
Endosomal mTORC2 Is Required for Phosphoinositide-Dependent AKT Activation in Platelet-Derived Growth Factor-Stimulated Glioma Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Transfection, and Reagents
2.2. Plasmids and siRNAs
2.3. Immunoblot Analysis and Antibodies
2.4. Analysis of PDGFR Endocytosis
2.5. In Vitro GST Pull-Down Assay for Purification of Rab5-GTP-Associated Endosomes
2.6. Rapamycin (RAPA)-Mediated Depletion of Phosphoinositide in Endosomes
2.7. Immunofluorescence and Live-Cell Confocal Imaging
2.8. Nikon-Structured Illumination Microscopy (N-SIM)
2.9. AKT Kinase Assay
2.10. CellMask Plasma Membrane Staining
2.11. Statistics and Reproducibility
3. Results
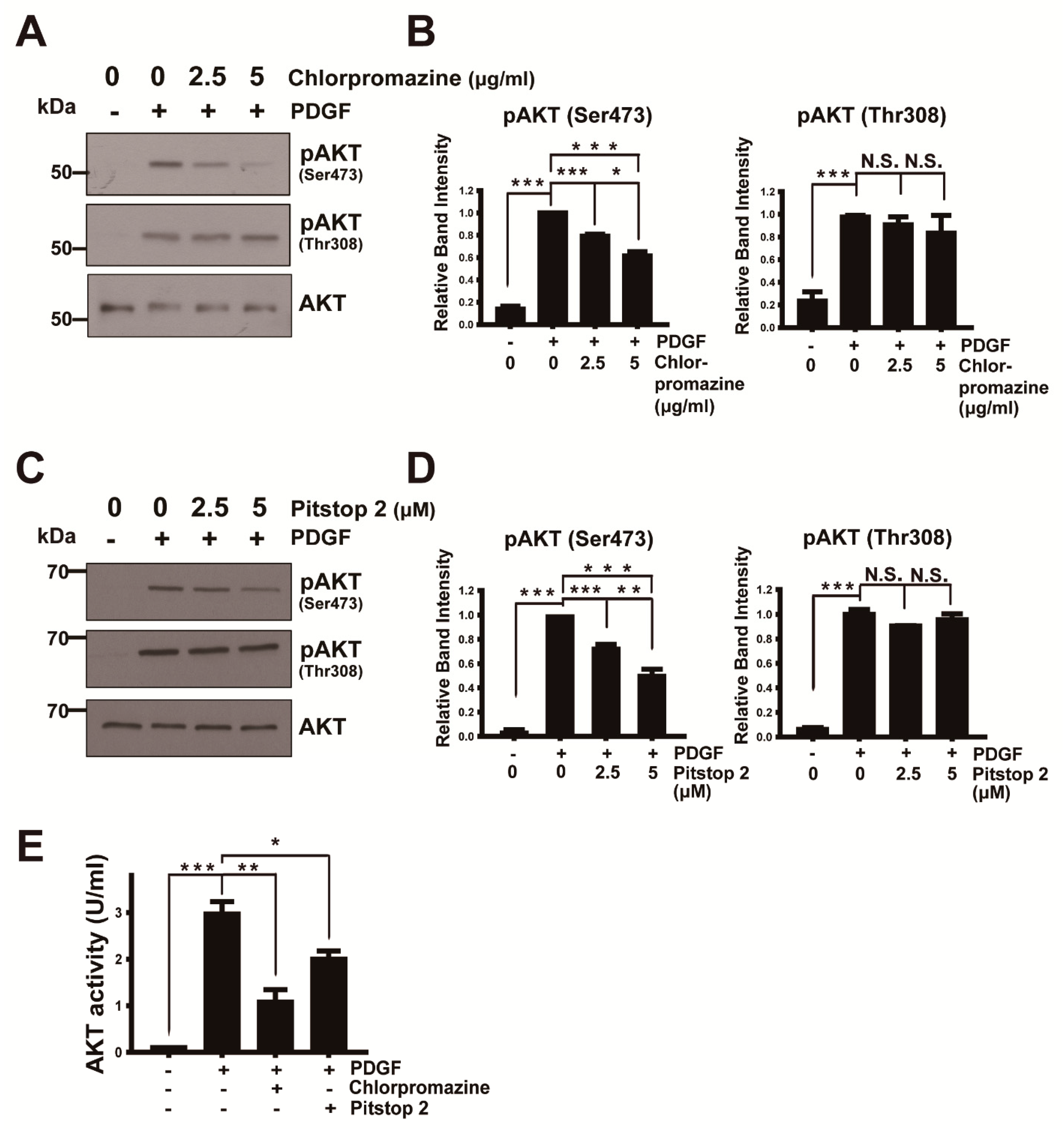
3.1. AKT Activation via Phosphorylation at Ser473 Requires Endocytosis
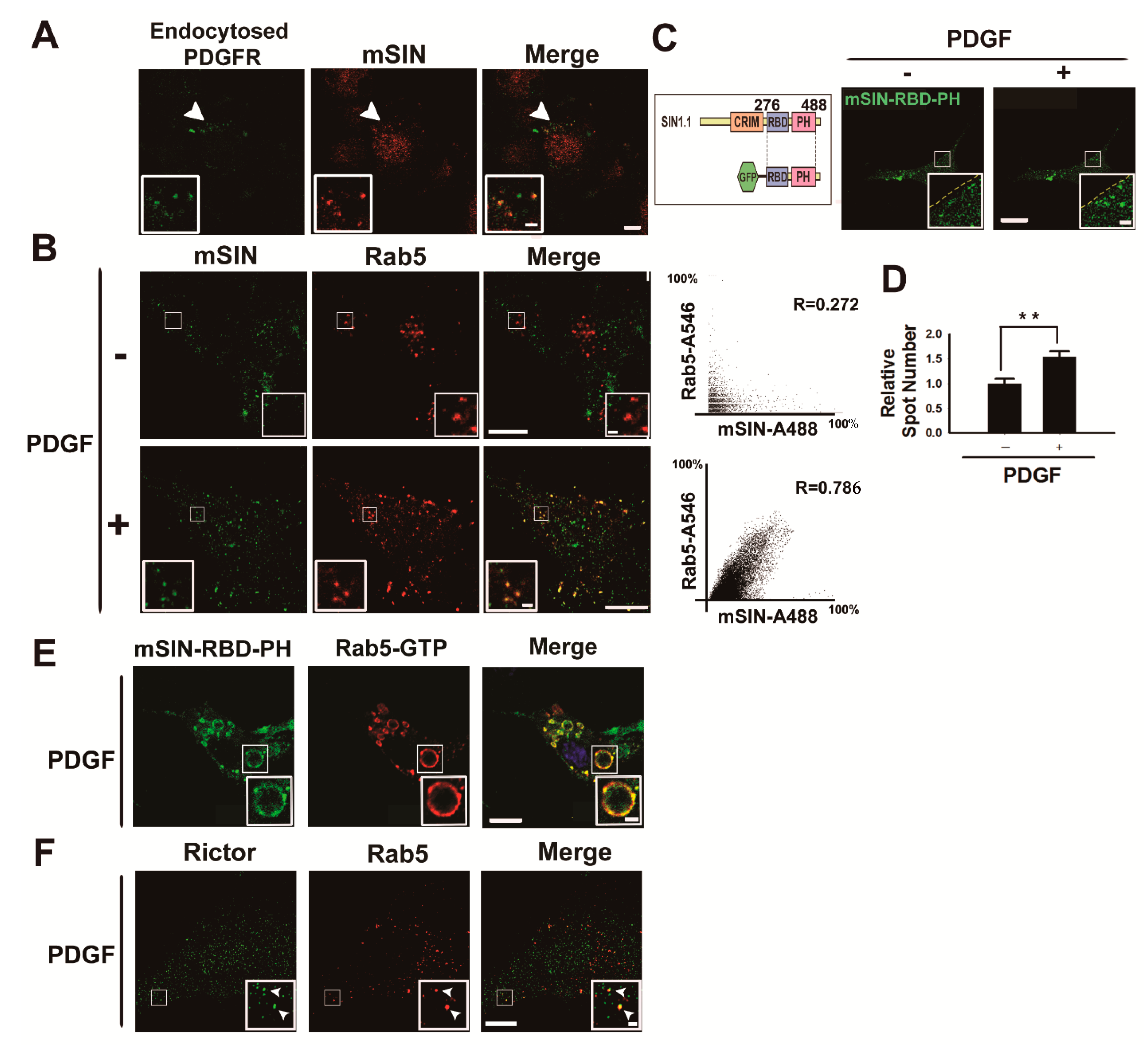
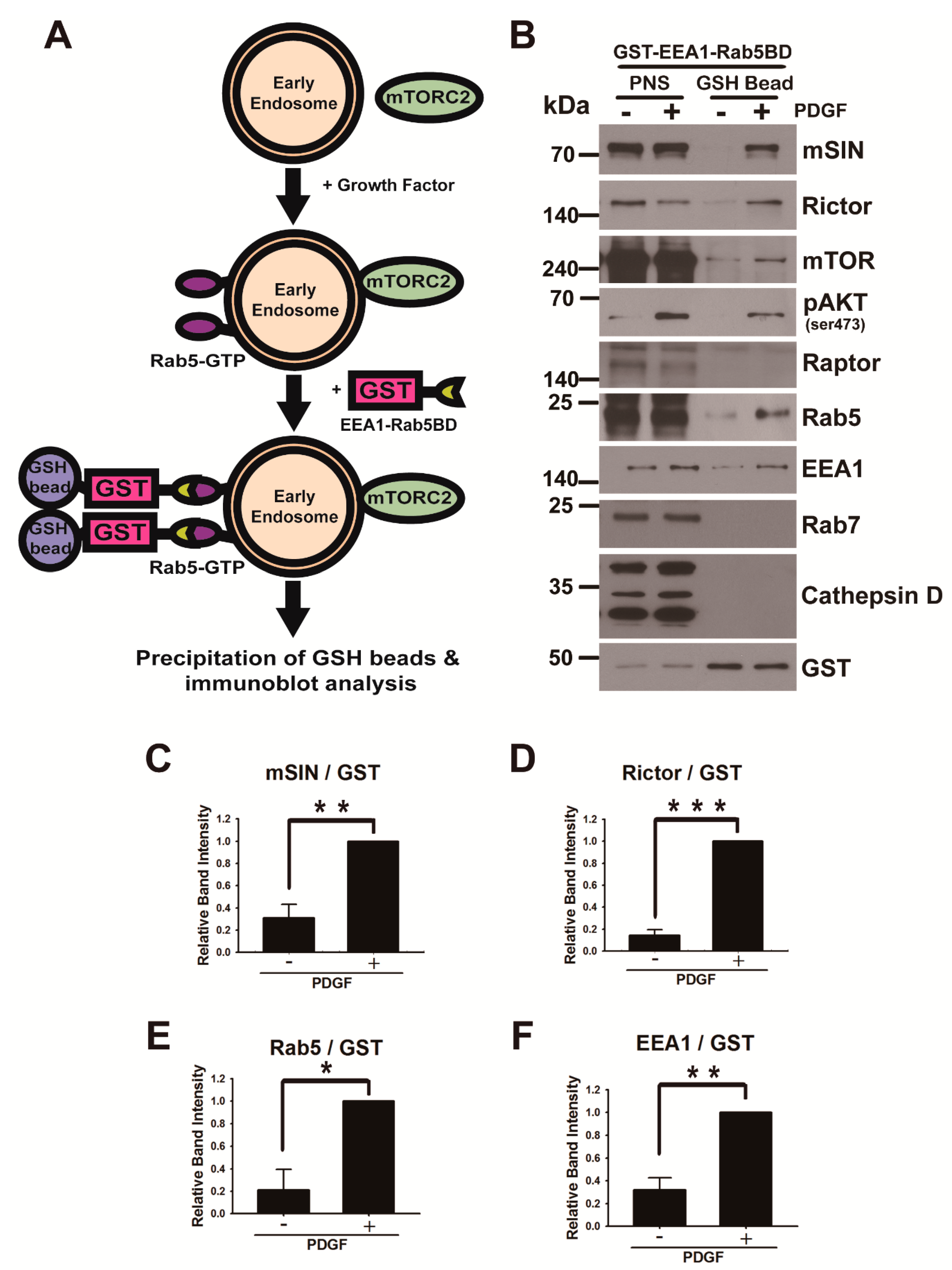
3.2. mTORC2 Is Recruited into the Early Endosome upon Growth Factor Stimulation
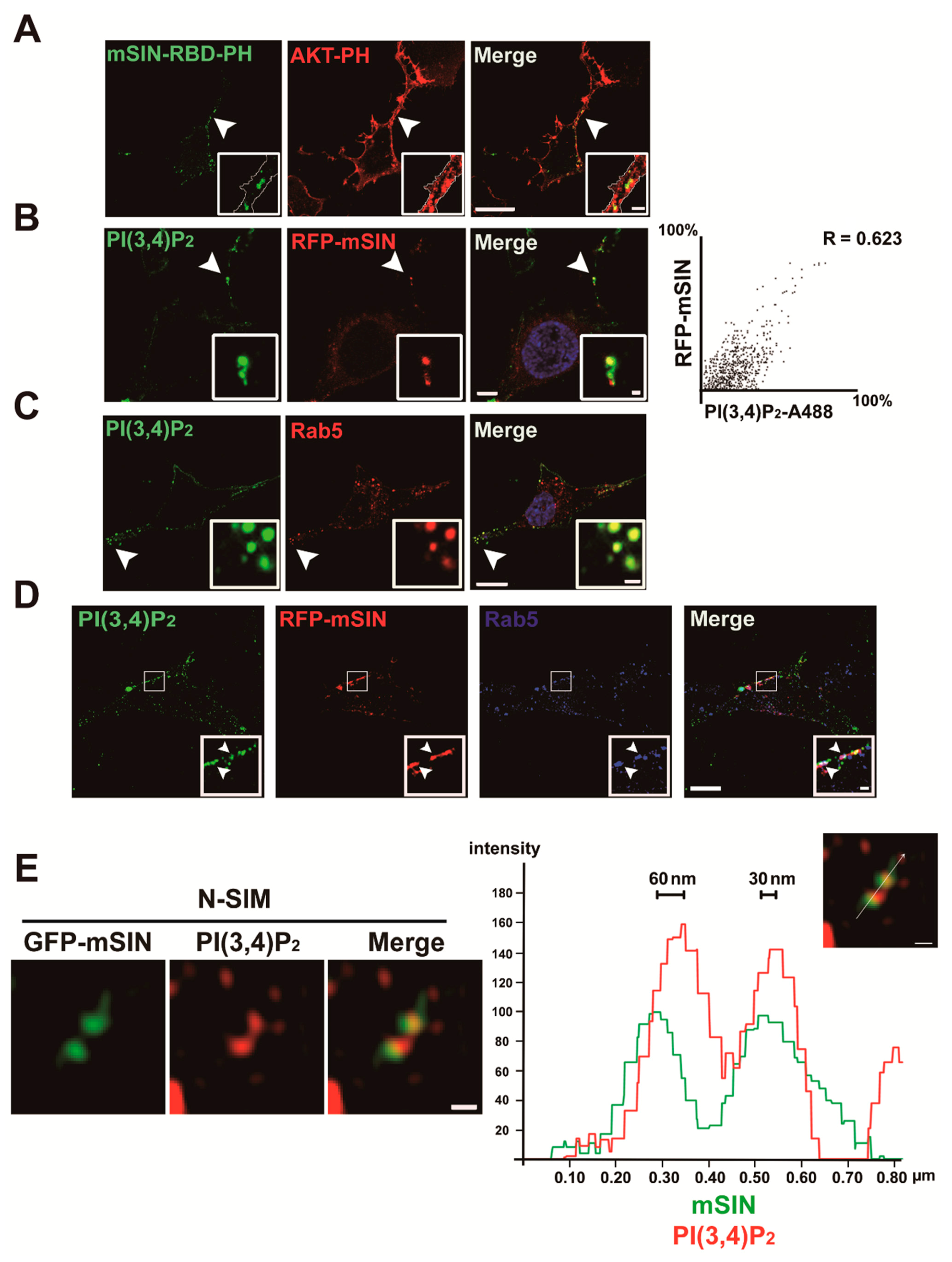
3.3. Localization of mTORC2 to Early Endosomes Is Mediated by PtdIns(3,4)P2
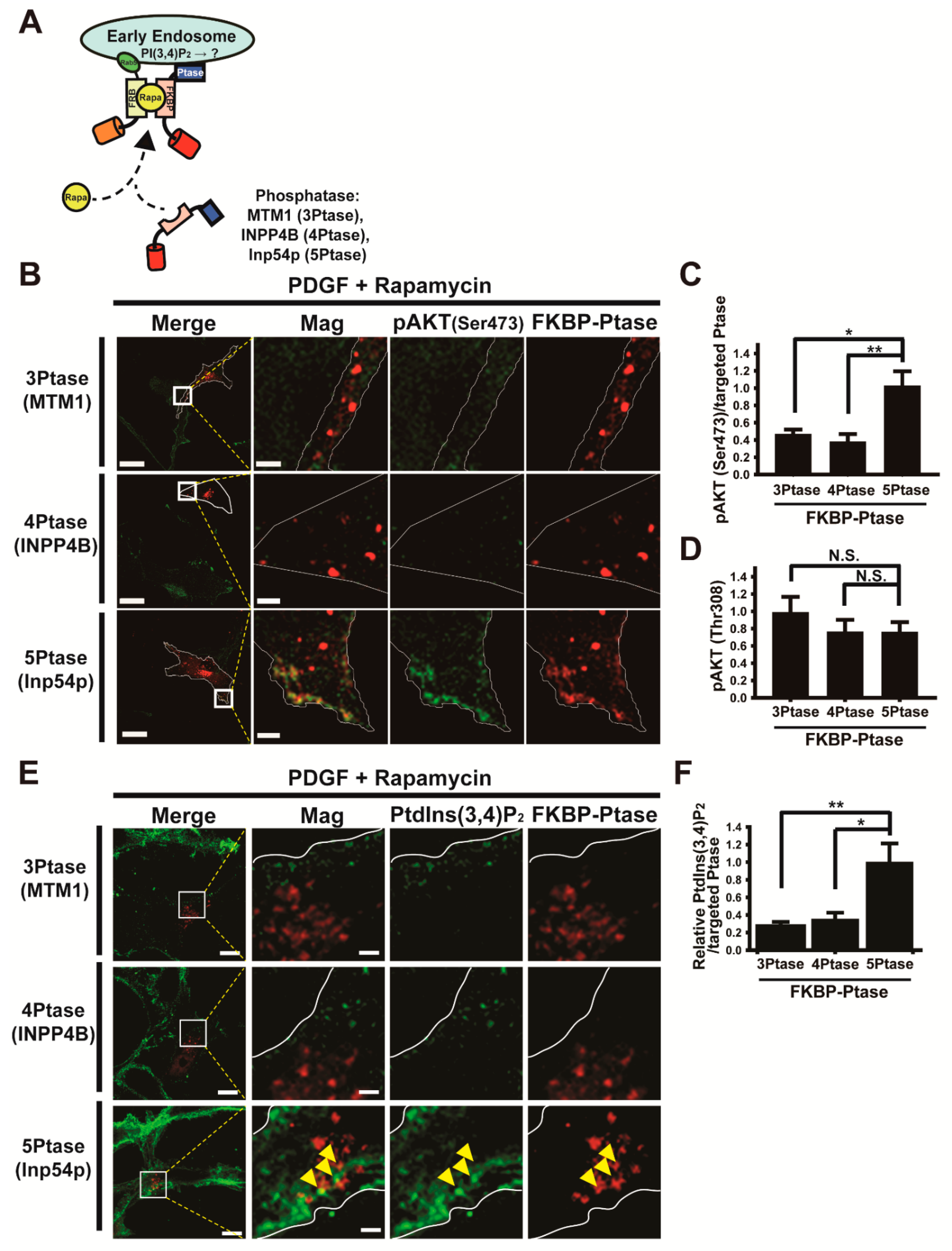
3.4. Forced Depletion of Endosomal PtdIns(3,4)P2 by Targeted Phosphatases Causes Reduced AKT Phosphorylation at Ser473
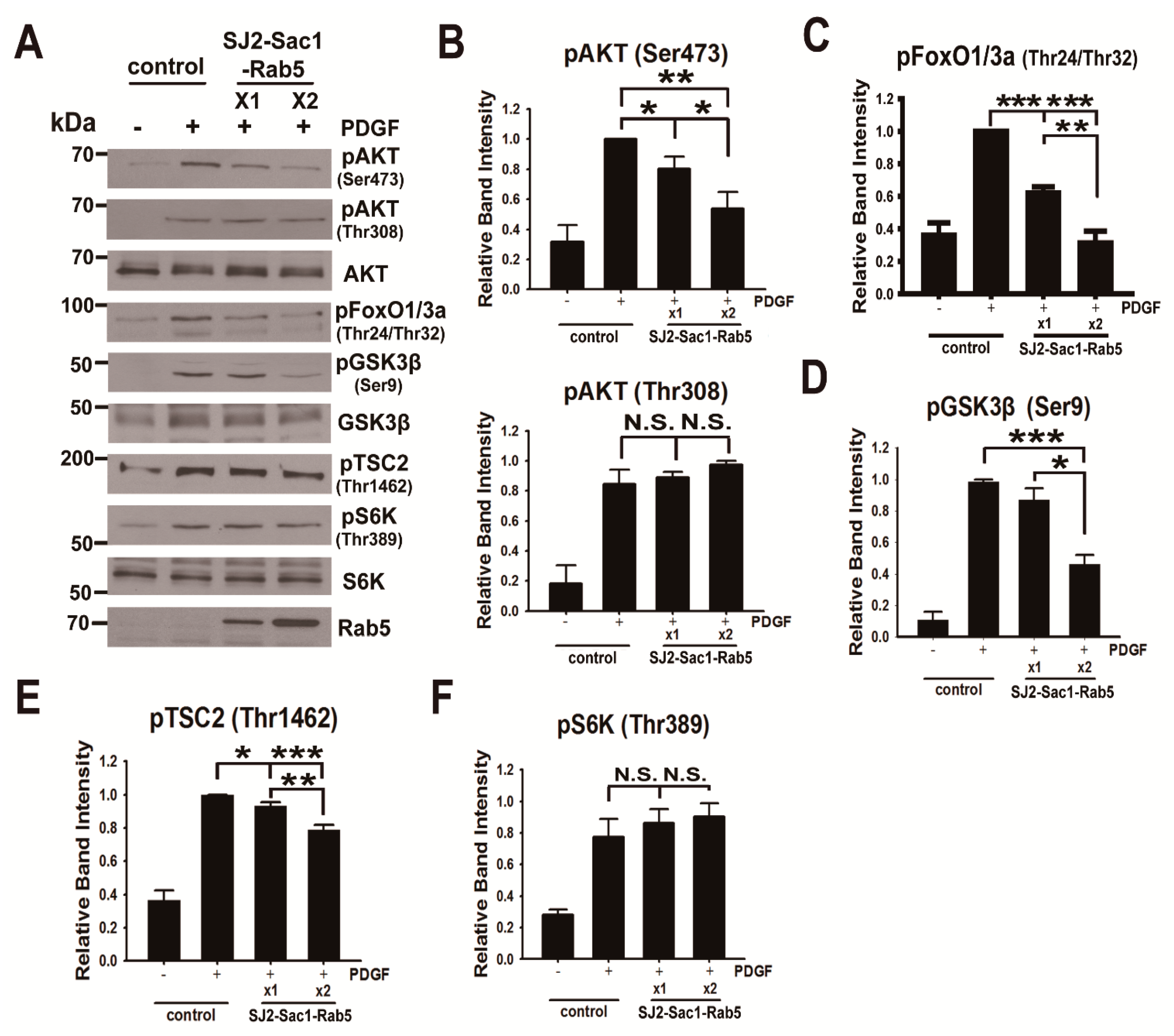
3.5. Endosomal Phosphoinositide Accumulation Is Required for the Activation of a Subset of AKT Substrates
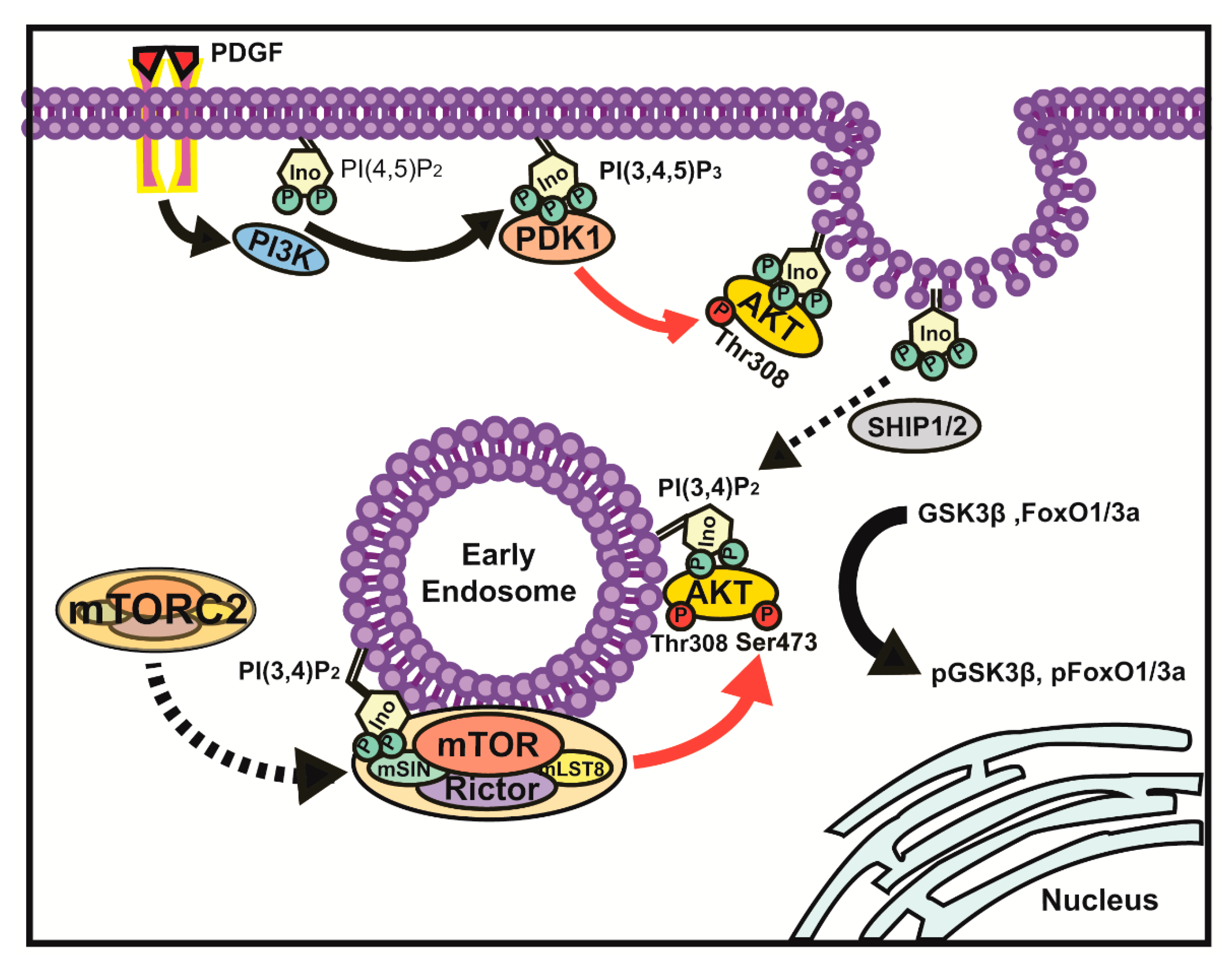
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bellacosa, A.; Testa, J.R.; Staal, S.P.; Tsichlis, P.N. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science 1991, 254, 274–277. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Stephens, L.; Anderson, K.; Stokoe, D.; Erdjument-Bromage, H.; Painter, G.F.; Holmes, A.B.; Gaffney, P.R.J.; Reese, C.B.; McCormick, F.; Tempst, P.; et al. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-Trisphosphate-dependent activation of protein kinase B. Science 1998, 279, 710–714. [Google Scholar] [CrossRef]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5)P3-dependent activation of the mTORC2 kinase complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef]
- Jia, R.; Bonifacino, J.S. Lysosome positioning influences mTORC2 and AKT signaling. Mol. Cell 2019, 75, 26–38.e3. [Google Scholar] [CrossRef]
- Thapa, N.; Chen, M.; Horn, H.T.; Choi, S.; Wen, T.; Anderson, R.A. Phosphatidylinositol 3-kinase signalling is spatially organized at endosomal compartments by microtubule-associated protein 4. Nat. Cell Biol. 2020, 22, 1357–1370. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Stanfel, M.N.; Shamieh, L.S.; Kaeberlein, M.; Kennedy, B.K. The TOR pathway comes of age. Biochim. Biophys. Acta BBA Gen. Subj. 2009, 1790, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Guan, K.-L. Expanding mTOR signaling. Cell Res. 2007, 17, 666–681. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The rag GTPases Bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef]
- García-Martínez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Ikenoue, T.; Inoki, K.; Yang, Q.; Zhou, X.; Guan, K.-L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008, 27, 1919–1931. [Google Scholar] [CrossRef]
- Jacinto, E.; Lorberg, A. TOR regulation of AGC kinases in yeast and mammals. Biochem. J. 2008, 410, 19–37. [Google Scholar] [CrossRef]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mTORC2 by association with the ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, D.; Walther, T.C. TORC2 plasma membrane localization is essential for cell viability and restricted to a distinct domain. Mol. Biol. Cell 2009, 20, 1565–1575. [Google Scholar] [CrossRef]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef]
- James, S.R.; Downes, C.P.; Gigg, R.; Grove, S.J.A.; Holmes, A.B.; Alessi, D.R. Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subsequent activation. Biochem. J. 1996, 315, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Dibble, C.C.; Matsuzaki, M.; Manning, B.D. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol. Cell. Biol. 2008, 28, 4104–4115. [Google Scholar] [CrossRef]
- Ebner, M.; Sinkovics, B.; Szczygieł, M.; Ribeiro, D.W.; Yudushkin, I. Localization of mTORC2 activity inside cells. J. Cell Biol. 2017, 216, 343–353. [Google Scholar] [CrossRef]
- Ma, K.; Cheung, S.M.; Marshall, A.J.; Duronio, V. PI(3,4,5)P3 and PI(3,4)P2 levels correlate with PKB/akt phosphorylation at Thr308 and Ser473, respectively; PI(3,4)P2 levels determine PKB activity. Cell. Signal. 2008, 20, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Scheid, M.P.; Huber, M.; Damen, J.E.; Hughes, M.; Kang, V.; Neilsen, P.; Prestwich, G.D.; Krystal, G.; Duronio, V. Phosphatidylinositol (3,4,5)P3 is essential but not sufficient for Protein Kinase B (PKB) activation; phosphatidylinositol (3,4)P2 Is Required for PKB phosphorylation at Ser-473. J. Biol. Chem. 2002, 277, 9027–9035. [Google Scholar] [CrossRef]
- Ghosh, D.; Srivastava, G.P.; Xu, D.; Schulz, L.C.; Roberts, R.M. A link between SIN1 (MAPKAP1) and poly(rC) binding protein 2 (PCBP2) in counteracting environmental stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11673–11678. [Google Scholar] [CrossRef]
- Kang, S.W.; Chae, H.Z.; Seo, M.S.; Kim, K.; Baines, I.C.; Rhee, S.G. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-α. J. Biol. Chem. 1998, 273, 6297–6302. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.; Jo, D.H.; Kim, J.H.; Kim, S.R.; Kang, D.; Hwang, D.; Chung, J. Bispecific anti-mPDGFRβ x cotinine scFv-Cκ-scFv fusion protein and cotinine-duocarmycin can form antibody-drug conjugate-like complexes that exert cytotoxicity against mPDGFRβ expressing cells. Methods 2019, 154, 125–135. [Google Scholar] [CrossRef]
- Lim, J.M.; Lee, K.S.; Woo, H.A.; Kang, D.; Rhee, S.G. Control of the pericentrosomal H2O2 level by peroxiredoxin I is critical for mitotic progression. J. Cell Biol. 2015, 210, 23–33. [Google Scholar] [CrossRef]
- Posor, Y.; Eichhorn-Gruenig, M.; Puchkov, D.; Schöneberg, J.; Ullrich, A.; Lampe, A.; Müller, R.; Zarbakhsh, S.; Gulluni, F.; Hirsch, E.; et al. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nat. Cell Biol. 2013, 499, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Oakley, F.D.; Abbott, D.; Li, Q.; Engelhardt, J.F. Signaling components of redox active endosomes: The redoxosomes. Antioxidants Redox Signal. 2009, 11, 1313–1333. [Google Scholar] [CrossRef]
- Villaseñor, R.; Kalaidzidis, Y.; Zerial, M. Signal processing by the endosomal system. Curr. Opin. Cell Biol. 2016, 39, 53–60. [Google Scholar] [CrossRef]
- Furnari, F.B.; Lin, H.; Huang, H.J.S.; Cavenee, W.K. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc. Natl. Acad. Sci. USA 1997, 94, 12479–12484. [Google Scholar] [CrossRef] [PubMed]
- Park, M.-J.; Kim, M.-S.; Park, I.-C.; Kang, H.-S.; Yoo, H.; Park, S.H.; Rhee, C.H.; Hong, S.-I.; Lee, S.-H. PTEN suppresses hyaluronic acid-induced matrix metalloproteinase-9 expression in U87MG glioblastoma cells through focal adhesion kinase dephosphorylation. Cancer Res. 2002, 62, 6318–6322. [Google Scholar] [PubMed]
- Wang, L.H.; Rothberg, K.G.; Anderson, R.G. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J. Cell Biol. 1993, 123, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Von Kleist, L.; Stahlschmidt, W.; Bulut, H.; Gromova, K.; Puchkov, D.; Robertson, M.J.; MacGregor, K.A.; Tomilin, N.; Pechstein, A.; Chau, N.; et al. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell 2011, 146, 471–484. [Google Scholar] [CrossRef]
- Sigismund, S.; Argenzio, E.; Tosoni, D.; Cavallaro, E.; Polo, S.; Di Fiore, P.P. Clathrin-mediated internalization is essential for sustained egfr signaling but dispensable for degradation. Dev. Cell 2008, 15, 209–219. [Google Scholar] [CrossRef]
- Vieira, A.V.; Lamaze, C.; Schmid, S.L. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science 1996, 274, 2086–2089. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 Maintains rictor-mTOR Complex integrity and regulates akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef]
- Barbieri, M.A.; Roberts, R.L.; Gumusboga, A.; Highfield, H.; Alvarez-Dominguez, C.; Wells, A.; Stahl, P.D. Epidermal growth factor and membrane trafficking. J. Cell Biol. 2000, 151, 539–550. [Google Scholar] [CrossRef]
- Pan, D.; Matsuura, Y. Structures of the pleckstrin homology domain ofSaccharomyces cerevisiaeAvo1 and its human orthologue Sin1, an essential subunit of TOR complex 2. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 386–392. [Google Scholar] [CrossRef]
- Zerial, M.; McBride, H.M. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2001, 2, 107–117. [Google Scholar] [CrossRef]
- Mishra, A.; Eathiraj, S.; Corvera, S.; Lambright, D.G. Structural basis for Rab GTPase recognition and endosome tethering by the C2H2 zinc finger of Early Endosomal Autoantigen 1 (EEA1). Proc. Natl. Acad. Sci. USA 2010, 107, 10866–10871. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in growth, metabolism, and disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Gewinner, C.; Wang, Z.C.; Richardson, A.; Teruya-Feldstein, J.; Etemadmoghadam, D.; Bowtell, D.; Barretina, J.; Lin, W.M.; Rameh, L.; Salmena, L.; et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits pi3k signaling. Cancer Cell 2009, 16, 115–125. [Google Scholar] [CrossRef]
- Braccini, L.; Ciraolo, E.; Campa, C.C.; Perino, A.; Longo, D.L.; Tibolla, G.; Pregnolato, M.; Cao, Y.; Tassone, B.; Damilano, F.; et al. PI3K-C2γ is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat. Commun. 2015, 6, 7400. [Google Scholar] [CrossRef]
- Hagiwara, A.; Cornu, M.; Cybulski, N.; Polak, P.; Betz, C.; Trapani, F.; Terracciano, L.; Heim, M.H.; Rüegg, M.A.; Hall, M.N. Hepatic mTORC2 activates glycolysis and lipogenesis through akt, glucokinase, and SREBP1c. Cell Metab. 2012, 15, 725–738. [Google Scholar] [CrossRef]
- Franke, T.F.; Kaplan, D.R.; Cantley, L.C.; Toker, A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 1997, 275, 665–668. [Google Scholar] [CrossRef]
- Jackson, T.; Stephens, L.; Hawkins, P. Receptor specificity of growth factor-stimulated synthesis of 3-phosphorylated inositol lipids in Swiss 3T3 cells. J. Biol. Chem. 1992, 267, 16627–16636. [Google Scholar] [CrossRef]
- Stephens, L.R.; Hughes, K.T.; Irvine, R.F. Pathway of phosphatidylinositol(3,4,5)-trisphosphate synthesis in activated neutrophils. Nat. Cell Biol. 1991, 351, 33–39. [Google Scholar] [CrossRef]
- Leslie, N.R.; Biondi, R.M.; Alessi, D.R.; Leslie, N. Phosphoinositide-regulated kinases and phosphoinositide phosphatases. Chem. Rev. 2001, 101, 2365–2380. [Google Scholar] [CrossRef]
- Dowler, S.; Currie, R.A.; Campbell, D.G.; Deak, M.; Kular, G.; Downes, C.P.; Alessi, D.R. Identification of pleckstrin-homology-domain-containing proteins with novel phosphoinositide-binding specificities. Biochem. J. 2000, 351, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.S.; Maehama, T.; Dixon, J.E. Myotubularin, a protein tyrosine phosphatase mutated in myotubular myopathy, dephosphorylates the lipid second messenger, phosphatidylinositol 3-phosphate. Proc. Natl. Acad. Sci. USA 2000, 97, 8910–8915. [Google Scholar] [CrossRef]
- Norris, F.A.; Auethavekiat, V.; Majerus, P.W. The isolation and characterization of cDNA encoding human and rat brain inositol polyphosphate 4-phosphatase. J. Biol. Chem. 1995, 270, 16128–16133. [Google Scholar] [CrossRef] [PubMed]
- Stolz, L.E.; Kuo, W.J.; Longchamps, J.; Sekhon, M.K.; York, J.D. INP51, a yeast inositol polyphosphate 5-phosphatase required for phosphatidylinositol 4,5-bisphosphate homeostasis and whose absence confers a cold-resistant phenotype. J. Biol. Chem. 1998, 273, 11852–11861. [Google Scholar] [CrossRef]
- Hammond, G.R.; Machner, M.P.; Balla, T. A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. J. Cell Biol. 2014, 205, 113–126. [Google Scholar] [CrossRef]
- Ebner, M.; Lučić, I.; Leonard, T.A.; Yudushkin, I. PI(3,4,5)P3 engagement restricts akt activity to cellular membranes. Mol. Cell 2017, 65, 416–431.e6. [Google Scholar] [CrossRef] [PubMed]
- Goulden, B.D.; Pacheco, J.; Dull, A.; Zewe, J.P.; Deiters, A.; Hammond, G.R. A high-avidity biosensor reveals plasma membrane PI(3,4)P2 is predominantly a class I PI3K signaling product. J. Cell Biol. 2018, 218, 1066–1079. [Google Scholar] [CrossRef]
- Kim, S.-J.; Jeong, M.-J.; Jo, H.-J.; Jung, J.H.; Kaang, B.-K.; Choi, Y.-B.; Kim, J.-H. Identification of postsynaptic phosphatidylinositol-4,5-bisphosphate (PIP2) roles for synaptic plasticity using chemically induced dimerization. Sci. Rep. 2017, 7, 3351. [Google Scholar] [CrossRef]
- Suh, B.-C.; Inoue, T.; Meyer, T.; Hille, B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science 2006, 314, 1454–1457. [Google Scholar] [CrossRef]
- Uchida, Y.; Rutaganira, F.U.; Jullié, D.; Shokat, K.M.; Von Zastrow, M. Endosomal phosphatidylinositol 3-kinase is essential for canonical GPCR signaling. Mol. Pharmacol. 2016, 91, 65–73. [Google Scholar] [CrossRef]
- Thoreen, C.C.; Chantranupong, L.; Keys, H.R.; Wang, T.; Gray, N.S.; Sabatini, D.M. A unifying model for mTORC1-mediated regulation of mRNA translation. Nat. Cell Biol. 2012, 485, 109–113. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.-H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Nemoto, Y.; Wenk, M.R.; Watanabe, M.; Daniell, L.; Murakami, T.; Ringstad, N.; Yamada, H.; Takei, K.; De Camilli, P. Identification and characterization of a synaptojanin 2 splice isoform predominantly expressed in nerve terminals. J. Biol. Chem. 2001, 276, 41133–41142. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P.T. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef]
- Rameh, L.E.; Cantley, L.C. The Role of phosphoinositide 3-kinase lipid products in cell function. J. Biol. Chem. 1999, 274, 8347–8350. [Google Scholar] [CrossRef] [PubMed]
- Schenck, A.; Goto-Silva, L.; Collinet, C.; Rhinn, M.; Giner, A.; Habermann, B.; Brand, M.; Zerial, M. The endosomal protein appl1 mediates akt substrate specificity and cell survival in vertebrate development. Cell 2008, 133, 486–497. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Inoki, K.; Ikenoue, T.; Guan, K.L. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev 2006, 20, 2820–2832. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Heo, S.; Brzostowski, J.; Kang, D. Endosomal mTORC2 Is Required for Phosphoinositide-Dependent AKT Activation in Platelet-Derived Growth Factor-Stimulated Glioma Cells. Cancers 2021, 13, 2405. https://doi.org/10.3390/cancers13102405
Kim S, Heo S, Brzostowski J, Kang D. Endosomal mTORC2 Is Required for Phosphoinositide-Dependent AKT Activation in Platelet-Derived Growth Factor-Stimulated Glioma Cells. Cancers. 2021; 13(10):2405. https://doi.org/10.3390/cancers13102405
Chicago/Turabian StyleKim, Suree, Sukyeong Heo, Joseph Brzostowski, and Dongmin Kang. 2021. "Endosomal mTORC2 Is Required for Phosphoinositide-Dependent AKT Activation in Platelet-Derived Growth Factor-Stimulated Glioma Cells" Cancers 13, no. 10: 2405. https://doi.org/10.3390/cancers13102405
APA StyleKim, S., Heo, S., Brzostowski, J., & Kang, D. (2021). Endosomal mTORC2 Is Required for Phosphoinositide-Dependent AKT Activation in Platelet-Derived Growth Factor-Stimulated Glioma Cells. Cancers, 13(10), 2405. https://doi.org/10.3390/cancers13102405