Cyclic Hypoxia: An Update on Its Characteristics, Methods to Measure It and Biological Implications in Cancer


Simple Summary
Abstract
1. Introduction
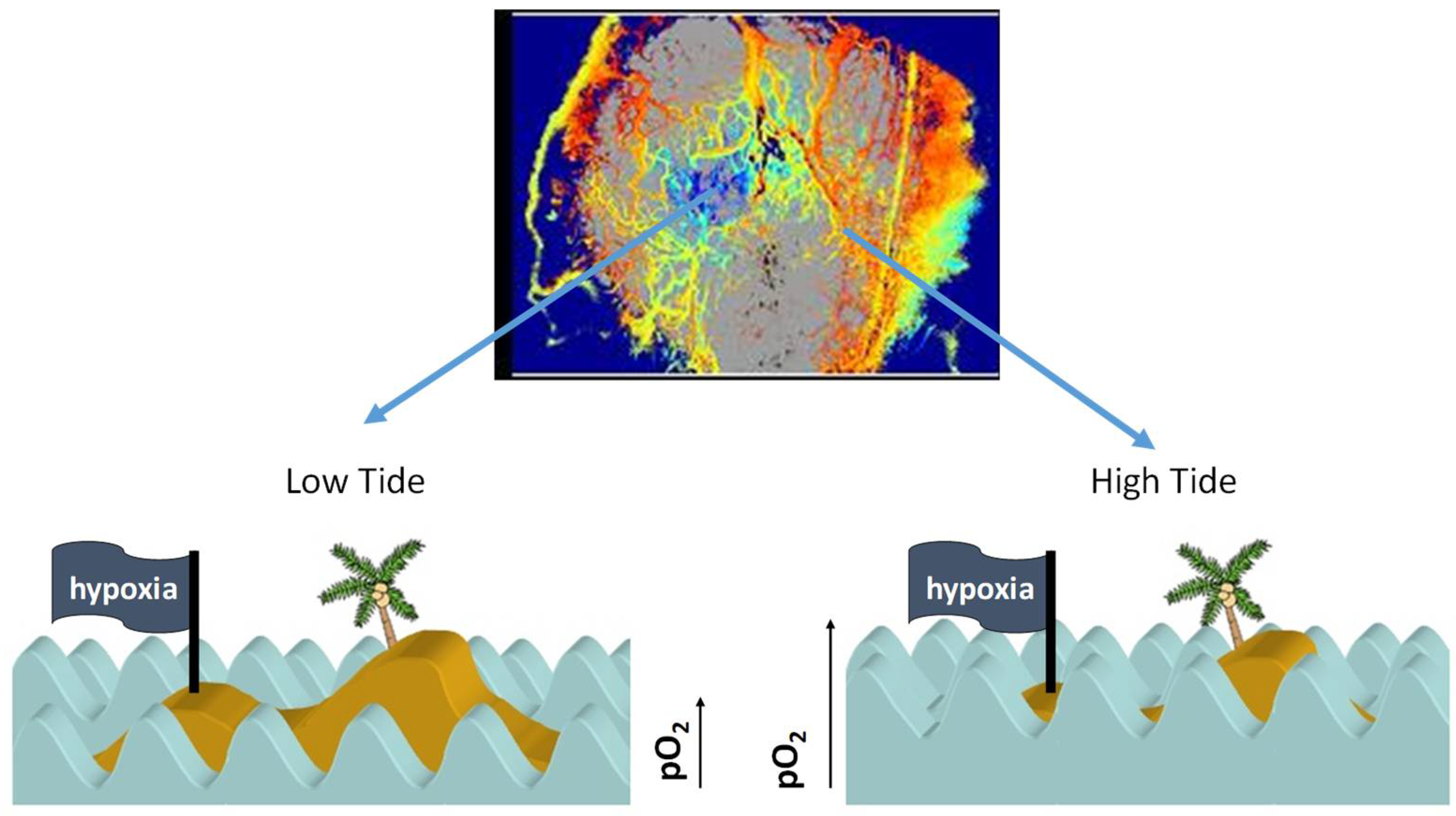
2. Physiological Characterization
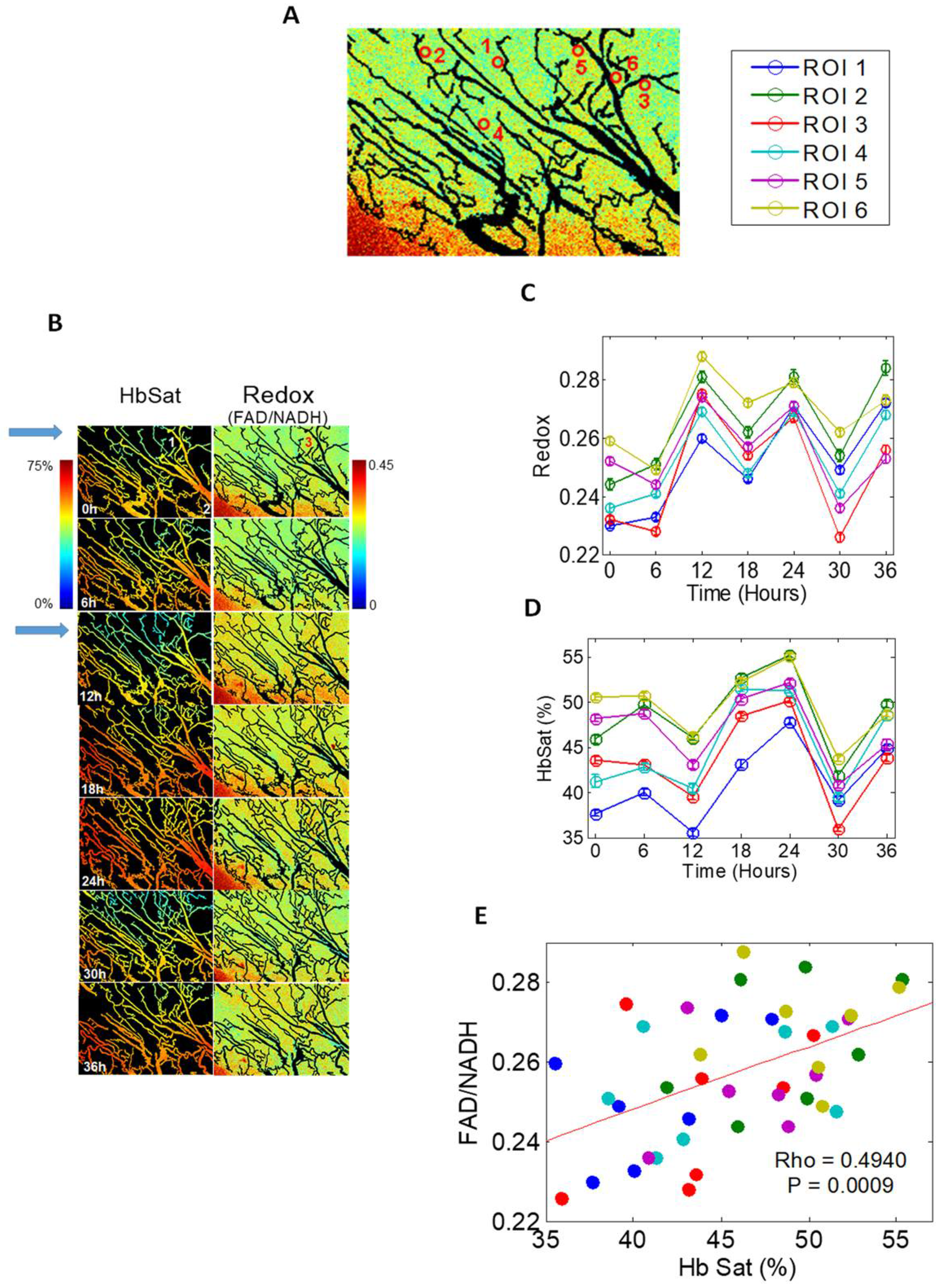
3. Does Cyclic Hypoxia Occur in Canine and Human Tumors?
4. Mechanisms Underlying Cyclic Hypoxia
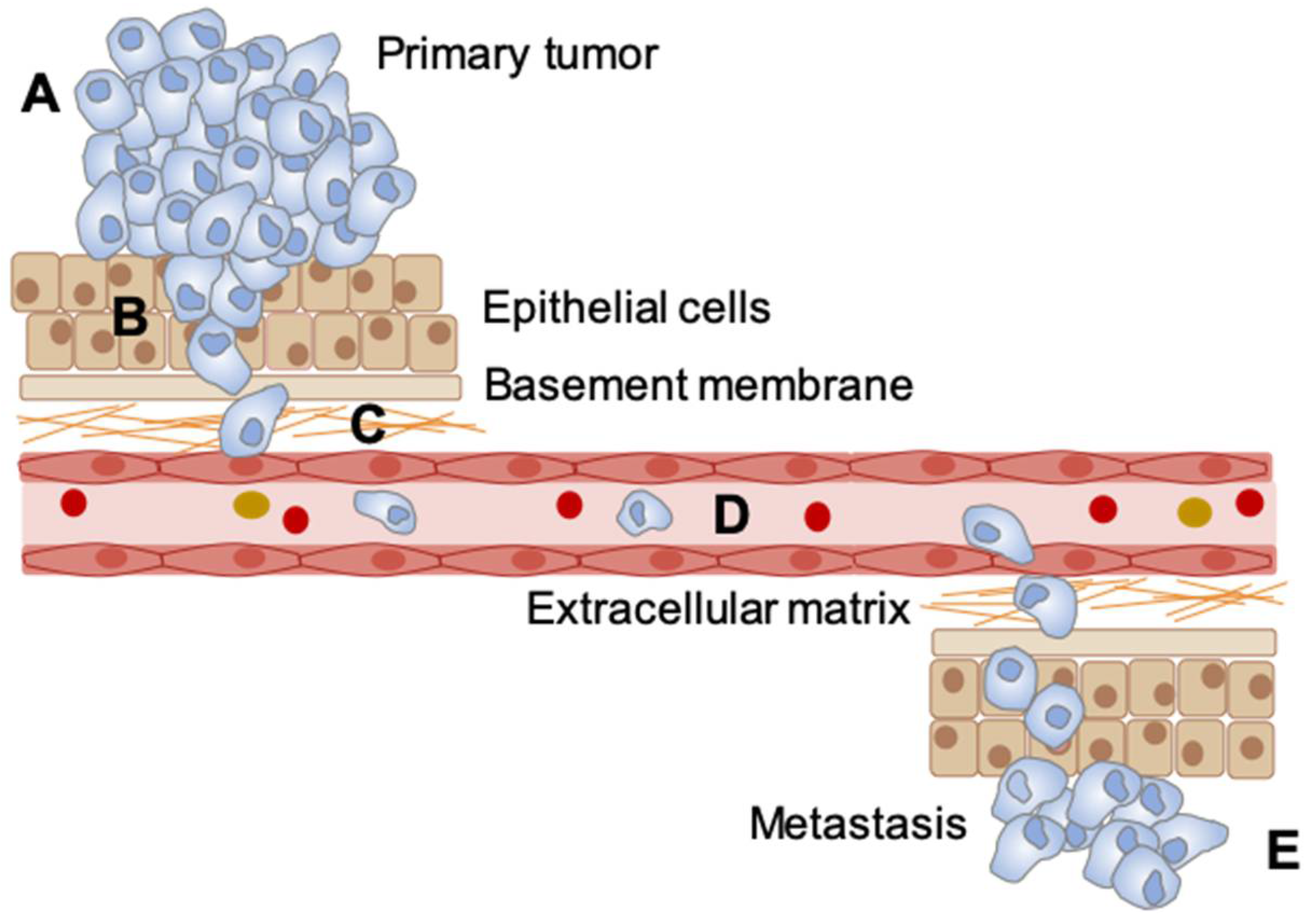
5. Cellular Consequences
6. Future Directions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ashcraft, K.A.; Betof Warner, A.; Nair, S.K.; Dewhirst, M.W. Can Exercise-Induced Modulation of the Tumor Physiologic Microenvironment Improve Antitumor Immunity? Cancer Res. 2019, 79, 2447–2456. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, M.W.; Cao, Y.; Moeller, B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer 2008, 8, 425–437. [Google Scholar] [CrossRef]
- Dewhirst, M.W. Relationships between Cycling Hypoxia, HIF-1, Angiogenesis and Oxidative Stress. Radiat. Res. 2009, 172, 653–665. [Google Scholar] [CrossRef]
- Bayer, C.; Vaupel, P. Acute versus chronic hypoxia in tumors. Controversial data concerning time frames and biological consequences. Strahlenther. Onkol. 2012, 188, 616–627. [Google Scholar] [CrossRef]
- Kukwa, W.; Migacz, E.; Druc, K.; Grzesiuk, E.; Czarnecka, A.M. Obstructive sleep apnea and cancer: Effects of intermittent hypoxia? Future Oncol. 2015, 11, 3285–3298. [Google Scholar] [CrossRef]
- Michiels, C.; Tellier, C.; Feron, O. Cycling hypoxia: A key feature of the tumor microenvironment. Biochim. Biophys. Acta Rev. Cancer 2016, 1866, 76–86. [Google Scholar] [CrossRef]
- Yamaura, H.; Matsuzawa, T. Tumor regrowth after irradiation—Experimental approach. Int. J. Radiat. Biol. 1979, 35, 201–219. [Google Scholar] [CrossRef]
- Brown, J.M. Evidence for acutely hypoxic cells in mouse-tumors, and a possible mechanism of re-oxygenation. Br. J. Radiol. 1979, 52, 650–656. [Google Scholar] [CrossRef]
- Chaplin, D.J.; Olive, P.L.; Durand, R.E. Intermittent blood-flow in a murine tumor—Radiobiological effects. Cancer Res. 1987, 47, 597–601. [Google Scholar]
- Braun, R.D.; Lanzen, J.L.; Snyder, S.A.; Dewhirst, M.W. Comparison of tumor and normal tissue oxygen tension measurements using OxyLite or microelectrodes in rodents. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2533–H2544. [Google Scholar] [CrossRef] [PubMed]
- Cardenas-Navia, L.I.; Mace, D.; Richardson, R.A.; Wilson, D.F.; Shan, S.; Dewhirst, M.W. The pervasive presence of fluctuating oxygenation in tumors. Cancer Res. 2008, 68, 5812–5819. [Google Scholar] [CrossRef] [PubMed]
- Skala, M.C.; Fontanella, A.; Lan, L.; Izatt, J.A.; Dewhirst, M.W. Longitudinal optical imaging of tumor metabolism and hemodynamics. J. Biomed. Opt. 2010, 15, 011112. [Google Scholar] [CrossRef]
- Nehmeh, S.A.; Lee, N.Y.; Schroder, H.; Squire, O.; Zanzonico, P.B.; Erdi, Y.E.; Greco, C.; Mageras, G.; Pham, H.S.; Larson, S.M.; et al. Reproducibility of intratumor distribution of (18)F-fluoromisonidazole in head and neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, M.R.; Johnson, S.P.; Ramasawmy, R.; Pedley, R.B.; Lythgoe, M.F.; Walker-Samuel, S. Decomposition of spontaneous fluctuations in tumour oxygenation using BOLD MRI and independent component analysis. Br. J. Cancer 2015, 113, 1168–1177. [Google Scholar] [CrossRef]
- O’Connor, J.P.; Boult, J.K.; Jamin, Y.; Babur, M.; Finegan, K.G.; Williams, K.J.; Little, R.A.; Jackson, A.; Parker, G.J.; Reynolds, A.R.; et al. Oxygen-Enhanced MRI Accurately Identifies, Quantifies, and Maps Tumor Hypoxia in Preclinical Cancer Models. Cancer Res. 2016, 76, 787–795. [Google Scholar] [CrossRef]
- Khan, N.; Mupparaju, S.; Hou, H.; Williams, B.B.; Swartz, H. Repeated assessment of orthotopic glioma pO(2) by multi-site EPR oximetry: A technique with the potential to guide therapeutic optimization by repeated measurements of oxygen. J. Neurosci. Methods 2012, 204, 111–117. [Google Scholar] [CrossRef]
- Matsumoto, S.; Yasui, H.; Mitchell, J.B.; Krishna, M.C. Imaging cycling tumor hypoxia. Cancer Res. 2010, 70, 10019–10023. [Google Scholar] [CrossRef]
- Magat, J.; Jordan, B.F.; Cron, G.O.; Gallez, B. Noninvasive mapping of spontaneous fluctuations in tumor oxygenation using 19F MRI. Med. Phys. 2010, 37, 5434–5441. [Google Scholar] [CrossRef]
- Bennewith, K.L.; Raleigh, J.A.; Durand, R.E. Orally administered pimonidazole to label hypoxic tumor cells. Cancer Res. 2002, 62, 6827–6830. [Google Scholar]
- Braun, R.D.; Lanzen, J.L.; Dewhirst, M.W. Fourier analysis of fluctuations of oxygen tension and blood flow in R3230Ac tumors and muscle in rats. Am. J. Physiol. Heart Circ. Physiol. 1999, 277, H551–H568. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, M.W.; Kimura, H.; Rehmus, S.W.E.; Braun, R.D.; Papahadjopoulos, D.; Hong, K.; Secomb, T.W. Microvascular studies on the origins of perfusion-limited hypoxia. Br. J. Cancer 1996, 74, S247–S251. [Google Scholar]
- Kimura, H.; Braun, R.D.; Ong, E.T.; Hsu, R.; Secomb, T.W.; Papahadjopoulos, D.; Hong, K.L.; Dewhirst, M.W. Fluctuations in red cell flux in tumor microvessels can lead to transient hypoxia and reoxygenation in tumor parenchyma. Cancer Res. 1996, 56, 5522–5528. [Google Scholar] [PubMed]
- Lanzen, J.; Braun, R.D.; Klitzman, B.; Brizel, D.; Secomb, T.W.; Dewhirst, M.W. Direct demonstration of instabilities in oxygen concentrations within the extravascular compartment of an experimental tumor. Cancer Res. 2006, 66, 2219–2223. [Google Scholar] [CrossRef] [PubMed]
- Palmer, G.M.; Fontanella, A.N.; Zhang, G.Q.; Hanna, G.; Fraser, C.L.; Dewhirst, M.W. Optical imaging of tumor hypoxia dynamics. J. Biomed. Opt. 2010, 15. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.T.; Boss, M.K.; Dewhirst, M.W. Imaging tumor hypoxia to advance radiation oncology. Antioxid. Redox Signal. 2014, 21, 313–337. [Google Scholar] [CrossRef]
- Rickard, A.G.; Palmer, G.M.; Dewhirst, M.W. Clinical and Pre-clinical Methods for Quantifying Tumor Hypoxia. Adv. Exp. Med. Biol. 2019, 1136, 19–41. [Google Scholar] [CrossRef]
- Thrall, D.E.; Maccarini, P.; Stauffer, P.; Macfall, J.; Hauck, M.; Snyder, S.; Case, B.; Linder, K.; Lan, L.; McCall, L.; et al. Thermal dose fractionation affects tumour physiological response. Int. J. Hyperth. 2012, 28, 431–440. [Google Scholar] [CrossRef][Green Version]
- Brurberg, K.G.; Skogmo, H.K.; Graff, B.A.; Olsen, D.R.; Rofstad, E.K. Fluctuations in pO2 in poorly and well-oxygenated spontaneous canine tumors before and during fractionated radiation therapy. Radiother. Oncol. 2005, 77, 220–226. [Google Scholar] [CrossRef]
- Brizel, D.M.; Rosner, G.L.; Prosnitz, L.R.; Dewhirst, M.W. Patterns and variability of tumor oxygenation in human soft tissue sarcomas, cervical carcinomas, and lymph node metastases. Int. J. Radiat. Oncol. Biol. Phys. 1995, 32, 1121–1125. [Google Scholar] [CrossRef]
- Shah, N.; Cerussi, A.; Eker, C.; Espinoza, J.; Butler, J.; Fishkin, J.; Hornung, R.; Tromberg, B. Noninvasive functional optical spectroscopy of human breast tissue. Proc. Natl. Acad. Sci. USA 2001, 98, 4420–4425. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Shah, A.; Wang, B.; Rajaram, N.; Wang, Q.; Ramanujam, N.; Palmer, G.M.; Dewhirst, M.W. Measuring tumor cycling hypoxia and angiogenesis using a side-firing fiber optic probe. J. Biophotonics 2014, 7, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Rich, L.J.; Miller, A.; Singh, A.K.; Seshadri, M. Photoacoustic Imaging as an Early Biomarker of Radio Therapeutic Efficacy in Head and Neck Cancer. Theranostics 2018, 8, 2064–2078. [Google Scholar] [CrossRef] [PubMed]
- Redler, G.; Epel, B.; Halpern, H.J. Principal component analysis enhances SNR for dynamic electron paramagnetic resonance oxygen imaging of cycling hypoxia in vivo. Magn. Reson. Med. 2014, 71, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Ellingsen, C.; Ovrebo, K.M.; Galappathi, K.; Mathiesen, B.; Rofstad, E.K. pO(2) Fluctuation Pattern and Cycling Hypoxia in Human Cervical Carcinoma and Melanoma Xenografts. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, 1317–1323. [Google Scholar] [CrossRef][Green Version]
- Baudelet, C.; Ansiaux, R.; Jordan, B.F.; Havaux, X.; Macq, B.; Gallez, B. Physiological noise in murine solid tumours using T2*-weighted gradient-echo imaging: A marker of tumour acute hypoxia? Phys. Med. Biol. 2004, 49, 3389–3411. [Google Scholar] [CrossRef]
- Pigott, K.H.; Hill, S.A.; Chaplin, D.J.; Saunders, M.I. Microregional fluctuations in perfusion within human tumours detected using laser Doppler flowmetry. Radiother. Oncol. 1996, 40, 45–50. [Google Scholar] [CrossRef]
- Panek, R.; Welsh, L.; Baker, L.C.J.; Schmidt, M.A.; Wong, K.H.; Riddell, A.M.; Koh, D.M.; Dunlop, A.; McQuaid, D.; d’Arcy, J.A.; et al. Noninvasive Imaging of Cycling Hypoxia in Head and Neck Cancer Using Intrinsic Susceptibility MRI. Clin. Cancer Res. 2017, 23, 4233–4241. [Google Scholar] [CrossRef]
- Wang, K.L.; Yorke, E.; Nehmeh, S.A.; Humm, J.L.; Ling, C.C. Modeling acute and chronic hypoxia using serial images of F-18-FMISO PET. Med. Phys. 2009, 36, 4400–4408. [Google Scholar] [CrossRef]
- Ellingsen, C.; Andersen, L.M.; Galappathi, K.; Rofstad, E.K. Hypoxia biomarkers in squamous cell carcinoma of the uterine cervix. BMC Cancer 2015, 15, 805. [Google Scholar] [CrossRef]
- Sobhanifar, S.; Aquino-Parsons, C.; Stanbridge, E.J.; Olive, P. Reduced expression of hypoxia-inducible factor-1alpha in perinecrotic regions of solid tumors. Cancer Res. 2005, 65, 7259–7266. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, B.; Aquino-Parsons, C.; Raleigh, J.A.; Stanbridge, E.J.; Durand, R.E.; Banath, J.P.; MacPhail, S.H.; Olive, P.L. Comparison between pimonidazole binding, oxygen electrode measurements, and expression of endogenous hypoxia markers in cancer of the uterine cervix. Cytom. B Clin. Cytom. 2006, 70, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.J.; Sonveaux, P.; Porporato, P.E.; Danhier, P.; Gallez, B.; Batinic-Haberle, I.; Nien, Y.C.; Schroeder, T.; Dewhirst, M.W. NADPH oxidase-mediated reactive oxygen species production activates hypoxia-inducible factor-1 (HIF-1) via the ERK pathway after hyperthermia treatment. Proc. Natl. Acad. Sci. USA 2010, 107, 20477–20482. [Google Scholar] [CrossRef] [PubMed]
- Chaudary, N.; Hill, R.P. Increased expression of metastasis-related genes in hypoxic cells sorted from cervical and lymph nodal xenograft tumors. Lab. Investig. 2009, 89, 587–596. [Google Scholar] [CrossRef]
- Koritzinsky, M.; Wouters, B.G. The roles of reactive oxygen species and autophagy in mediating the tolerance of tumor cells to cycling hypoxia. Semin. Radiat. Oncol. 2013, 23, 252–261. [Google Scholar] [CrossRef]
- Moeller, B.J.; Cao, Y.; Li, C.Y.; Dewhirst, M.W. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: Role of reoxygenation, free radicals, and stress granules. Cancer Cell 2004, 5, 429–441. [Google Scholar] [CrossRef]
- Lee, G.; Won, H.S.; Lee, Y.M.; Choi, J.W.; Oh, T.I.; Jang, J.H.; Choi, D.K.; Lim, B.O.; Kim, Y.J.; Park, J.W.; et al. Oxidative Dimerization of PHD2 is Responsible for its Inactivation and Contributes to Metabolic Reprogramming via HIF-1alpha Activation. Sci. Rep. 2016, 6, 18928. [Google Scholar] [CrossRef]
- Ljungkvist, A.S.; Bussink, J.; Kaanders, J.H.; van der Kogel, A.J. Dynamics of tumor hypoxia measured with bioreductive hypoxic cell markers. Radiat. Res. 2007, 167, 127–145. [Google Scholar] [CrossRef]
- Yasui, H.; Matsumoto, S.; Devasahayam, N.; Munasinghe, J.P.; Choudhuri, R.; Saito, K.; Subramanian, S.; Mitchell, J.B.; Krishna, M.C. Low-Field Magnetic Resonance Imaging to Visualize Chronic and Cycling Hypoxia in Tumor-Bearing Mice. Cancer Res. 2010, 70, 6427–6436. [Google Scholar] [CrossRef]
- Podtaev, S.; Morozov, M.; Frick, P. Wavelet-based correlations of skin temperature and blood flow oscillations. Cardiovasc. Eng. 2008, 8, 185–189. [Google Scholar] [CrossRef]
- Arciero, J.C.; Secomb, T.W. Spontaneous oscillations in a model for active control of microvessel diameters. Math. Med. Biol. J. IMA 2012, 29, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Forouzan, O.; Yang, X.X.; Sosa, J.M.; Burns, J.M.; Shevkoplyas, S.S. Spontaneous oscillations of capillary blood flow in artificial microvascular networks. Microvasc. Res. 2012, 84, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Lemons, D.E.; Chien, S.; Crawshaw, L.I.; Weinbaum, S.; Jiji, L.M. Significance of vessel size and type in vascular heat-transfer. Am. J. Physiol. 1987, 253, R128–R135. [Google Scholar] [CrossRef] [PubMed]
- Wetsel, W.C. Sensing hot and cold with TRP channels. Int. J. Hyperth. 2011, 27, 388–398. [Google Scholar] [CrossRef]
- Huang, Y.H.; Fliegert, R.; Guse, A.H.; Lu, W.; Du, J. A structural overview of the ion channels of the TRPM family. Cell Calcium 2020, 85. [Google Scholar] [CrossRef]
- Tan, C.L.; Cooke, E.K.; Leib, D.E.; Lin, Y.C.; Daly, G.E.; Zimmerman, C.A.; Knight, Z.A. Warm-Sensitive Neurons that Control Body Temperature. Cell 2016, 167, 47–59. [Google Scholar] [CrossRef]
- Liu, X.B.; Ong, H.L.; Ambudkar, I. TRP Channel Involvement in Salivary Glands-Some Good, Some Bad. Cells 2018, 7, 74. [Google Scholar] [CrossRef]
- Mergler, S.; Valtink, M.; Coulson-Thomas, V.J.; Lindemann, D.; Reinach, P.S.; Engelmann, K.; Pleyer, U. TRPV channels mediate temperature-sensing in human corneal endothelial cells. Exp. Eye Res. 2010, 90, 758–770. [Google Scholar] [CrossRef]
- Caterina, M.J. Transient receptor potential ion channels as participants in thermosensation and thermoregulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R64–R76. [Google Scholar] [CrossRef]
- Roemer, R.B.; Oleson, J.R.; Cetas, T.C. Oscillatory temperature response to constant power applied to canine muscle. Am. J. Physiol. 1985, 249, R153–R158. [Google Scholar] [CrossRef]
- Dewhirst, M.W.; Ong, E.T.; Braun, R.D.; Smith, B.; Klitzman, B.; Evans, S.M.; Wilson, D. Quantification of longitudinal tissue pO(2) gradients in window chamber tumours: Impact on tumour hypoxia. Br. J. Cancer 1999, 79, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Cardenas-Navia, L.I.; Yu, D.H.; Braun, R.D.; Brizel, D.M.; Secomb, T.W.; Dewhirst, M.W. Tumor-dependent kinetics of partial pressure of oxygen fluctuations during air and oxygen breathing. Cancer Res. 2004, 64, 6010–6017. [Google Scholar] [CrossRef] [PubMed]
- Baudelet, C.; Cron, G.O.; Ansiaux, R.; Crokart, N.; DeWever, J.; Feron, O.; Gallez, B. The role of vessel maturation and vessel functionality in spontaneous fluctuations of T-2*-weighted GRE signal within tumors. NMR Biomed. 2006, 19, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, Z.; Xu, B.; Chen, K.; Sun, J.; Ren, L. Intermittent hypoxia reduces microglia proliferation and induces DNA damage in vitro. Iran. J. Basic Med. Sci. 2016, 19, 497–502. [Google Scholar] [CrossRef][Green Version]
- Rouschop, K.M.; Dubois, L.J.; Keulers, T.G.; van den Beucken, T.; Lambin, P.; Bussink, J.; van der Kogel, A.J.; Koritzinsky, M.; Wouters, B.G. PERK/eIF2alpha signaling protects therapy resistant hypoxic cells through induction of glutathione synthesis and protection against ROS. Proc. Natl. Acad. Sci. USA 2013, 110, 4622–4627. [Google Scholar] [CrossRef]
- Rouschop, K.M.; Ramaekers, C.H.; Schaaf, M.B.; Keulers, T.G.; Savelkouls, K.G.; Lambin, P.; Koritzinsky, M.; Wouters, B.G. Autophagy is required during cycling hypoxia to lower production of reactive oxygen species. Radiother. Oncol. 2009, 92, 411–416. [Google Scholar] [CrossRef]
- Hammond, E.M.; Dorie, M.J.; Giaccia, A.J. ATR/ATM targets are phosphorylated by ATR in response to hypoxia and ATM in response to reoxygenation. J. Biol. Chem. 2003, 278, 12207–12213. [Google Scholar] [CrossRef]
- Toffoli, S.; Roegiers, A.; Feron, O.; Van Steenbrugge, M.; Ninane, N.; Raes, M.; Michiels, C. Intermittent hypoxia is an angiogenic inducer for endothelial cells: Role of HIF-1. Angiogenesis 2009, 12, 47–67. [Google Scholar] [CrossRef]
- Martinive, P.; Defresne, F.; Quaghebeur, E.; Daneau, G.; Crokart, N.; Gregoire, V.; Gallez, B.; Dessy, C.; Feron, O. Impact of cyclic hypoxia on HIF-1alpha regulation in endothelial cells-new insights for anti-tumor treatments. FEBS J. 2009, 276, 509–518. [Google Scholar] [CrossRef]
- Malec, V.; Gottschald, O.R.; Li, S.; Rose, F.; Seeger, W.; Hanze, J. HIF-1 alpha signaling is augmented during intermittent hypoxia by induction of the Nrf2 pathway in NOX1-expressing adenocarcinoma A549 cells. Free Radic. Biol. Med. 2010, 48, 1626–1635. [Google Scholar] [CrossRef]
- Martinive, P.; Defresne, F.; Bouzin, C.; Saliez, J.; Lair, F.; Gregoire, V.; Michiels, C.; Dessy, C.; Feron, O. Preconditioning of the tumor vasculature and tumor cells by intermittent hypoxia: Implications for anticancer therapies. Cancer Res. 2006, 66, 11736–11744. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Lee, C.H.; Liang, J.A.; Yu, C.Y.; Shyu, W.C. Cycling hypoxia increases U87 glioma cell radioresistance via ROS induced higher and long-term HIF-1 signal transduction activity. Oncol. Rep. 2010, 24, 1629–1636. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.H.; Shyu, W.C.; Chiang, C.Y.; Kuo, J.W.; Shen, W.C.; Liu, R.S. NADPH oxidase subunit 4-mediated reactive oxygen species contribute to cycling hypoxia-promoted tumor progression in glioblastoma multiforme. PLoS ONE 2011, 6, e23945. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.H.; Wu, C.P.; Lee, H.T.; Liang, J.A.; Yu, C.Y.; Lin, Y.J. NADPH oxidase subunit 4 mediates cycling hypoxia-promoted radiation resistance in glioblastoma multiforme. Free Radic. Biol. Med. 2012, 53, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Nanduri, J.; Khan, S.; Semenza, G.L.; Prabhakar, N.R. Induction of HIF-1alpha expression by intermittent hypoxia: Involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J. Cell Physiol. 2008, 217, 674–685. [Google Scholar] [CrossRef]
- Chen, W.L.; Wang, C.C.; Lin, Y.J.; Wu, C.P.; Hsieh, C.H. Cycling hypoxia induces chemoresistance through the activation of reactive oxygen species-mediated B-cell lymphoma extra-long pathway in glioblastoma multiforme. J. Transl. Med. 2015, 13, 389. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, X.; Wang, X.; Wei, L.; Liu, X.; Yuan, S.; Lv, L. Effect of chronic intermittent hypoxia on biological behavior and hypoxia-associated gene expression in lung cancer cells. J. Cell. Biochem. 2010, 111, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Nambiar, D.K.; Ramteke, A.; Kumar, R.; Dhar, D.; Agarwal, C.; Bergman, B.; Graner, M.; Maroni, P.; Singh, R.P.; et al. Hypoxia induces triglycerides accumulation in prostate cancer cells and extracellular vesicles supporting growth and invasiveness following reoxygenation. Oncotarget 2015, 6, 22836–22856. [Google Scholar] [CrossRef]
- Bhaskara, V.K.; Mohanam, I.; Rao, J.S.; Mohanam, S. Intermittent hypoxia regulates stem-like characteristics and differentiation of neuroblastoma cells. PLoS ONE 2012, 7, e30905. [Google Scholar] [CrossRef]
- Miao, Z.F.; Zhao, T.T.; Wang, Z.N.; Xu, Y.Y.; Mao, X.Y.; Wu, J.H.; Liu, X.Y.; Xu, H.; You, Y.; Xu, H.M. Influence of different hypoxia models on metastatic potential of SGC-7901 gastric cancer cells. Tumor Biol. 2014, 35, 6801–6808. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, D.; Zhang, L.; Xie, X.; Wu, Y.; Liu, Y.; Shao, G.; Su, Z. Upregulation of autophagy by hypoxia-inducible factor-1alpha promotes EMT and metastatic ability of CD133+ pancreatic cancer stem-like cells during intermittent hypoxia. Oncol. Rep. 2014, 32, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, W.; Wang, L.; Zhu, T.; Zhong, J.; Xie, N. Hypoxia-inducible factor 1 mediates intermittent hypoxia-induced migration of human breast cancer MDA-MB-231 cells. Oncol. Lett. 2017, 14, 7715–7722. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.W.; Wang, C.C.; Wu, C.P.; Lin, Y.J.; Lee, Y.C.; Cheng, Y.W.; Hsieh, C.H. Tumor cycling hypoxia induces chemoresistance in glioblastoma multiforme by upregulating the expression and function of ABCB1. Neuro Oncol. 2012, 14, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Alhawarat, F.M.; Hammad, H.M.; Hijjawi, M.S.; Sharab, A.S.; Abuarqoub, D.A.; Al Shhab, M.A.; Zihlif, M.A. The effect of cycling hypoxia on MCF-7 cancer stem cells and the impact of their microenvironment on angiogenesis using human umbilical vein endothelial cells (HUVECs) as a model. PeerJ 2019, 7, e5990. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Chetty, C.; Bhoopathi, P.; Lakka, S.; Mohanam, S.; Rao, J.S.; Dinh, D.E. Downregulation of uPA/uPAR inhibits intermittent hypoxia-induced epithelial-mesenchymal transition (EMT) in DAOY and D283 medulloblastoma cells. Int. J. Oncol. 2011, 38, 733–744. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, A.; Sceneay, J.; Godde, N.; Kinwel, T.; Ham, S.; Thompson, E.W.; Humbert, P.O.; Moller, A. Intermittent hypoxia induces a metastatic phenotype in breast cancer. Oncogene 2018, 37, 4214–4225. [Google Scholar] [CrossRef] [PubMed]
- Martinez, C.A.; Kerr, B.; Jin, C.; Cistulli, P.A.; Cook, K.M. Obstructive Sleep Apnea Activates HIF-1 in a Hypoxia Dose-Dependent Manner in HCT116 Colorectal Carcinoma Cells. Int. J. Mol. Sci. 2019, 20, 445. [Google Scholar] [CrossRef]
- Louie, E.; Nik, S.; Chen, J.S.; Schmidt, M.; Song, B.; Pacson, C.; Chen, X.F.; Park, S.; Ju, J.; Chen, E.I. Identification of a stem-like cell population by exposing metastatic breast cancer cell lines to repetitive cycles of hypoxia and reoxygenation. Breast Cancer Res. 2010, 12, R94. [Google Scholar] [CrossRef]
- Daneau, G.; Boidot, R.; Martinive, P.; Feron, O. Identification of cyclooxygenase-2 as a major actor of the transcriptomic adaptation of endothelial and tumor cells to cyclic hypoxia: Effect on angiogenesis and metastases. Clin. Cancer Res. 2010, 16, 410–419. [Google Scholar] [CrossRef]
- Delprat, V.; Tellier, C.; Demazy, C.; Raes, M.; Feron, O.; Michiels, C. Cycling hypoxia promotes a pro-inflammatory phenotype in macrophages via JNK/p65 signaling pathway. Sci. Rep. 2020, 10, 882. [Google Scholar] [CrossRef]
- Hlouschek, J.; Ritter, V.; Wirsdorfer, F.; Klein, D.; Jendrossek, V.; Matschke, J. Targeting SLC25A10 alleviates improved antioxidant capacity and associated radioresistance of cancer cells induced by chronic-cycling hypoxia. Cancer Lett. 2018, 439, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Hlouschek, J.; Hansel, C.; Jendrossek, V.; Matschke, J. The Mitochondrial Citrate Carrier (SLC25A1) Sustains Redox Homeostasis and Mitochondrial Metabolism Supporting Radioresistance of Cancer Cells With Tolerance to Cycling Severe Hypoxia. Front. Oncol. 2018, 8, 170. [Google Scholar] [CrossRef] [PubMed]
- Kumareswaran, R.; Chaudary, N.; Jaluba, K.; Meng, A.; Sykes, J.; Borhan, A.; Hill, R.P.; Bristow, R.G. Cyclic hypoxia does not alter RAD51 expression or PARP inhibitor cell kill in tumor cells. Radiother. Oncol. 2015, 116, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Matschke, J.; Riffkin, H.; Klein, D.; Handrick, R.; Ludemann, L.; Metzen, E.; Shlomi, T.; Stuschke, M.; Jendrossek, V. Targeted Inhibition of Glutamine-Dependent Glutathione Metabolism Overcomes Death Resistance Induced by Chronic Cycling Hypoxia. Antioxid. Redox Signal. 2016, 25, 89–107. [Google Scholar] [CrossRef] [PubMed]
- Tellier, C.; Desmet, D.; Petit, L.; Finet, L.; Graux, C.; Raes, M.; Feron, O.; Michiels, C. Cycling hypoxia induces a specific amplified inflammatory phenotype in endothelial cells and enhances tumor-promoting inflammation in vivo. Neoplasia 2015, 17, 66–78. [Google Scholar] [CrossRef]
- Verduzco, D.; Lloyd, M.; Xu, L.; Ibrahim-Hashim, A.; Balagurunathan, Y.; Gatenby, R.A.; Gillies, R.J. Intermittent hypoxia selects for genotypes and phenotypes that increase survival, invasion, and therapy resistance. PLoS ONE 2015, 10, e0120958. [Google Scholar] [CrossRef]
- Nanduri, J.; Wang, N.; Yuan, G.; Khan, S.A.; Souvannakitti, D.; Peng, Y.J.; Kumar, G.K.; Garcia, J.A.; Prabhakar, N.R. Intermittent hypoxia degrades HIF-2alpha via calpains resulting in oxidative stress: Implications for recurrent apnea-induced morbidities. Proc. Natl. Acad. Sci. USA 2009, 106, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Jarrar, Y.; Zihlif, M.; Al Bawab, A.Q.; Sharab, A. Effects of Intermittent Hypoxia on Expression of Glucose Metabolism Genes in MCF7 Breast Cancer Cell Line. Curr. Cancer Drug Targets 2020, 20, 216–222. [Google Scholar] [CrossRef]
- Li, L.; Ren, F.; Qi, C.; Xu, L.; Fang, Y.; Liang, M.; Feng, J.; Chen, B.; Ning, W.; Cao, J. Intermittent hypoxia promotes melanoma lung metastasis via oxidative stress and inflammation responses in a mouse model of obstructive sleep apnea. Respir. Res. 2018, 19, 28. [Google Scholar] [CrossRef]
- Snyder, B.; Shell, B.; Cunningham, J.T.; Cunningham, R.L. Chronic intermittent hypoxia induces oxidative stress and inflammation in brain regions associated with early-stage neurodegeneration. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef]
- Kalliomaki, T.M.; McCallum, G.; Lunt, S.J.; Wells, P.G.; Hill, R.P. Analysis of the effects of exposure to acute hypoxia on oxidative lesions and tumour progression in a transgenic mouse breast cancer model. BMC Cancer 2008, 8, 151. [Google Scholar] [CrossRef] [PubMed]
- Kalliomaki, T.M.; McCallum, G.; Wells, P.G.; Hill, R.P. Progression and metastasis in a transgenic mouse breast cancer model: Effects of exposure to in vivo hypoxia. Cancer Lett. 2009, 282, 98–108. [Google Scholar] [CrossRef]
- Cairns, R.A.; Kalliomaki, T.; Hill, R.P. Acute (cyclic) hypoxia enhances spontaneous metastasis of KHT murine tumors. Cancer Res. 2001, 61, 8903–8908. [Google Scholar] [PubMed]
- Cairns, R.A.; Hill, R.P. Acute hypoxia enhances spontaneous lymph node metastasis in an orthotopic murine model of human cervical carcinoma. Cancer Res. 2004, 64, 2054–2061. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cao, J.; Ma, S.; Dong, R.; Meng, W.; Ying, M.; Weng, Q.; Chen, Z.; Ma, J.; Fang, Q.; et al. Tumor hypoxia enhances Non-Small Cell Lung Cancer metastasis by selectively promoting macrophage M2 polarization through the activation of ERK signaling. Oncotarget 2014, 5, 9664–9677. [Google Scholar] [CrossRef] [PubMed]
- Rofstad, E.K.; Gaustad, J.V.; Egeland, T.A.; Mathiesen, B.; Galappathi, K. Tumors exposed to acute cyclic hypoxic stress show enhanced angiogenesis, perfusion and metastatic dissemination. Int. J. Cancer 2010, 127, 1535–1546. [Google Scholar] [CrossRef]
- Nanduri, J.; Vaddi, D.R.; Khan, S.A.; Wang, N.; Makerenko, V.; Prabhakar, N.R. Xanthine oxidase mediates hypoxia-inducible factor-2alpha degradation by intermittent hypoxia. PLoS ONE 2013, 8, e75838. [Google Scholar] [CrossRef]
- Peng, Y.J.; Yuan, G.; Khan, S.; Nanduri, J.; Makarenko, V.V.; Reddy, V.D.; Vasavda, C.; Kumar, G.K.; Semenza, G.L.; Prabhakar, N.R. Regulation of hypoxia-inducible factor-alpha isoforms and redox state by carotid body neural activity in rats. J. Physiol. 2014, 592, 3841–3858. [Google Scholar] [CrossRef]
- Li, J.; Bosch-Marce, M.; Nanayakkara, A.; Savransky, V.; Fried, S.K.; Semenza, G.L.; Polotsky, V.Y. Altered metabolic responses to intermittent hypoxia in mice with partial deficiency of hypoxia-inducible factor-1alpha. Physiol. Genom. 2006, 25, 450–457. [Google Scholar] [CrossRef]
- Galanis, A.; Pappa, A.; Giannakakis, A.; Lanitis, E.; Dangaj, D.; Sandaltzopoulos, R. Reactive oxygen species and HIF-1 signalling in cancer. Cancer Lett. 2008, 266, 12–20. [Google Scholar] [CrossRef]
- Raedschelders, K.; Ansley, D.M.; Chen, D.D. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol. Ther. 2012, 133, 230–255. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Nathan, C.; Cunningham-Bussel, A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 2013, 13, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Reczek, C.R.; Chandel, N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Reyes, I.; Cardona, L.R.; Kong, H.; Vasan, K.; McElroy, G.S.; Werner, M.; Kihshen, H.; Reczek, C.R.; Weinberg, S.E.; Gao, P.; et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 2020. [Google Scholar] [CrossRef]
- Lee, S.R.; Yang, K.S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tabar, S.S.; Malec, V.; Eul, B.G.; Klepetko, W.; Weissmann, N.; Grimminger, F.; Seeger, W.; Rose, F.; Hanze, J. NOX4 regulates ROS levels under normoxic and hypoxic conditions, triggers proliferation, and inhibits apoptosis in pulmonary artery adventitial fibroblasts. Antioxid. Redox Signal. 2008, 10, 1687–1698. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1alpha and HIF2alpha: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Brahimi-Horn, M.C.; Pouyssegur, J. HIF at a glance. J. Cell Sci. 2009, 122, 1055–1057. [Google Scholar] [CrossRef] [PubMed]
- Dobrynin, G.; McAllister, T.E.; Leszczynska, K.B.; Ramachandran, S.; Krieg, A.J.; Kawamura, A.; Hammond, E.M. KDM4A regulates HIF-1 levels through H3K9me3. Sci. Rep. 2017, 7, 11094. [Google Scholar] [CrossRef]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef]
- Koumenis, C.; Wouters, B.G. “Translating” tumor hypoxia: Unfolded protein response (UPR)-dependent and UPR-independent pathways. Mol. Cancer Res. 2006, 4, 423–436. [Google Scholar] [CrossRef]
- Poon, E.; Harris, A.L.; Ashcroft, M. Targeting the hypoxia-inducible factor (HIF) pathway in cancer. Expert Rev. Mol. Med. 2009, 11, e26. [Google Scholar] [CrossRef]
- Duan, C. Hypoxia-inducible factor 3 biology: Complexities and emerging themes. Am. J. Physiol. Cell Physiol. 2016, 310, C260–C269. [Google Scholar] [CrossRef]
- Hagen, T. Oxygen versus Reactive Oxygen in the Regulation of HIF-1alpha: The Balance Tips. Biochem. Res. Int. 2012, 2012, 436981. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Lai, U.H.; Zhu, L.; Singh, A.; Ahmed, M.; Forsyth, N.R. Reactive Oxygen Species Formation in the Brain at Different Oxygen Levels: The Role of Hypoxia Inducible Factors. Front. Cell Dev. Biol. 2018, 6, 132. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.W.; Ashcroft, M. Exploring the molecular interface between hypoxia-inducible factor signalling and mitochondria. Cell. Mol. Life Sci. 2019, 76, 1759–1777. [Google Scholar] [CrossRef] [PubMed]
- Niecknig, H.; Tug, S.; Reyes, B.D.; Kirsch, M.; Fandrey, J.; Berchner-Pfannschmidt, U. Role of reactive oxygen species in the regulation of HIF-1 by prolyl hydroxylase 2 under mild hypoxia. Free Radic. Res. 2012, 46, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, K.D.; Guzy, R.D.; Pan, Y.; Young, R.M.; Cash, T.P.; Schumacker, P.T.; Simon, M.C. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005, 1, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Buchan, J.R.; Parker, R. Eukaryotic stress granules: The ins and outs of translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef]
- Cavadas, M.A.; Nguyen, L.K.; Cheong, A. Hypoxia-inducible factor (HIF) network: Insights from mathematical models. Cell Commun. Signal. 2013, 11, 42. [Google Scholar] [CrossRef]
- Leedale, J.; Herrmann, A.; Bagnall, J.; Fercher, A.; Papkovsky, D.; See, V.; Bearon, R.N. Modeling the dynamics of hypoxia inducible factor-1alpha (HIF-1alpha) within single cells and 3D cell culture systems. Math. Biosci. 2014, 258, 33–43. [Google Scholar] [CrossRef]
- Ardaseva, A.; Gatenby, R.A.; Anderson, A.R.A.; Byrne, H.M.; Maini, P.K.; Lorenzi, T. A Mathematical Dissection of the Adaptation of Cell Populations to Fluctuating Oxygen Levels. Bull. Math. Biol. 2020, 82, 81. [Google Scholar] [CrossRef]
- Palcic, B.; Brosing, J.W.; Skarsgard, L.D. Survival measurements at low doses: Oxygen enhancement ratio. Br. J. Cancer 1982, 46, 980–984. [Google Scholar] [CrossRef][Green Version]
- Hammond, E.M.; Asselin, M.C.; Forster, D.; O’Connor, J.P.; Senra, J.M.; Williams, K.J. The meaning, measurement and modification of hypoxia in the laboratory and the clinic. Clin. Oncol. 2014, 26, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Horsman, M.R.; Mortensen, L.S.; Petersen, J.B.; Busk, M.; Overgaard, J. Imaging hypoxia to improve radiotherapy outcome. Nat. Rev. Clin. Oncol. 2012, 9, 674–687. [Google Scholar] [CrossRef]
- Skwarski, M.; Bowler, E.; Wilson, J.D.; Higgins, G.S.; Hammond, E.M. Targeting Tumor Hypoxia. In Molecular Targeted Radiosensitizers: Opportunities and Challenges; Willers, H., Eke, I., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 265–299. [Google Scholar]
- Garcia-Barros, M.; Paris, F.; Cordon-Cardo, C.; Lyden, D.; Rafii, S.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 2003, 300, 1155–1159. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, J.; Wang, X.; Li, Y.; Chen, Y.; Li, K.; Zhang, J.; Yao, L.; Guo, G. HIF-1 and NDRG2 contribute to hypoxia-induced radioresistance of cervical cancer Hela cells. Exp. Cell Res. 2010, 316, 1985–1993. [Google Scholar] [CrossRef] [PubMed]
- Harada, H. Hypoxia-inducible factor 1-mediated characteristic features of cancer cells for tumor radioresistance. J. Radiat. Res. 2016, 57 (Suppl. S1), i99–i105. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Paget, J.T.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef]
- Moeller, B.J.; Dreher, M.R.; Rabbani, Z.N.; Schroeder, T.; Cao, Y.; Li, C.Y.; Dewhirst, M.W. Pleiotropic effects of HIF-1 blockade on tumor radiosensitivity. Cancer Cell 2005, 8, 99–110. [Google Scholar] [CrossRef]
- Moeller, B.J.; Dewhirst, M.W. HIF-1 and tumour radiosensitivity. Br. J. Cancer 2006, 95, 1–5. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Wadsworth, B.J.; Cederberg, R.A.; Lee, C.M.; Firmino, N.S.; Franks, S.E.; Pan, J.; Colpo, N.; Lin, K.S.; Benard, F.; Bennewith, K.L. Angiotensin II type 1 receptor blocker telmisartan inhibits the development of transient hypoxia and improves tumour response to radiation. Cancer Lett. 2020, 493, 31–40. [Google Scholar] [CrossRef]
- Cosse, J.P.; Michiels, C. Tumour hypoxia affects the responsiveness of cancer cells to chemotherapy and promotes cancer progression. Anticancer Agents Med. Chem. 2008, 8, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Monti, E.; Gariboldi, M.B. HIF-1 as a target for cancer chemotherapy, chemosensitization and chemoprevention. Curr. Mol. Pharmacol. 2011, 4, 62–77. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef]
- Lu, X.; Kang, Y. Hypoxia and hypoxia-inducible factors: Master regulators of metastasis. Clin. Cancer Res. 2010, 16, 5928–5935. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Rofstad, E.K.; Galappathi, K.; Mathiesen, B.; Ruud, E.B. Fluctuating and diffusion-limited hypoxia in hypoxia-induced metastasis. Clin. Cancer Res. 2007, 13, 1971–1978. [Google Scholar] [CrossRef]
- Gutsche, K.; Randi, E.B.; Blank, V.; Fink, D.; Wenger, R.H.; Leo, C.; Scholz, C.C. Intermittent hypoxia confers pro-metastatic gene expression selectively through NF-kappaB in inflammatory breast cancer cells. Free Radic. Biol. Med. 2016, 101, 129–142. [Google Scholar] [CrossRef]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Zeng, W.; Liu, P.; Pan, W.; Singh, S.R.; Wei, Y. Hypoxia and hypoxia inducible factors in tumor metabolism. Cancer Lett. 2015, 356, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Drager, L.F.; Jun, J.C.; Polotsky, V.Y. Metabolic consequences of intermittent hypoxia: Relevance to obstructive sleep apnea. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Chiang, A.A. Obstructive sleep apnea and chronic intermittent hypoxia: A review. Chin. J. Physiol. 2006, 49, 234–243. [Google Scholar] [PubMed]
- Ryan, S. Adipose tissue inflammation by intermittent hypoxia: Mechanistic link between obstructive sleep apnoea and metabolic dysfunction. J. Physiol. 2017, 595, 2423–2430. [Google Scholar] [CrossRef] [PubMed]
- Zannella, V.E.; Dal Pra, A.; Muaddi, H.; McKee, T.D.; Stapleton, S.; Sykes, J.; Glicksman, R.; Chaib, S.; Zamiara, P.; Milosevic, M.; et al. Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clin. Cancer Res. 2013, 19, 6741–6750. [Google Scholar] [CrossRef] [PubMed]
- Ashton, T.M.; Fokas, E.; Kunz-Schughart, L.A.; Folkes, L.K.; Anbalagan, S.; Huether, M.; Kelly, C.J.; Pirovano, G.; Buffa, F.M.; Hammond, E.M.; et al. The anti-malarial atovaquone increases radiosensitivity by alleviating tumour hypoxia. Nat. Commun. 2016, 7, 12308. [Google Scholar] [CrossRef]
- Benej, M.; Hong, X.; Vibhute, S.; Scott, S.; Wu, J.; Graves, E.; Le, Q.T.; Koong, A.C.; Giaccia, A.J.; Yu, B.; et al. Papaverine and its derivatives radiosensitize solid tumors by inhibiting mitochondrial metabolism. Proc. Natl. Acad. Sci. USA 2018, 115, 10756–10761. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Measurement Techniques | Requires Chemical Probe | Direct pO2 Measurement | Frequency of Detection | Spatial Resolution | Ref. |
|---|---|---|---|---|---|
| Polographic O₂ microelectrodes | No | Yes | <<1 s Suitable for rapid cycles | 20–30 μm | [11] |
| Oxylite optical probe | No | Yes | <<1 s Suitable for rapid cycles | 250 μm | [11] |
| Phosphorescence lifetime imaging | Pd-porphyrin dendrimer, IV | Yes | 2–2.5 min Suitable for rapid cycles | <mm-cm | [12] |
| Optical Spectroscopy * | No | No | 30 s Suitable for rapid and slow cycles | mm-cm | [13] |
| PET/SPECT * | Yes | No | >24 h Suitable for slow cycles | Several mm | [14] |
| BOLD MRI * | No | No | 4 s Suitable for rapid cycles | Sub mm-mm | [15] |
| OE-MRI * | No | No | 2.5 min Suitable for rapid cycles | 0.24 mm | [16] |
| EPR | Implantable O2 sensitive spin probe with resonator | Yes | 10–60 s Suitable for rapid and slow cycles | mm-> cm | [17] |
| EPRI | O2 sensitive spin probe, IV | Yes | 2–3 min Suitable for rapid and slow cycles | 1.6–1.8 mm | [18] |
| F-19 MRI | Intratumoral injection of hexafluorobenzene | Yes | 1.5 min Suitable for rapid and slow cycles | 2 mm | [19] |
| Dual hypoxia marker drugs * | 2 hypoxia marker drugs given at various intervals from each other | No | These probes average extent of hypoxia over 60–90 min | μm | [20] |
| Cycling Conditions (% O2 and Length of Exposure) | Number of Cycles | Ref. |
|---|---|---|
| 1% O2 (6.67 min)—21% O2 (6.67 min) | 72 | [64] |
| <0.02% O2 (1 h)—21% O2 (1 h) | 2–5 | [65] |
| <0.02% O2 (1 h)—21% O2 (1 h) | 2–5 | [66] |
| <0.02% O2 (16 h)—21% O2 (1 h) | 1 | [67] |
| 1% O2 (1 h)—21% O2 (30 min) | 4 | [68] |
| <1% O2 (1 h)—21% O2 (30 min) | 3 | [69] |
| 1% O2 (2 h)—21% O2 (2 h) | 1–3 | [70] |
| 0.5–1% O2 (1 h)—21% O2 (30 min) | 3 | [71] |
| 0.5–1% O2 (1 h)—21% O2 (30 min) | 3 | [72] |
| 0.5–1% O2 (10 min)—21% O2 (10 min) | 12 | [73] |
| 0.5–1% O2 (10 min)—21% O2 (10 min) | 12 | [74] |
| 1.5% O2 (30 s)—21% O2 (5 min) | 60 | [75] |
| 0.5–1% O2 (1 h)—21% O2 (30 min) | 3 | [76] |
| 0.1% O2 (24 h)—21% O2 (72 h) | 20 | [77] |
| 1% O2 (48 h)—21% O2 (24, 48, 72 h) | 1 | [78] |
| 1% O2 (24 h)—21% O2 (24 h) | 1, 5, 10 | [79] |
| 1% O2 (12 h)—21% O2 (12 h) | 7 | [80] |
| 1% O2 (12 h)—21% O2 (12 h) | 5 | [81] |
| 1% O2 (12 h)—21% O2 (12 h) | 5, 10, 15, 20 | [82] |
| 0.5–1% O2 (1 h)—21% O2 (30 min) | 3 | [83] |
| <1% O2 (‘shots’ for 8, 72 h)—21% O2 (n/a) | 1,3/week | [84] |
| 1% O2 (48 h)—21% O2 (48 h) | 18–20 | [85] |
| 1% O2 (24 h)—21% O2 (24 h) | 5 | [86] |
| <0.1% O2 (5 min)—7.76% O2 (5 min) | 36 or 108 | [87] |
| 1% O2 + nutrient deprivation (7 days)—21% O2 (1–3 weeks) | 1–3 | [88] |
| 0.5% O2 (1 h)—21% O2 (30 min) | 3 | [89] |
| 1% O2 (1 h)—21% O2 (30 min) | 4 | [90] |
| <1% O2 (48 h)—21% O2 (120 h) | 16 or 25 | [91] |
| <1% O2 (48 h)—21% O2 (120 h) | 16 or 25 | [92] |
| <0.1% O2 (30 min or 3 h)—7% O2 (30 min) | 21 or 72 | [93] |
| <0.1% O2 (48 h)—21% O2 (120 h) | 16 or 25 | [94] |
| 1% O2 (1 h)—21% O2 (30 min) | 4 | [95] |
| 0.2 or 1% O2 (8, 16, 24 h)—21% O2 (8, 16, 24 h) | 3, 6, 50 | [96] |
| 1.5% O2 (30 s)—21% O2 (5 min) | 15, 30, 60 | [97] |
| <1% O2 (8, 72 h)—21% O2 (undefined) | 30, 60 | [98] |
| 7% O2 (1 h)—21% O2 (30 min) | 3 | [72] |
| 5% O2 (30 s)—21% O2 (1 min) | 180/day (21 days) | [99] |
| 7% O2 (10 min)—21% O2 (10 min) | 12 | [73] |
| 10% O2 (~3 min)—21% O2 (~3 min) | 80/day (7 days) | [100] |
| 7% O2 (10 min)—21% O2 (10 min) | 12/day (5/week for 6 weeks) | [101] |
| 7% O2 (10 min)—21% O2 (10 min) | 6 or 12/day (5/week for 6 weeks) | [102] |
| 7% O2 (1 h)—21% O2 (30 min) | 3 | [71] |
| 5–7% O2 (10 min)—21% O2 (10 min) | 12/day | [103] |
| 7% O2 (10 min)—21% O2 (10 min) | 12/day (21 days) | [104] |
| 7% O2 (10 min)—21% O2 (10 min) | 12 | [76] |
| 8% O2 (10 min)—21% O2 (10 min) | 12/day (35 days) | [105] |
| 8% O2 (10 min)—21% O2 (10 min) | 12/day | [106] |
| 7% O2 (1 h)—21% O2 (30 min) | 3/day (1 day) | [95] |
| 7% O2 (10 min)—21% O2 (10 min) | 12/day (21 days) | [44] |
| 5% O2 (15 s)—21% O2 (5 min) | 9/day (10 days) | [97] |
| 5% O2 (15 s)—21% O2 (5 min) | 9/day (10 days) | [107] |
| 5% O2 (15 s)—21% O2 (5 min) | 9/day (10 days) | [108] |
| 5% O2 (30 s)—21% O2 (30 s) | 5 days | [109] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bader, S.B.; Dewhirst, M.W.; Hammond, E.M. Cyclic Hypoxia: An Update on Its Characteristics, Methods to Measure It and Biological Implications in Cancer. Cancers 2021, 13, 23. https://doi.org/10.3390/cancers13010023
Bader SB, Dewhirst MW, Hammond EM. Cyclic Hypoxia: An Update on Its Characteristics, Methods to Measure It and Biological Implications in Cancer. Cancers. 2021; 13(1):23. https://doi.org/10.3390/cancers13010023
Chicago/Turabian StyleBader, Samuel B., Mark W. Dewhirst, and Ester M. Hammond. 2021. "Cyclic Hypoxia: An Update on Its Characteristics, Methods to Measure It and Biological Implications in Cancer" Cancers 13, no. 1: 23. https://doi.org/10.3390/cancers13010023
APA StyleBader, S. B., Dewhirst, M. W., & Hammond, E. M. (2021). Cyclic Hypoxia: An Update on Its Characteristics, Methods to Measure It and Biological Implications in Cancer. Cancers, 13(1), 23. https://doi.org/10.3390/cancers13010023