
Identification of Candidate Protein Biomarkers for CIN2+ Lesions from Self-Sampled, Dried Cervico–Vaginal Fluid Using LC-MS/MS

 , ,
, ,
Abstract
Simple Summary
Abstract
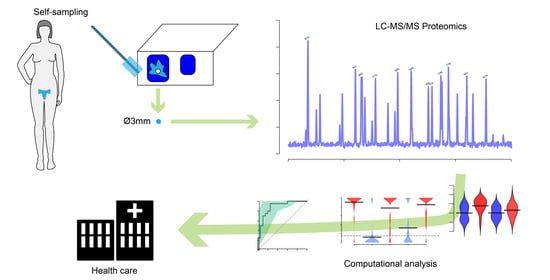
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Mass Spectrometry Analysis and Quantification
2.2.1. Sample Preparation
2.2.2. LC-MS/MS
2.2.3. MS Data Handling
2.3. Statistical Analysis
3. Results
3.1. Dried CVF Is Suitable for Proteomic Analyses with Mass Spectrometry
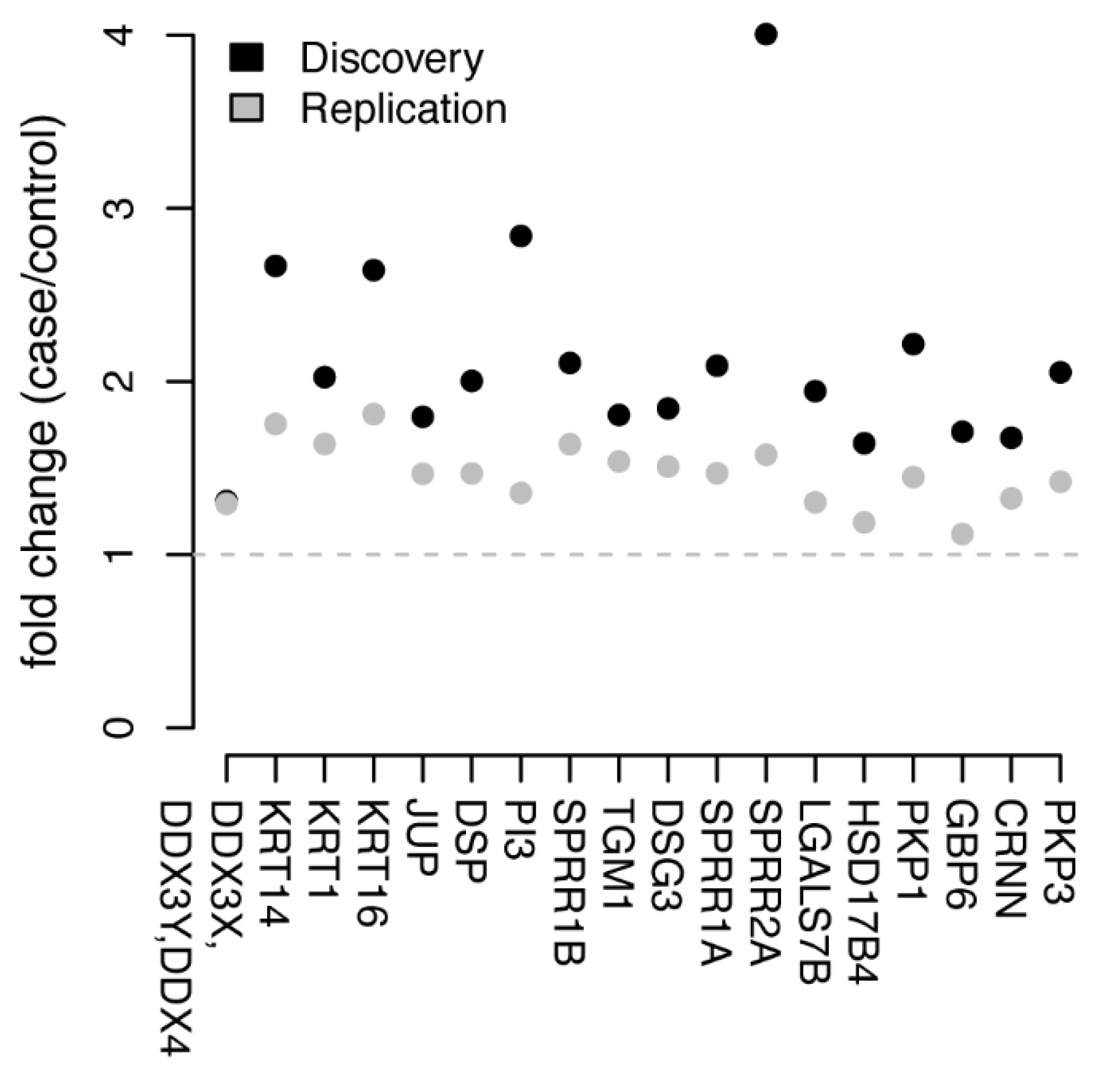
3.2. Univariate Protein Biomarkers for CIN2+
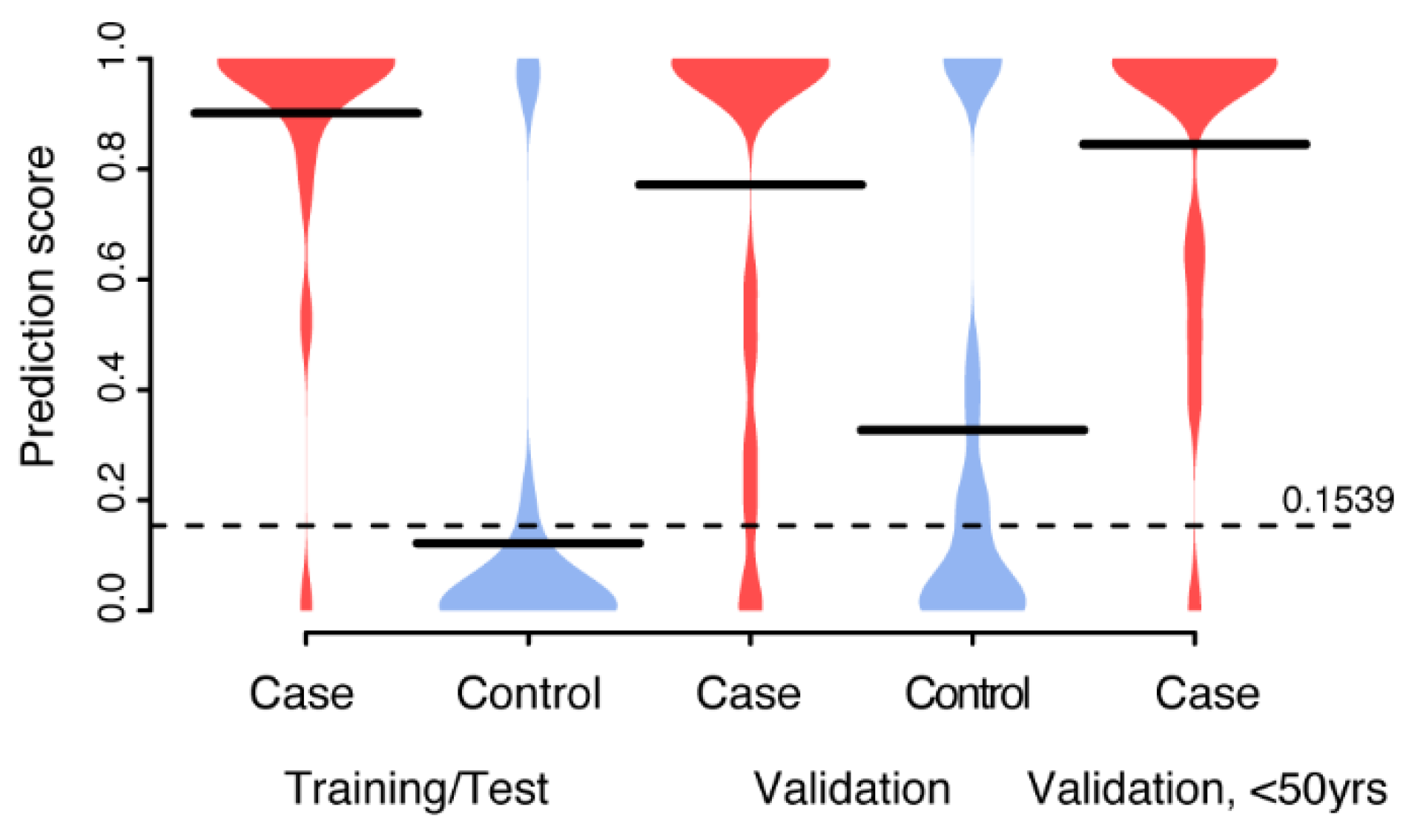
3.3. Multivariate Biomarker Signature for CIN2+
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Harper, D.M.; Demars, L.R. HPV vaccines—A review of the first decade. Gynecol. Oncol. 2017, 146, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Tsikouras, P.; Zervoudis, S.; Manav, B.; Tomara, E.; Iatrakis, G.; Romanidis, C.; Bothou, A.; Galazios, G. Cervical cancer: Screening, diagnosis and staging. J. BUON 2016, 21, 320–325. [Google Scholar] [PubMed]
- Goodman, A. HPV testing as a screen for cervical cancer. BMJ 2015, 350, 1–11. [Google Scholar] [CrossRef]
- World Health Organization. Comprehensive Cervical Cancer Control a Guide to Essential Practice, 2nd ed.; World Health Organization: Geneva, Switzerland, 2014; ISBN 978-92-4-154895-3. [Google Scholar]
- Solomon, D. The 2001 Bethesda System.Terminology for Reporting Results of Cervical Cytology. JAMA 2002, 287, 2114. [Google Scholar] [CrossRef]
- Good, D.M.; Thongboonkerd, V.; Novak, J.; Bascands, J.-L.; Schanstra, J.P.; Coon, J.J.; Dominiczak, A.; Mischak, H. Body Fluid Proteomics for Biomarker Discovery: Lessons from the Past Hold the Key to Success in the Future. J. Proteome Res. 2007, 6, 4549–4555. [Google Scholar] [CrossRef] [PubMed]
- Berggrund, M.; Enroth, S.; Lundberg, M.; Assarsson, E.; Stålberg, K.; Lindquist, D.; Hallmans, G.; Grankvist, K.; Olovsson, M.; Gyllensten, U. Identification of Candidate Plasma Protein Biomarkers for Cervical Cancer Using the Multiplex Proximity Extension Assay. Mol. Cell. Proteom. 2019, 18, 735–743. [Google Scholar] [CrossRef]
- Aarnio, R.; Östensson, E.; Olovsson, M.; Gustavsson, I.; Gyllensten, U. Cost-effectiveness analysis of repeated self-sampling for HPV testing in primary cervical screening: A randomized study. BMC Cancer 2020, 20, 645. [Google Scholar] [CrossRef] [PubMed]
- Burd, E.M.; Dean, C.L. Human Papillomavirus. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Van Raemdonck, G.A.A.; Tjalma, W.A.A.; Coen, E.P.; Depuydt, C.E.; Van Ostade, X.W.M. Identification of Protein Biomarkers for Cervical Cancer Using Human Cervicovaginal Fluid. PLoS ONE 2014, 9, e106488. [Google Scholar] [CrossRef]
- Zegels, G.; Van Raemdonck, G.A.A.; Tjalma, W.A.A.; Van Ostade, X.W.M. Use of cervicovaginal fluid for the identification of biomarkers for pathologies of the female genital tract. Proteome Sci. 2010, 8, 63. [Google Scholar] [CrossRef]
- Boylan, K.L.M.; Afiuni-Zadeh, S.; Geller, M.A.; Hickey, K.; Griffin, T.J.; Pambuccian, S.E.; Skubitz, A.P.N. A feasibility study to identify proteins in the residual Pap test fluid of women with normal cytology by mass spectrometry-based proteomics. Clin. Proteom. 2014, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, I.; Lindell, M.; Wilander, E.; Strand, A.; Gyllensten, U. Use of FTA card for dry collection, transportation and storage of cervical cell specimen to detect high-risk HPV. J. Clin. Virol. 2009, 46, 112–116. [Google Scholar] [CrossRef]
- Berggrund, M.; Ekman, D.; Gustavsson, I.; Sundfeldt, K.; Olovsson, M.; Enroth, S.; Gyllensten, U. Protein Detection Using the Multiplexed Proximity Extension Assay (PEA) from Plasma and Vaginal Fluid Applied to the Indicating FTA Elute Micro CardTM. J. Circ. Biomarkers 2016, 5, 9. [Google Scholar] [CrossRef]
- Gustavsson, I.; Aarnio, R.; Berggrund, M.; Hedlund-Lindberg, J.; Strand, A.S.; Sanner, K.; Wikström, I.; Enroth, S.; Olovsson, M.; Gyllensten, U. Randomised study shows that repeated selfsampling and HPV test has more than twofold higher detection rate of women with CIN2+ histology than Pap smear cytology. Br. J. Cancer 2018, 118, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Berggrund, M.; Gustavsson, I.; Aarnio, R.; Lindberg, J.H.; Sanner, K.; Wikström, I.; Enroth, S.; Bunikis, I.; Olovsson, M.; Gyllensten, U. Temporal changes in the vaginal microbiota in self-samples and its association with persistent HPV16 infection and CIN2+. Virol. J. 2020, 17, 147. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, I.; Sanner, K.; Lindell, M.; Strand, A.; Olovsson, M.; Wikström, I.; Wilander, E.; Gyllensten, U. Type-specific detection of high-risk human papillomavirus (HPV) in self-sampled cervicovaginal cells applied to FTA elute cartridge. J. Clin. Virol. 2011, 51, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, I.; Aarnio, R.; Myrnäs, M.; Hedlund-Lindberg, J.; Taku, O.; Meiring, T.; Wikström, I.; Enroth, S.; Williamson, A.L.; Olovsson, M.; et al. Clinical validation of the HPVIR high-risk HPV test on cervical samples according to the international guidelines for human papillomavirus DNA test requirements for cervical cancer screening. Virol. J. 2019, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, A.P.; Hjort, K.; Hillered, L.; Sjödin, M.O.D.; Bergquist, J.; Wetterhall, M. Multiplexed quantification of proteins adsorbed to surface-modified and non-modified microdialysis membranes. Anal. Bioanal. Chem. 2012, 402, 2057–2067. [Google Scholar] [CrossRef]
- Urgen Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Kuhn, M. Building Predictive Models in R Using the Caret Package. J. Stat. Softw. 2020, 28. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Kampstra, P. Beanplot: A Boxplot Alternative for VisualComparison of Distributions. J. Stat. Softw. 2008, 28, 1–9. [Google Scholar] [CrossRef]
- Assarsson, E.; Lundberg, M.; Holmquist, G.; Björkesten, J.; Bucht Thorsen, S.; Ekman, D.; Eriksson, A.; Rennel Dickens, E.; Ohlsson, S.; Edfeldt, G.; et al. Homogenous 96-Plex PEA Immunoassay Exhibiting High Sensitivity, Specificity, and Excellent Scalability. PLoS ONE 2014, 9, e95192. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347. [Google Scholar] [CrossRef] [PubMed]
- Chokchaichamnankit, D.; Watcharatanyatip, K.; Subhasitanont, P.; Weeraphan, C.; Keeratichamroen, S.; Sritana, N.; Kantathavorn, N.; Ayudthaya, P.D.N.; Saharat, K.; Chantaraamporn, J.; et al. Urinary biomarkers for the diagnosis of cervical cancer by quantitative label-free mass spectrometry analysis. Oncol. Lett. 2019, 17, 5453–5468. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Hu, J.; Zhang, T.; Jiang, C.; Wang, H.Y. TRIM29 overexpression is associated with poor prognosis and promotes tumor progression by activating wnt/β-catenin pathway in cervical cancer. Oncotarget 2016, 7, 28579–28591. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort | N | Age a | HPV-Status b | No of Proteins c | NPTCP % Overlap d |
|---|---|---|---|---|---|
| Discovery | |||||
| Case | 20 | 54.2 (50–60) | 16 (45%), 33/52/58 (35%), 31 (15%), 39 (10%), 51 (10%), 18/45 (5%), 59 (5%) | 2411 (360) | 97.4 (56.2) |
| Control | 20 | 53.8 (50–59) | Negative | 2724 (494) | 96.7 (66.0) |
| Replication | |||||
| Case1 | 20 | 50.8 (47–60) | 16 (35%), 33/52/58 (35%), 51 (15%), 39 (10%), 56 (10%), 18/45 (5%), 31 (5%), 59 (5%) | 2137 (332) | 96.1 (53.6) |
| Case2 | 19 | 35.6 (30–45) | 16 (100%) | 1630 (247) | 96.1 (43.1) |
| Control | 20 | 50.9 (47–59) | Negative | 2322 (329) | 97.4 (53.6) |
| 3697 (169) | 98.7 (37.9) |
| Model Size | AUC a | AUC Difference b | Sensitivity c | Specificity d | |
|---|---|---|---|---|---|
| Continuous | |||||
| Train | 6 proteins | 0.76 (0.61–0.92) | 0.95 (0.85–1.00) | 0.60 (0.40–0.80) | |
| Validation | 0.67 (0.50–0.84) | 0.44 | 0.75 (0.55–0.95) | 0.45 (0.25–0.65) | |
| Binomial | |||||
| Train | 90 proteins + age | 0.81 (0.66–0.95) | 0.75 (0.55–0.95) | 0.85 (0.70–1.00) | |
| Validation | 0.65 (0.48–0.83) | 0.19 | 0.65 (0.45–0.85) | 0.55 (0.35–0.75) | |
| Combined | |||||
| Train | 7 proteins | 0.88 (0.78–0.99) | 0.95 (0.85–1.00) | 0.75 (0.55–0.90) | |
| Validation | 0.74 (0.58–0.91) | 0.17 | 0.90 (0.75–1.00) | 0.55 (0.35–0.75) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutiérrez, A.L.; Lindberg, J.H.; Shevchenko, G.; Gustavsson, I.; Bergquist, J.; Gyllensten, U.; Enroth, S. Identification of Candidate Protein Biomarkers for CIN2+ Lesions from Self-Sampled, Dried Cervico–Vaginal Fluid Using LC-MS/MS. Cancers 2021, 13, 2592. https://doi.org/10.3390/cancers13112592
Gutiérrez AL, Lindberg JH, Shevchenko G, Gustavsson I, Bergquist J, Gyllensten U, Enroth S. Identification of Candidate Protein Biomarkers for CIN2+ Lesions from Self-Sampled, Dried Cervico–Vaginal Fluid Using LC-MS/MS. Cancers. 2021; 13(11):2592. https://doi.org/10.3390/cancers13112592
Chicago/Turabian StyleGutiérrez, Ariadna Lara, Julia Hedlund Lindberg, Ganna Shevchenko, Inger Gustavsson, Jonas Bergquist, Ulf Gyllensten, and Stefan Enroth. 2021. "Identification of Candidate Protein Biomarkers for CIN2+ Lesions from Self-Sampled, Dried Cervico–Vaginal Fluid Using LC-MS/MS" Cancers 13, no. 11: 2592. https://doi.org/10.3390/cancers13112592
APA StyleGutiérrez, A. L., Lindberg, J. H., Shevchenko, G., Gustavsson, I., Bergquist, J., Gyllensten, U., & Enroth, S. (2021). Identification of Candidate Protein Biomarkers for CIN2+ Lesions from Self-Sampled, Dried Cervico–Vaginal Fluid Using LC-MS/MS. Cancers, 13(11), 2592. https://doi.org/10.3390/cancers13112592