrossRef]
- Lu, R.; Hu, X.; Zhou, J.; Sun, J.; Zhu, A.Z.; Xu, X.; Zheng, H.; Gao, X.; Wang, X.; Jin, H. COPS5 amplification and overexpression confers tamoxifen-resistance in ERα-positive breast cancer by degradation of NCoR. Nat. Commun. 2016, 7, 12044. [Google Scholar] [CrossRef]
- Doyon, G.; St-Jean, S.; Darsigny, M.; Asselin, C.; Boudreau, F. Nuclear receptor co-repressor is required to maintain proliferation of normal intestinal epithelial cells in culture and down-modulates the expression of pigment epithelium-derived factor. J. Biol. Chem. 2009, 284, 25220–25229. [Google Scholar] [CrossRef]
- Zhang, D.; Cho, E.; Wong, J. A critical role for the co-repressor N-CoR in erythroid differentiation and heme synthesis. Cell Res. 2007, 17, 804–814. [Google Scholar] [CrossRef]
- Noblejas-López, M.d.M.; Morcillo-García, S.; Nieto-Jiménez, C.; Nuncia-Cantarero, M.; Győrffy, B.; Galan-Moya, E.M.; Pandiella, A.; Ocaña, A. Evaluation of transcriptionally regulated genes identifies NCOR1 in hormone receptor negative breast tumors and lung adenocarcinomas as a potential tumor suppressor gene. PLoS ONE 2018, 13, e0207776. [Google Scholar] [CrossRef]
- Kashima, H.; Horiuchi, A.; Uchikawa, J.; Miyamoto, T.; Suzuki, A.; Ashida, T.; Konishi, I.; Shiozawa, T. Up-regulation of nuclear receptor corepressor (NCoR) in progestin-induced growth suppression of endometrial hyperplasia and carcinoma. Anticancer Res. 2009, 29, 1023–1029. [Google Scholar]
- Fujioka, S.; Schmidt, C.; Sclabas, G.M.; Li, Z.; Pelicano, H.; Peng, B.; Yao, A.; Niu, J.; Zhang, W.; Evans, D.B. Stabilization of p53 is a novel mechanism for proapoptotic function of NF-κB. J. Biol. Chem. 2004, 279, 27549–27559. [Google Scholar] [CrossRef]
- Lavin, M.a.; Gueven, N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006, 13, 941–950. [Google Scholar] [CrossRef]
- Cheng, Q.; Chen, J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle 2010, 9, 472–478. [Google Scholar] [CrossRef]
- Konduri, S.D.; Medisetty, R.; Liu, W.; Kaipparettu, B.A.; Srivastava, P.; Brauch, H.; Fritz, P.; Swetzig, W.M.; Gardner, A.E.; Khan, S.A. Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc. Natl. Acad. Sci. USA 2010, 107, 15081–15086. [Google Scholar] [CrossRef]
- Chang, M. Tamoxifen resistance in breast cancer. Biomol. Ther. 2012, 20, 256. [Google Scholar] [CrossRef]
- Ali, S.; Rasool, M.; Chaoudhry, H.; Pushparaj, P.N.; Jha, P.; Hafiz, A.; Mahfooz, M.; Sami, G.A.; Kamal, M.A.; Bashir, S. Molecular mechanisms and mode of tamoxifen resistance in breast cancer. Bioinformation 2016, 12, 135. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Park, S.-H.; Lee, J.; Sung, G.-J.; Song, J.-H.; Kwak, S.; Jeong, J.-H.; Kong, M.-J.; Hwang, J.-T.; Choi, H.-K.; et al. TNFα Enhances Tamoxifen Sensitivity through Dissociation of ERα-p53-NCOR1 Complexes in ERα-Positive Breast Cancer. Cancers 2021, 13, 2601. https://doi.org/10.3390/cancers13112601
Kim H, Park S-H, Lee J, Sung G-J, Song J-H, Kwak S, Jeong J-H, Kong M-J, Hwang J-T, Choi H-K, et al. TNFα Enhances Tamoxifen Sensitivity through Dissociation of ERα-p53-NCOR1 Complexes in ERα-Positive Breast Cancer. Cancers. 2021; 13(11):2601. https://doi.org/10.3390/cancers13112601
Chicago/Turabian StyleKim, Hyunhee, Seung-Ho Park, Jangho Lee, Gi-Jun Sung, Ji-Hye Song, Sungmin Kwak, Ji-Hoon Jeong, Min-Jeong Kong, Jin-Taek Hwang, Hyo-Kyoung Choi, and et al. 2021. "TNFα Enhances Tamoxifen Sensitivity through Dissociation of ERα-p53-NCOR1 Complexes in ERα-Positive Breast Cancer" Cancers 13, no. 11: 2601. https://doi.org/10.3390/cancers13112601
APA StyleKim, H., Park, S.-H., Lee, J., Sung, G.-J., Song, J.-H., Kwak, S., Jeong, J.-H., Kong, M.-J., Hwang, J.-T., Choi, H.-K., & Choi, K.-C. (2021). TNFα Enhances Tamoxifen Sensitivity through Dissociation of ERα-p53-NCOR1 Complexes in ERα-Positive Breast Cancer. Cancers, 13(11), 2601. https://doi.org/10.3390/cancers13112601