
The Combination of Single-Cell and Next-Generation Sequencing Can Reveal Mosaicism for BRCA2 Mutations and the Fine Molecular Details of Tumorigenesis


 and
and
Abstract
Simple Summary
Abstract
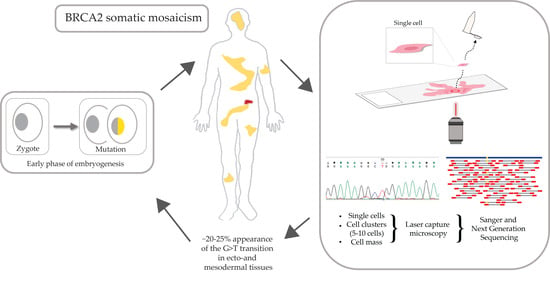
1. Introduction
2. Materials and Methods
2.1. Patient History and the Subjects of the Experiments
- Ovarian cancer patient
- Analyzed tissue types from the cancer patient
- ·
- FFPE block with ovarian serous adenocarcinoma WHO grade 2. for BRCA 2 mutation analysis with a diagnostic purpose;
- ·
- FFPE block with tumor-free ovarian tissue (chosen by an expert) from the cancer patient (called normal);
- ·
- Blood (venous) sample from the cancer patient;
- ·
- Buccal mucosal tissue (swab) from the cancer patient.
- Analyzed tissue type from the offspring of the patient
- ·
- Buccal mucosal tissue.
2.2. Overview of the Workflow
2.3. gDNA Isolation from Tissue and Single Cells/Bulks of 5–10 Cells
2.4. Amplification and Sequencing of the Examined BRCA2 Region
2.5. Data Analysis
3. Results
3.1. Bulk Tissue Sample Sequencing—Comparison of Sanger Sequencing and NGS
3.2. Decreasing the Cell Number and Taking a Deeper Insight—Sanger and NGS Comparison on Microdissected Single Cells/Clusters of 5–10 Cells
3.3. Amplificability and Sequencability of FFPE Samples
3.4. Examination of Single Lymphocyte Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Apostoli, A.J.; Ailles, L. Clonal evolution and tumor-initiating cells: New dimensions in cancer patient treatment. Crit. Rev. Clin. Lab. Sci. 2016, 53, 40–51. [Google Scholar] [CrossRef]
- Black, J.R.M.; McGranahan, N. Genetic and non-genetic clonal diversity in cancer evolution. Nat. Rev. Cancer 2021. [Google Scholar] [CrossRef]
- Scully, R.; Livingston, D.M. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature 2000, 408, 429–432. [Google Scholar] [CrossRef]
- Jasin, M. Homologous repair of DNA damage and tumorigenesis: The BRCA connection. Oncogene 2002, 21, 8981–8993. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002, 108, 171–182. [Google Scholar] [CrossRef]
- Zamborszky, J.; Szikriszt, B.; Gervai, J.Z.; Pipek, O.; Poti, A.; Krzystanek, M.; Ribli, D.; Szalai-Gindl, J.M.; Csabai, I.; Szallasi, Z.; et al. Loss of BRCA1 or BRCA2 markedly increases the rate of base substitution mutagenesis and has distinct effects on genomic deletions. Oncogene 2017, 36, 5085–5086. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef]
- Trenner, A.; Sartori, A.A. Harnessing DNA double-strand break repair for cancer treatment. Front. Oncol. 2019, 9, 1388. [Google Scholar] [CrossRef]
- Mita, P.; Sun, X.; Fenyo, D.; Kahler, D.J.; Li, D.; Agmon, N.; Wudzinska, A.; Keegan, S.; Bader, J.S.; Yun, C.; et al. BRCA1 and S phase DNA repair pathways restrict LINE-1 retrotransposition in human cells. Nat. Struct. Mol. Biol. 2020, 27, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; McCorvie, T.J.; Yates, L.A.; Zhang, X. Structural basis of homologous recombination. Cell Mol. Life Sci. 2020, 77, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Kenemans, P.; Verstraeten, R.A.; Verheijen, R.H. Oncogenic pathways in hereditary and sporadic breast cancer. Maturitas 2008, 61, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Van der Groep, P.; Bouter, A.; van der Zanden, R.; Siccama, I.; Menko, F.H.; Gille, J.J.P.; van Kalken, C.; van der Wall, E.; Verheijen, R.H.M.; van Diest, P.J. Distinction between hereditary and sporadic breast cancer on the basis of clinicopathological data. J. Clin. Pathol. 2006, 59, 611–617. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef]
- Mafficini, A.; Simbolo, M.; Parisi, A.; Rusev, B.; Luchini, C.; Cataldo, I.; Piazzola, E.; Sperandio, N.; Turri, G.; Franchi, M.; et al. BRCA somatic and germline mutation detection in paraffin embedded ovarian cancers by next-generation sequencing. Oncotarget 2016, 7, 1076–1083. [Google Scholar] [CrossRef]
- Koczkowska, M.; Zuk, M.; Gorczynski, A.; Ratajska, M.; Lewandowska, M.; Biernat, W.; Limon, J.; Wasag, B. Detection of somatic BRCA1/2 mutations in ovarian cancer-next-generation sequencing analysis of 100 cases. Cancer Med. 2016, 5, 1640–1646. [Google Scholar] [CrossRef]
- Sunada, S.; Nakanishi, A.; Miki, Y. Crosstalk of DNA double-strand break repair pathways in poly(ADP-ribose) polymerase inhibitor treatment of breast cancer susceptibility gene 1/2-mutated cancer. Cancer Sci. 2018, 109, 893–899. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; van Attikum, H. PARP inhibitor resistance: A Tug-of-war in BRCA-mutated cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef]
- Osher, D.J.; Kushner, Y.B.; Arseneau, J.; Foulkes, W.D. Melphalan as a treatment for BRCA-related ovarian carcinoma: Can you teach an old drug new tricks? J. Clin. Pathol. 2011, 64, 924–926. [Google Scholar] [CrossRef]
- Fan, F.S.; Yang, C.F. Complete response to orally administered melphalan in malignant pleural effusion from an occult female genital organ primary neoplasm with BRCA1/2 mutations: A case report. J. Med. Case Rep. 2018, 12, 122. [Google Scholar] [CrossRef] [PubMed]
- Altschuler, S.J.; Wu, L.F. Cellular heterogeneity: Do differences make a difference? Cell 2010, 141, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Pelkmans, L. Cell Biology. Using cell-to-cell variability--a new era in molecular biology. Science 2012, 336, 425–426. [Google Scholar] [CrossRef] [PubMed]
- Almendro, V.; Marusyk, A.; Polyak, K. Cellular heterogeneity and molecular evolution in cancer. Ann. Rev. Pathol. 2013, 8, 277–302. [Google Scholar] [CrossRef]
- Lim, S.B.; Yeo, T.; Lee, W.D.; Bhagat, A.A.S.; Tan, S.J.; Tan, D.S.W.; Lim, W.T.; Lim, C.T. Addressing cellular heterogeneity in tumor and circulation for refined prognostication. Proc. Natl. Acad. Sci. USA 2019, 116, 17957–17962. [Google Scholar] [CrossRef]
- Espina, V.; Heiby, M.; Pierobon, M.; Liotta, L.A. Laser capture microdissection technology. Exp. Rev. Mol. Diagn. 2007, 7, 647–657. [Google Scholar] [CrossRef]
- Brasko, C.; Smith, K.; Molnar, C.; Farago, N.; Hegedus, L.; Balind, A.; Balassa, T.; Szkalisity, A.; Sukosd, F.; Kocsis, K.; et al. Intelligent image-based in situ single-cell isolation. Nat. Commun. 2018, 9, 226. [Google Scholar] [CrossRef]
- Liu, X.; Harada, S. DNA isolation from mammalian samples. Curr. Protoc. Mol. Biol. 2013, 102. [Google Scholar] [CrossRef]
- Cline, M.S.; Liao, R.G.; Parsons, M.T.; Paten, B.; Alquaddoomi, F.; Antoniou, A.; Baxter, S.; Brody, L.; Cook-Deegan, R.; Coffin, A.; et al. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet. 2018, 14, e1007752. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; McPherson, A.; Laks, E.; Biele, J.; Yap, D.; Wan, A.; Smith, M.A.; Nielsen, C.B.; McAlpine, J.N.; Aparicio, S.; et al. Clonal genotype and population structure inference from single-cell tumor sequencing. Nat. Methods 2016, 13, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.V.; Bisgaard, M.L.; Jonson, L.; Albrechtsen, A.; Filtenborg-Barnkob, B.; Eiberg, H.; Ejlertsen, B.; Nielsen, F.C. Novel de novo BRCA2 mutation in a patient with a family history of breast cancer. BMC Med. Genet. 2008, 9, 58. [Google Scholar] [CrossRef] [PubMed]
- Edwards, E.; Yearwood, C.; Sillibourne, J.; Baralle, D.; Eccles, D. Identification of a de novo BRCA1 mutation in a woman with early onset bilateral breast cancer. Fam. Cancer 2009, 8, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Kwong, A.; Ng, E.K.; Tang, E.Y.; Wong, C.L.; Law, F.B.; Leung, C.P.; Chan, A.; Cheung, M.T.; To, M.Y.; Ma, E.S.; et al. A novel de novo BRCA1 mutation in a Chinese woman with early onset breast cancer. Fam. Cancer 2011, 10, 233–237. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Alhopuro, P.; Vainionpaa, R.; Anttonen, A.K.; Aittomaki, K.; Nevanlinna, H.; Poyhonen, M. Constitutional mosaicism for a BRCA2 mutation as a cause of early-onset breast cancer. Fam. Cancer 2020, 19, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Azzollini, J.; Pesenti, C.; Ferrari, L.; Fontana, L.; Calvello, M.; Peissel, B.; Portera, G.; Tabano, S.; Carcangiu, M.L.; Riva, P.; et al. Revertant mosaicism for family mutations is not observed in BRCA1/2 phenocopies. PLoS ONE 2017, 12, e0171663. [Google Scholar] [CrossRef] [PubMed]
- Lai-Cheong, J.E.; McGrath, J.A.; Uitto, J. Revertant mosaicism in skin: Natural gene therapy. Trends Mol. Med. 2011, 17, 140–148. [Google Scholar] [CrossRef]
- Pasmooij, A.M.; Jonkman, M.F.; Uitto, J. Revertant mosaicism in heritable skin diseases: Mechanisms of natural gene therapy. Discov. Med. 2012, 14, 167–179. [Google Scholar]
- Gajecka, M. Unrevealed mosaicism in the next-generation sequencing era. Mol. Genet. Genom. 2016, 291, 513–530. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Tissue Type | Sanger Sequencing (Yellow Box Indicates Position 7795 of BRCA2) | Next-Generation Sequencing (% of Mutant T Instead of Wild-Type G at Position 7795 of BRCA2) |
|---|---|---|
| Tumor 1 |  | 78% |
| Tumor 2 |  | 77% |
| Buccal musoca 1 |  | 23% |
| Buccal musoca 2 |  | 21% |
| Normal |  | 17% |
| Blood |  | 26% |
| Buccal mucosa (daughter) |  | 0% |
| Sample Type (Approximate Number of Cells) | Sanger Sequencing (Yellow Box Indicates Position 7795 of BRCA2) | Presenting Allele(s) (At Position 7795 of BRCA2) |
|---|---|---|
| Normal (5–10 cells) |  | G/T |
| Normal (single cell) |  | G |
| Normal (single cell) |  | G |
| Normal (single cell) |  | G/T |
| Normal (single cell) |  | G |
| Normal (single cell) |  | G |
| Tumor (5–10 cells) |  | T |
| ID | Tissue/Cell | T | G |
|---|---|---|---|
| Norm-01 | Normal 1 | - | 100% |
| Norm-02 | Normal 1 | 7% | 92% |
| Norm-03 | Normal 1 | 9% | 90% |
| Norm-04 | Normal 1 | 79% | 21% |
| Norm-05 | Normal 1 | 57% | 42% |
| Norm-06 | Normal 1 | 94% | 5% |
| Norm-07 | Normal 1 | - | 100% |
| Norm-08 | Normal 1 | - | 100% |
| Norm-09 | Normal 1 | 53% | 46% |
| Norm-10 | Normal 1 | - | 100% |
| Norm-11 | Normal 1 | - | 100% |
| Norm-12 | Normal 1 | 7% | 92% |
| Norm_cl-01 | Normal 5–10 | 43% | 57% |
| Tum_cl-01 | Tumor 5–10 | 82% | 18% |
| Tum_cl-02 | Tumor 5–10 | 2% | 97% |
| Tum_cl-03 | Tumor 5–10 | 87% | 12% |
| Tum_cl-04 | Tumor 5–10 | 99% | - |
| Tum_cl-05 | Tumor 5–10 | 4% | 95% |
| Tissue Type | Cell Number | Sample Number | Amplification | Sequencing | ||
|---|---|---|---|---|---|---|
| FFPE | pcs | pcs | pcs | % | pcs | % |
| Normal tissue | 5–10 | 2 | 1 | 50 | 1 | 100 |
| Normal tissue | 1 | 20 | 12 | 60 | 12 | 100 |
| Tumor tissue | 5–10 | 10 | 5 | 50 | 5 | 100 |
| Tissue Type | Cell Number | Sample Number | Amplification | Sequencing | ||
|---|---|---|---|---|---|---|
| pcs | pcs | pcs | % | pcs | % | |
| Lymphocyte | 1 | 40 | 16 | 40 | 13 | 80 |
| Lymphocyte ID | G | T |
|---|---|---|
| Lymp_Taq-01 | 88% | 12% |
| Lymp_Taq-02 | 99% | - |
| Lymp_Taq-03 | 99% | - |
| Lymp_Taq-04 | 99% | - |
| Lymp_Taq-05 * | - | - |
| Lymp_Taq-06 * | - | - |
| Lymp_Taq-07 | 65% | 34% |
| Lymp_Taq-08 | - | 99% |
| Lymp_Taq-09 | 99% | - |
| Lymp_Taq-10 * | - | - |
| Lymp_Taq-11 | 99% | - |
| Lymp_Phu-01 | 99% | - |
| Lymp_Phu-02 | 4% | 96% |
| Lymp_Phu-03 | 58% | 41% |
| Lymp_Phu-04 | - | 99% |
| Lymp_Phu-05 | 34% | 65% |
| Sum | 64.80% | 34.30% |
| Tissue Type | Microdissection | Macrodissection | |||
|---|---|---|---|---|---|
| Cell Number/ Reaction | Number of Samples | Genotype | |||
| Mutant Allele | |||||
| T (G) | |||||
| FFPE | Ovarian normal tissue | 1 | 12 | 26% | 17% |
| Ovarian tumor tissue | 5–10 | 5 | 55% | 78% | |
| Native | Lymphocyte | 1 | 13 | 35% | 26% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gráf, A.; Enyedi, M.Z.; Pintér, L.; Kriston-Pál, É.; Jaksa, G.; Bálind, Á.; Ezer, É.; Horváth, P.; Sükösd, F.; Kiss, E.; et al. The Combination of Single-Cell and Next-Generation Sequencing Can Reveal Mosaicism for BRCA2 Mutations and the Fine Molecular Details of Tumorigenesis. Cancers 2021, 13, 2354. https://doi.org/10.3390/cancers13102354
Gráf A, Enyedi MZ, Pintér L, Kriston-Pál É, Jaksa G, Bálind Á, Ezer É, Horváth P, Sükösd F, Kiss E, et al. The Combination of Single-Cell and Next-Generation Sequencing Can Reveal Mosaicism for BRCA2 Mutations and the Fine Molecular Details of Tumorigenesis. Cancers. 2021; 13(10):2354. https://doi.org/10.3390/cancers13102354
Chicago/Turabian StyleGráf, Alexandra, Márton Zsolt Enyedi, Lajos Pintér, Éva Kriston-Pál, Gábor Jaksa, Árpád Bálind, Éva Ezer, Péter Horváth, Farkas Sükösd, Ernő Kiss, and et al. 2021. "The Combination of Single-Cell and Next-Generation Sequencing Can Reveal Mosaicism for BRCA2 Mutations and the Fine Molecular Details of Tumorigenesis" Cancers 13, no. 10: 2354. https://doi.org/10.3390/cancers13102354
APA StyleGráf, A., Enyedi, M. Z., Pintér, L., Kriston-Pál, É., Jaksa, G., Bálind, Á., Ezer, É., Horváth, P., Sükösd, F., Kiss, E., & Haracska, L. (2021). The Combination of Single-Cell and Next-Generation Sequencing Can Reveal Mosaicism for BRCA2 Mutations and the Fine Molecular Details of Tumorigenesis. Cancers, 13(10), 2354. https://doi.org/10.3390/cancers13102354