
Discovery and Evaluation of Protein Biomarkers as a Signature of Wellness in Late-Stage Cancer Patients in Early Phase Clinical Trials

 , , ,
, , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Sample Collection
2.2. Proteomic Sample Collection and Preparation
2.3. SWATH-MS Analysis
2.4. Statistical Analysis
2.5. Clinical Prognostic Parameters
3. Results
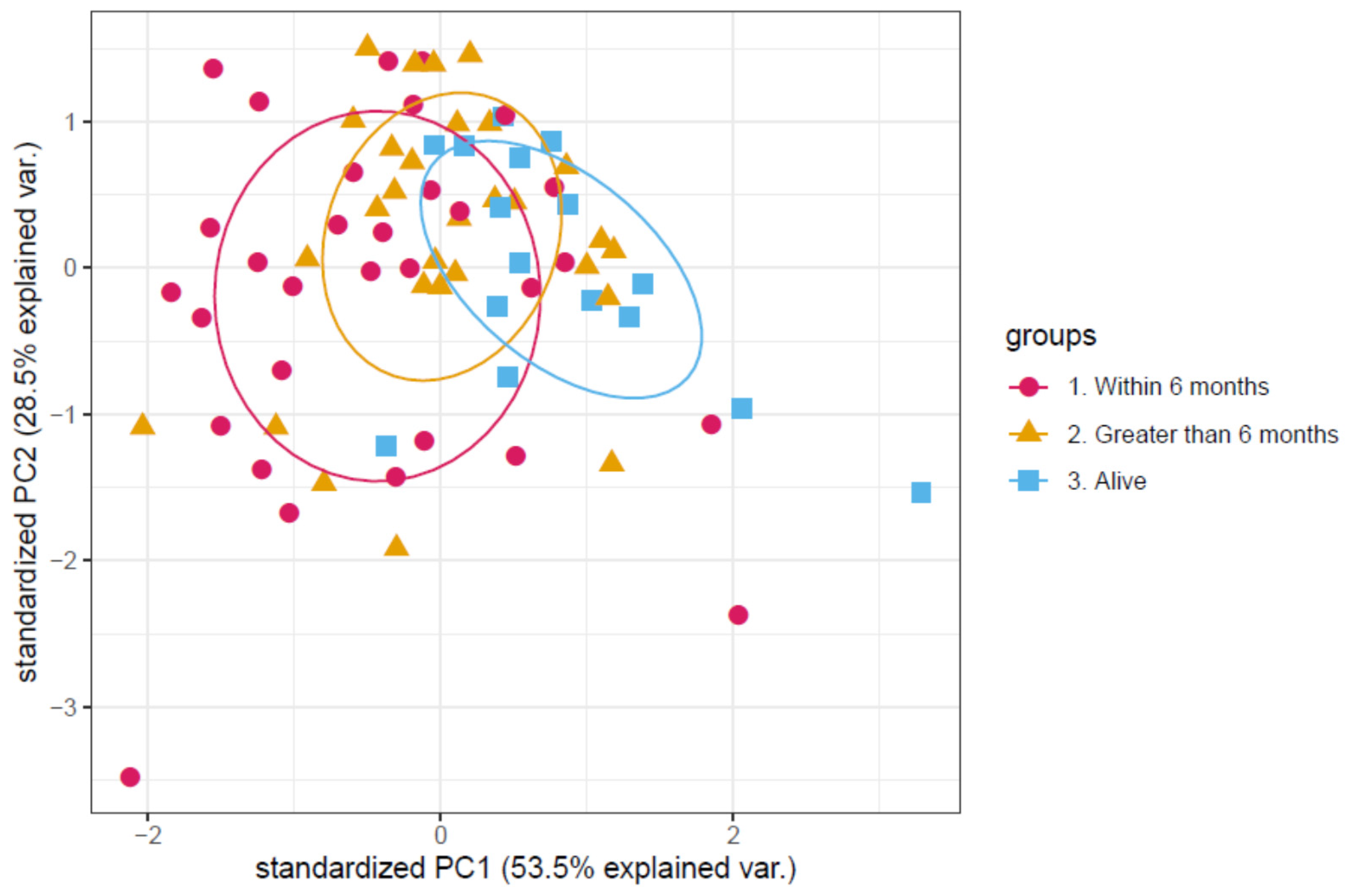
3.1. Determination of a Discriminatory Panel
3.2. Validation of the Proteomic Signature
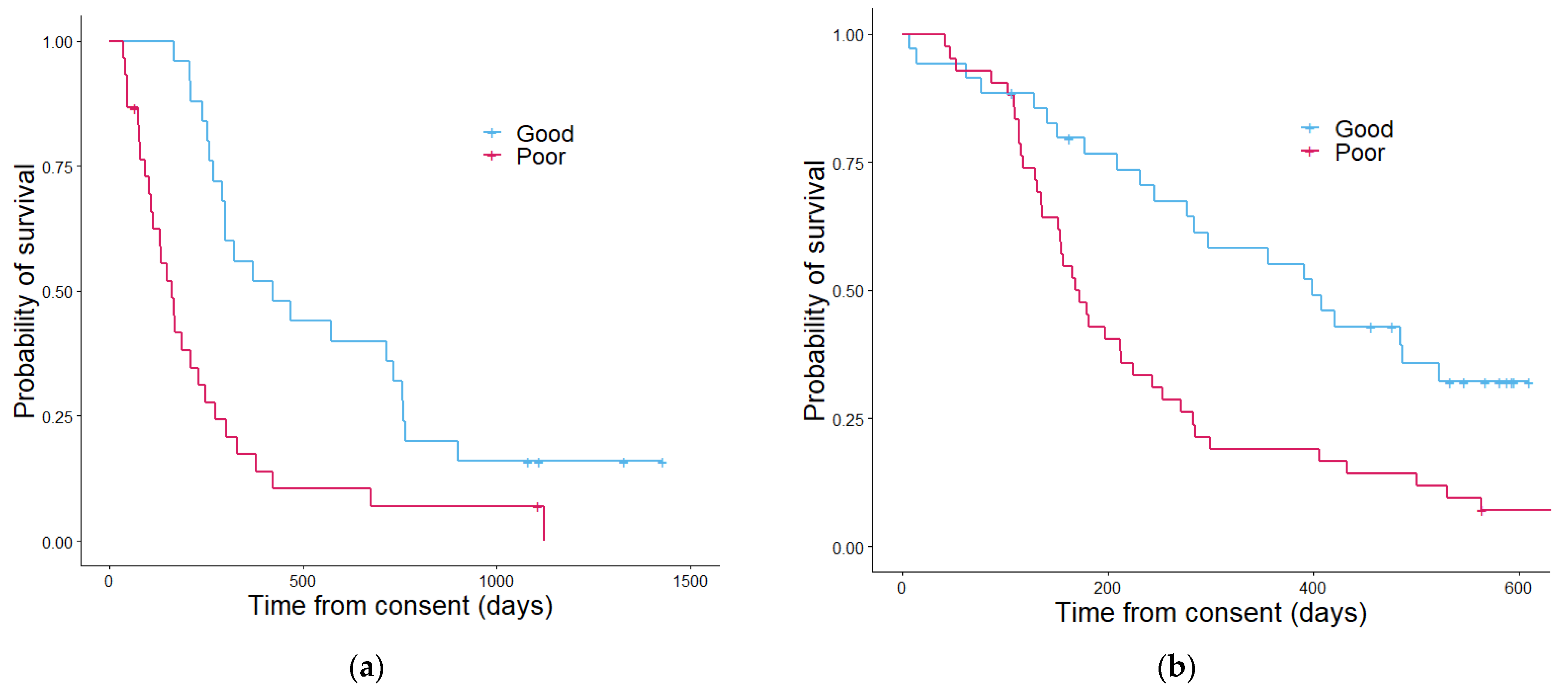
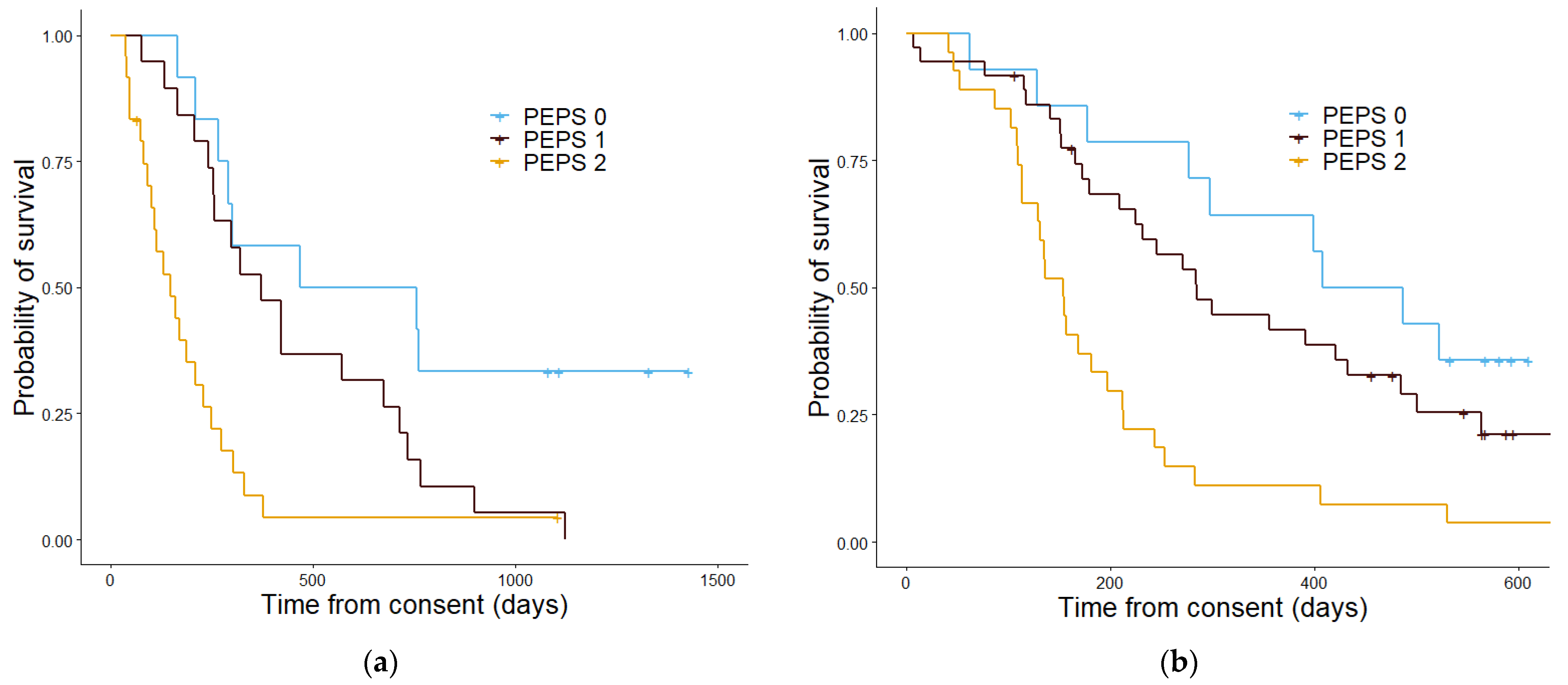
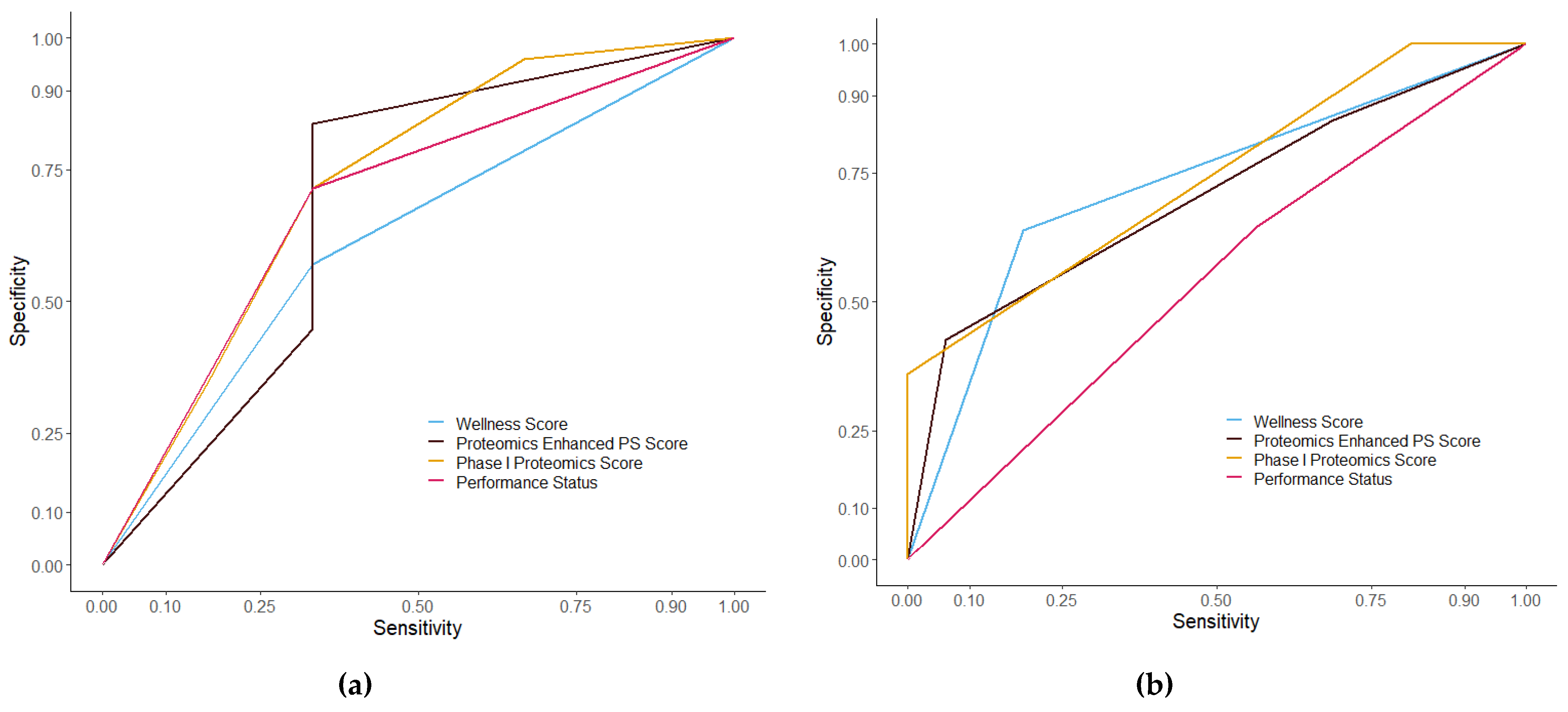
3.3. Assessment of Prognostic Scoring Methods
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, C.R. Predictive biomarkers and companion diagnostics. The future of immunohistochemistry: “in situ proteomics,” or just a “stain”? Appl. Immunohistochem. Mol. Morphol. 2014, 22, 555–561. [Google Scholar] [CrossRef]
- Aebersold, R.; Anderson, L.; Caprioli, R.; Druker, B.; Hartwell, L.; Smith, R. Perspective: A program to improve protein biomarker discovery for cancer. J. Proteome Res. 2005, 4, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Pepe, M.S.; Etzioni, R.; Feng, Z.; Potter, J.D.; Thompson, M.L.; Thornquist, M.; Winget, M.; Yasui, Y. Phases of biomarker development for early detection of cancer. J. Natl. Cancer Inst. 2001, 93, 1054–1061. [Google Scholar] [CrossRef]
- Viganó, A.; Bruera, E.; Jhangri, G.S.; Newman, S.C.; Fields, A.L.; Suarez-Almazor, M.E. Clinical survival predictors in patients with advanced cancer. Arch. Intern. Med. 2002, 160, 861–868. [Google Scholar] [CrossRef]
- Hoster, E.; Dreyling, M.; Klapper, W.; Gisselbrecht, C.; Van Hoof, A.; Kluin-Nelemans, H.C.; Pfreundschuh, M.; Reiser, M.; Metzner, B.; Einsele, H.; et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008, 111, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Molife, L.R.; Alam, S.; Olmos, D.; Puglisi, M.; Shah, K.; Fehrmann, R.; Trani, L.; Tjokrowidjaja, A.; de Bono, J.S.; Banerji, U.; et al. Defining the risk of toxicity in phase I oncology trials of novel molecularly targeted agents: A single centre experience. Ann. Oncol. 2012, 23, 1968–1973. [Google Scholar] [CrossRef] [PubMed]
- Wright, I.; Van Eyk, J.E. A Roadmap to Successful Clinical Proteomics. Clin. Chem. 2017, 63, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Holm, M.; Saraswat, M.; Joenväärä, S.; Ristimäki, A.; Haglund, C.; Renkonen, R. Colorectal cancer patients with different C-reactive protein levels and 5-year survival times can be differentiated with quantitative serum proteomics. PLoS ONE 2018, 13, e0195354. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Naba, A.; Bhutkar, A.; Guardia, T.; Miller, K.M.; Li, C.M.C.; Dayton, T.L.; Sanchez-Rivera, F.J.; Kim-Kiselak, C.; Jailkhani, N.; et al. Quantitative proteomics identify Tenascin-C as a promoter of lung cancer progression and contributor to a signature prognostic of patient survival. Proc. Natl. Acad. Sci. USA 2017, 114, E5625–E5634. [Google Scholar] [CrossRef]
- Becker, T.; Weberpals, J.; Jegg, A.M.; So, W.V.; Fischer, A.; Weisser, M.; Schmich, F.; Rüttinger, D.; Bauer-Mehren, A. An enhanced prognostic score for overall survival of patients with cancer derived from a large real-world cohort. Ann. Oncol. 2020, 31, 1561–1568. [Google Scholar] [CrossRef]
- Arkenau, H.-T.; Barriuso, J.; Olmos, D.; Ang, J.E.; de Bono, J.; Judson, I.; Kaye, S. Prospective validation of a prognostic score to improve patient selection for oncology phase I trials. J. Clin. Oncol. 2009, 27, 2692–2696. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, A.; Onoda, H.; Imai, N.; Iwaku, A.; Oishi, M.; Tanaka, K.; Fushiya, N.; Koike, K.; Nishino, H.; Matsushima, M.; et al. The Glasgow Prognostic Score, an inflammation based prognostic score, predicts survival in patients with hepatocellular carcinoma. BMC Cancer 2013, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.J.; Xie, W.; Kroeger, N.; Lee, J.L.; Rini, B.I.; Knox, J.J.; Bjarnason, G.A.; Srinivas, S.; Pal, S.K.; Yuasa, T.; et al. The international metastatic renal cell carcinoma database consortium model as a prognostic tool in patients with metastatic renal cell carcinoma previously treated with first-line targeted therapy: A population-based study. Lancet Oncol. 2015, 16, 293–300. [Google Scholar] [CrossRef]
- Karnofsky, D.A.; Abelmann, W.H.; Craver, L.F.; Burchenal, J.H. The use of the nitrogen mustards in the palliative treatment of carcinoma. With particular reference to bronchogenic carcinoma. Cancer 1948, 1, 634–656. [Google Scholar] [CrossRef]
- Erhart, F.; Hackl, M.; Hahne, H.; Buchroithner, J.; Meng, C.; Klingenbrunner, S.; Reitermaier, R.; Fischhuber, K.; Skalicky, S.; Berger, W.; et al. Combined proteomics/miRNomics of dendritic cell immunotherapy-treated glioblastoma patients as a screening for survival-associated factors. npj Vaccines 2020, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Price, N.D.; Magis, A.T.; Earls, J.C.; Glusman, G.; Levy, R.; Lausted, C.; McDonald, D.T.; Kusebauch, U.; Moss, C.L.; Zhou, Y.; et al. A wellness study of 108 individuals using personal, dense, dynamic data clouds. Nat. Biotechnol. 2017, 35, 747–756. [Google Scholar] [CrossRef]
- Zhang, B.; Whiteaker, J.R.; Hoofnagle, A.N.; Baird, G.S.; Rodland, K.D.; Paulovich, A.G. Clinical potential of mass spectrometry-based proteogenomics. Nat. Rev. Clin. Oncol. 2019, 16, 256–268. [Google Scholar] [CrossRef]
- Simpson, K.L.; Whetton, A.D.; Dive, C. Quantitative mass spectrometry-based techniques for clinical use: Biomarker identification and quantification. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Kosorok, M.R.; Zeng, D. Reinforcement learning design for cancer clinical trials. Stat. Med. 2009, 28, 3294–3315. [Google Scholar] [CrossRef]
- Shortliffe, E.H.; Sepúlveda, M.J. Clinical Decision Support in the Era of Artificial Intelligence. JAMA J. Am. Med. Assoc. 2018, 320, 2199–2200. [Google Scholar] [CrossRef]
- Russell, M.R.; Graham, C.; D’Amato, A.; Gentry-Maharaj, A.; Ryan, A.; Kalsi, J.K.; Ainley, C.; Whetton, A.D.; Menon, U.; Jacobs, I.; et al. A combined biomarker panel shows improved sensitivity for the early detection of ovarian cancer allowing the identification of the most aggressive type II tumours. Br. J. Cancer 2017, 117, 666–674. [Google Scholar] [CrossRef]
- Surinova, S.; Radova, L.; Choi, M.; Srovnal, J.; Brenner, H.; Vitek, O.; Hajduch, M.; Aebersold, R.; Brenner, H.; Kloor, M.; et al. Non-invasive prognostic protein biomarker signatures associated with colorectal cancer. EMBO Mol. Med. 2015, 7, 1153–1165. [Google Scholar] [CrossRef]
- Høgdall, E.; Fung, E.T.; Christensen, I.J.; Yip, C.; Nedergaard, L.; Engelholm, S.A.; Risum, S.; Petri, A.L.; Lundvall, L.; Lomas, L.; et al. Proteomic biomarkers for overall and progression-free survival in ovarian cancer patients. Proteom. Clin. Appl. 2010, 4, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [PubMed]
- Wulfkuhle, J.; Espina, V.; Liotta, L.; Petricoin, E. Genomic and proteomic technologies for individualisation and improvement of cancer treatment. Eur. J. Cancer 2004, 40, 2623–2632. [Google Scholar] [CrossRef]
- Burns, J.; Wilding, C.P.; Jones, R.L.; Huang, P.H. Proteomic research in sarcomas—Current status and future opportunities. Semin. Cancer Biol. 2020, 61, 56–70. [Google Scholar] [CrossRef]
- Esteva, F.J.; Hortobagyi, G.N.; Eifel, P.; Axelson, J.; Costa, J.; Crowley, J.; Curran, W.; Deshler, A.; Fulton, S.; Hendricks, C.; et al. Prognostic molecular markers in early breast cancer. Breast Cancer Res. 2004, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.C.; Aebersold, R. Data-independent acquisition-based SWATH—MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2018, 14, e8126. [Google Scholar] [CrossRef]
- López-Sánchez, L.M.; Jiménez-Izquierdo, R.; Peñarando, J.; Mena, R.; Guil-Luna, S.; Toledano, M.; Conde, F.; Villar, C.; Díaz, C.; Ortea, I.; et al. SWATH-based proteomics reveals processes associated with immune evasion and metastasis in poor prognosis colorectal tumours. J. Cell. Mol. Med. 2019, 23, 8219–8232. [Google Scholar] [CrossRef]
- Bouchal, P.; Schubert, O.T.; Faktor, J.; Capkova, L.; Imrichova, H.; Zoufalova, K.; Paralova, V.; Hrstka, R.; Liu, Y.; Ebhardt, H.A.; et al. Breast Cancer Classification Based on Proteotypes Obtained by SWATH Mass Spectrometry. Cell Rep. 2019, 28, 832–843. [Google Scholar] [CrossRef]
- Njoku, K.; Chiasserini, D.; Whetton, A.D.; Crosbie, E.J. Proteomic biomarkers for the detection of endometrial cancer. Cancers 2019, 11, 1572. [Google Scholar] [CrossRef]
- Russell, M.R.; Walker, M.J.; Williamson, A.J.K.; Gentry-Maharaj, A.; Ryan, A.; Kalsi, J.; Skates, S.; D’Amato, A.; Dive, C.; Pernemalm, M.; et al. Protein Z: A putative novel biomarker for early detection of ovarian cancer. Int. J. Cancer 2016, 138, 2984–2992. [Google Scholar] [CrossRef]
- Geary, B.; Walker, M.J.; Snow, J.T.; Lee, D.C.H.; Pernemalm, M.; Maleki-Dizaji, S.; Azadbakht, N.; Apostolidou, S.; Barnes, J.; Krysiak, P.; et al. Identification of a Biomarker Panel for Early Detection of Lung Cancer Patients. J. Proteome Res. 2019, 18, 3369–3382. [Google Scholar] [CrossRef]
- Malipatil, N.; Fachim, H.A.; Siddals, K.; Geary, B.; Wark, G.; Porter, N.; Anderson, S.; Donn, R.; Harvie, M.; Whetton, A.D.; et al. Data Independent Acquisition Mass Spectrometry Can Identify Circulating Proteins That Predict Future Weight Loss with a Diet and Exercise Programme. J. Clin. Med. 2019, 8, 141. [Google Scholar] [CrossRef]
- Collins, B.C.; Gillet, L.C.; Rosenberger, G.; Röst, H.L.; Vichalkovski, A.; Gstaiger, M.; Aebersold, R. Quantifying protein interaction dynamics by SWATH mass spectrometry: Application to the 14-3-3 system. Nat. Methods 2013, 10, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, J.; Sethi, A.; Li, Q.K.; Chen, L.; Collins, B.; Gillet, L.C.J.; Wollscheid, B.; Zhang, H.; Aebersold, R. Glycoproteomic Analysis of Prostate Cancer Tissues by SWATH Mass Spectrometry Discovers N-acylethanolamine Acid Amidase and Protein Tyrosine Kinase 7 as Signatures for Tumor Aggressiveness. Mol. Cell. Proteom. 2014, 13, 1753–1768. [Google Scholar] [CrossRef]
- Shao, S.; Guo, T.; Aebersold, R. Mass spectrometry-based proteomic quest for diabetes biomarkers. Biochim. Biophys. Acta Proteins Proteom. 2015, 1854, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Ortea, I.; Rodríguez-Ariza, A.; Chicano-Gálvez, E.; Arenas Vacas, M.S.; Jurado Gámez, B. Discovery of potential protein biomarkers of lung adenocarcinoma in bronchoalveolar lavage fluid by SWATH MS data-independent acquisition and targeted data extraction. J. Proteom. 2016, 138, 106–114. [Google Scholar] [CrossRef]
- Rothwell, D.G.; Ayub, M.; Cook, N.; Thistlethwaite, F.; Carter, L.; Dean, E.; Smith, N.; Villa, S.; Dransfield, J.; Clipson, A.; et al. Utility of ctDNA to support patient selection for early phase clinical trials: The TARGET study. Nat. Med. 2019, 25, 738–743. [Google Scholar] [CrossRef]
- Magnuson, A.; Bruinooge, S.S.; Singh, H.; Wilner, K.D.; Jalal, S.; Lichtman, S.M.; Kluetz, P.G.; Lyman, G.H.; Klepin, H.D.; Fleury, M.E.; et al. Modernizing Clinical Trial Eligibility Criteria: Recommendations of the ASCO- Friends of Cancer Research Performance Status Work Group. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Simcock, R.; Wright, J. Beyond Performance Status. Clin. Oncol. 2020, 32, 553–561. [Google Scholar] [CrossRef]
- Liu, Y.; Buil, A.; Collins, B.C.; Gillet, L.C.; Blum, L.C.; Cheng, L.-Y.; Vitek, O.; Mouritsen, J.; Lachance, G.; Spector, T.D.; et al. Quantitative variability of 342 plasma proteins in a human twin population. Mol. Syst. Biol. 2015, 11, 786. [Google Scholar] [CrossRef]
- Zhang, Y.-S.; Han, L.; Yang, C.; Liu, Y.-J.; Zhang, X.-M. Prognostic Value of LRG1 in Breast Cancer: A Retrospective Study. Oncol. Res. Treat. 2021, 44, 36–42. [Google Scholar] [CrossRef]
- Wang, M.; Wang, J.; Jiang, H. Diagnostic value of apolipoprotein C-I, transthyretin and apolipoprotein C-III in gastric cancer. Oncol. Lett. 2019, 17, 3227–3232. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Jia, D.; Wong, C.-M.; Oi-Lin Ng, I.; Zhang, Z.; Liu, L.; Wang, Q.; Zhao, F.; Li, J.; Yao, M.; et al. SERPINA5 inhibits tumor cell migration by modulating the fibronectin-integrin β1 signaling pathway in hepatocellular carcinoma. Mol. Oncol. 2014, 8, 366–377. [Google Scholar] [CrossRef]
- Zeng, W.J.; Yang, Y.L.; Wen, Z.P.; Chen, P.; Chen, X.P.; Gong, Z.C. Identification of gene expression and DNA methylation of SERPINA5 and TIMP1 as novel prognostic markers in lower-grade gliomas. PeerJ 2020, 2020. [Google Scholar] [CrossRef]
- Messner, C.B.; Demichev, V.; Wendisch, D.; Michalick, L.; White, M.; Freiwald, A.; Textoris-Taube, K.; Vernardis, S.I.; Egger, A.S.; Kreidl, M.; et al. Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cell Syst. 2020, 11, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Collins, B.C.; Hunter, C.L.; Liu, Y.; Schilling, B.; Rosenberger, G.; Bader, S.L.; Chan, D.W.; Gibson, B.W.; Gingras, A.C.; Held, J.M.; et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name (Correlation) | p Value | Hazard Ratio | Lower 95% CI | Upper 95% CI |
|---|---|---|---|---|
| A2GL (+) | 0.001 | 2.328 | 1.394 | 3.887 |
| APOC3 (−) | 0.023 | 0.7 | 0.514 | 0.952 |
| IPSP (−) | 0.013 | 0.437 | 0.227 | 0.84 |
| Predictive Values | Discovery | Validation |
|---|---|---|
| WS PPV | 96% | 74% |
| PS PPV | 47% | 54% |
| WS NPV | 60% | 59% |
| PS NPV | 60% | 59% |
| Scoring System | Score | Discovery n | Discovery Median OS (Days) | Validation n | Validation Median OS (Days) |
|---|---|---|---|---|---|
| Wellness Score | 0 | 34 | 377 | 35 | 407 |
| 1 | 21 | 148 | 42 | 167 | |
| p value | 5.5 × 10−4 | 4.5 × 10−4 | |||
| PPM | 0 | 6 | 1052 | 3 | 593 |
| 1 | 16 | 572 | 16 | 407 | |
| 2 | 18 | 257 | 36 | 283 | |
| 3 | 15 | 112 | 22 | 153 | |
| p value | 4.4 × 10−6 | 7.4 × 10−7 | |||
| PEPS | 0 | 16 | 467 | 14 | 486 |
| 1 | 20 | 377 | 36 | 298 | |
| 2 | 19 | 130 | 27 | 153 | |
| p value | 5.3 × 10−5 | 7.5 × 10−5 | |||
| PS | 0 | 18 | 628 | 29 | 298 |
| 1 (+2) | 37 | 296 | 49 | 196 | |
| p value | 0.001 | 0.084 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geary, B.; Peat, E.; Dransfield, S.; Cook, N.; Thistlethwaite, F.; Graham, D.; Carter, L.; Hughes, A.; Krebs, M.G.; Whetton, A.D. Discovery and Evaluation of Protein Biomarkers as a Signature of Wellness in Late-Stage Cancer Patients in Early Phase Clinical Trials. Cancers 2021, 13, 2443. https://doi.org/10.3390/cancers13102443
Geary B, Peat E, Dransfield S, Cook N, Thistlethwaite F, Graham D, Carter L, Hughes A, Krebs MG, Whetton AD. Discovery and Evaluation of Protein Biomarkers as a Signature of Wellness in Late-Stage Cancer Patients in Early Phase Clinical Trials. Cancers. 2021; 13(10):2443. https://doi.org/10.3390/cancers13102443
Chicago/Turabian StyleGeary, Bethany, Erin Peat, Sarah Dransfield, Natalie Cook, Fiona Thistlethwaite, Donna Graham, Louise Carter, Andrew Hughes, Matthew G. Krebs, and Anthony D. Whetton. 2021. "Discovery and Evaluation of Protein Biomarkers as a Signature of Wellness in Late-Stage Cancer Patients in Early Phase Clinical Trials" Cancers 13, no. 10: 2443. https://doi.org/10.3390/cancers13102443
APA StyleGeary, B., Peat, E., Dransfield, S., Cook, N., Thistlethwaite, F., Graham, D., Carter, L., Hughes, A., Krebs, M. G., & Whetton, A. D. (2021). Discovery and Evaluation of Protein Biomarkers as a Signature of Wellness in Late-Stage Cancer Patients in Early Phase Clinical Trials. Cancers, 13(10), 2443. https://doi.org/10.3390/cancers13102443