Stromal Protein-Mediated Immune Regulation in Digestive Cancers




Abstract
Simple Summary
Abstract
1. Introduction
2. Extracellular Matrix Proteins
3. Immune Regulation by ECM Proteins in Digestive Cancers
3.1. Collagens
3.2. Fibrous and Non-Fibrous Glycoproteins
3.2.1. Fibronectin
3.2.2. Laminin
3.2.3. Elastin
3.3. Matricellular Glycoproteins
3.3.1. SPARC/Osteonectin
3.3.2. Osteopontin
3.3.3. Periostin
3.3.4. βig-h3/TGF-β-Induced Protein
3.4. Proteoglycans
Versican
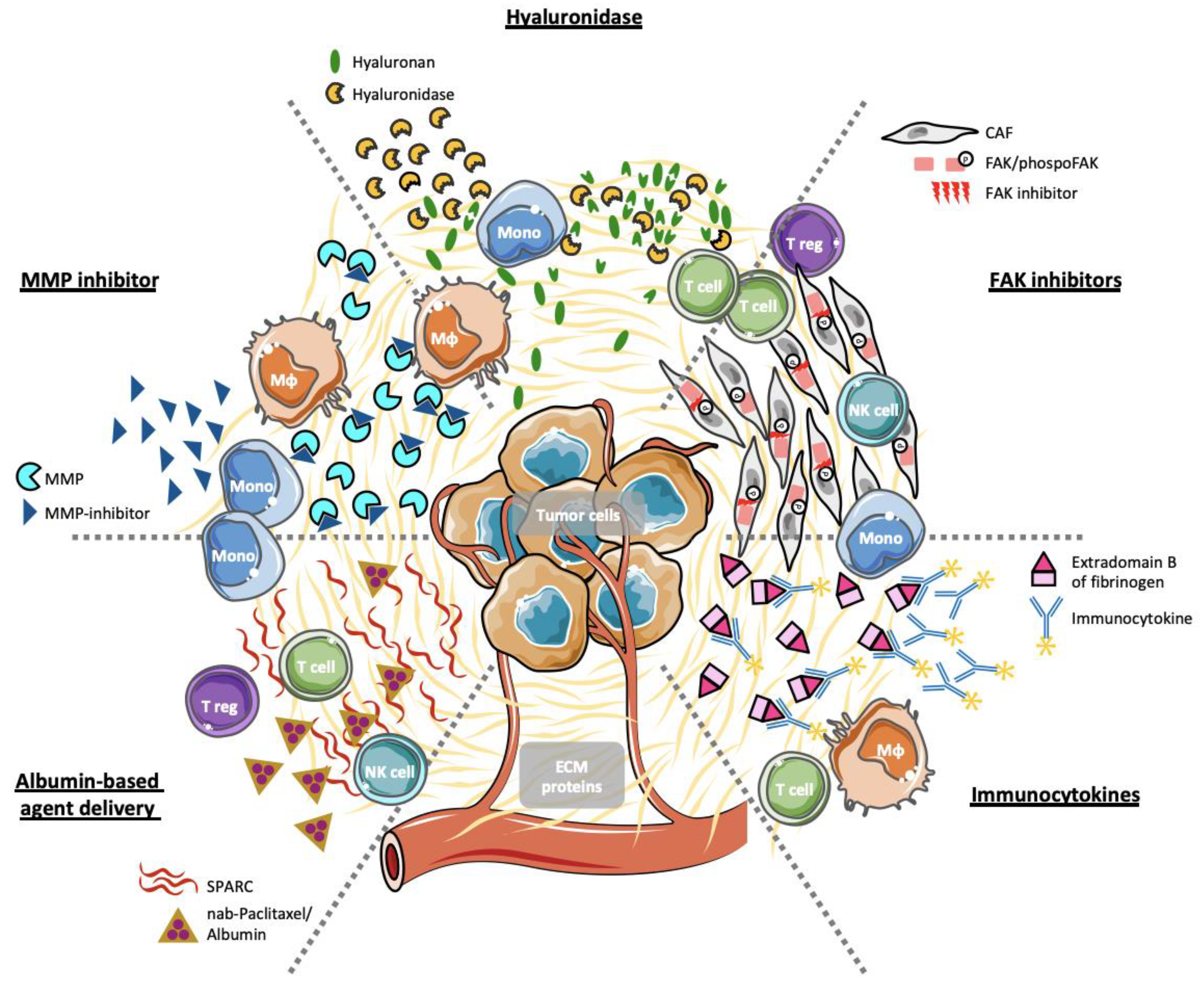
4. Stromal ECM Proteins as Selective Therapeutic Targets
4.1. Hyaluronan and Hyaluronidases
4.2. Immunocytokines
4.3. Inhibition of FAK Activity
4.4. Inhibition of Matrix Metalloproteases
4.5. Albumin-Based Agent Delivery
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Available online: https://gco.iarc.fr/today/home (accessed on 15 November 2020).
- Ducreux, M.; Cuhna, A.S.; Caramella, C.; Hollebecque, A.; Burtin, P.; Goere, D.; Seufferlein, T.; Haustermans, K.; Van Laethem, J.L.; Conroy, T.; et al. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26 (Suppl. 5), v56–v68. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Verheij, M.; Allum, W.; Cunningham, D.; Cervantes, A.; Arnold, D.; Committee, E.G. Gastric cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v38–v49. [Google Scholar] [CrossRef] [PubMed]
- Pentheroudakis, G.; Committee, E.G. Recent eUpdates to the ESMO Clinical Practice Guidelines on hepatocellular carcinoma, cancer of the pancreas, soft tissue and visceral sarcomas, cancer of the prostate and gastric cancer. Ann. Oncol. 2019, 30, 1395–1397. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Torphy, R.J.; Schulick, R.D.; Zhu, Y. Understanding the immune landscape and tumor microenvironment of pancreatic cancer to improve immunotherapy. Mol. Carcinog. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pearce, O.M.T.; Delaine-Smith, R.M.; Maniati, E.; Nichols, S.; Wang, J.; Bohm, S.; Rajeeve, V.; Ullah, D.; Chakravarty, P.; Jones, R.R.; et al. Deconstruction of a Metastatic Tumor Microenvironment Reveals a Common Matrix Response in Human Cancers. Cancer Discov. 2018, 8, 304–319. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Oya, Y.; Hayakawa, Y.; Koike, K. Tumor microenvironment in gastric cancers. Cancer Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Varga, J.; Wang, T.C.; Greten, F.R. The gastrointestinal tumor microenvironment. Gastroenterology 2013, 145, 63–78. [Google Scholar] [CrossRef]
- Socovich, A.M.; Naba, A. The cancer matrisome: From comprehensive characterization to biomarker discovery. Semin. Cell Dev. Biol. 2019, 89, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Hastings, J.F.; Skhinas, J.N.; Fey, D.; Croucher, D.R.; Cox, T.R. The extracellular matrix as a key regulator of intracellular signalling networks. Br. J. Pharmacol. 2019, 176, 82–92. [Google Scholar] [CrossRef]
- Hamidi, H.; Pietilä, M.; Ivaska, J. The complexity of integrins in cancer and new scopes for therapeutic targeting. Br. J. Cancer 2016, 115, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Kai, F.; Drain, A.P.; Weaver, V.M. The Extracellular Matrix Modulates the Metastatic Journey. Dev. Cell 2019, 49, 332–346. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef]
- Hartmann, N.; Giese, N.A.; Giese, T.; Poschke, I.; Offringa, R.; Werner, J.; Ryschich, E. Prevailing role of contact guidance in intrastromal T-cell trapping in human pancreatic cancer. Clin. Cancer Res. 2014, 20, 3422–3433. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Tannock, I.F. Targeting tumor architecture to favor drug penetration: A new weapon to combat chemoresistance in pancreatic cancer? Cancer Cell 2012, 21, 327–329. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef]
- Chong, H.C.; Tan, C.K.; Huang, R.-L.; Tan, N.S. Matricellular Proteins: A Sticky Affair with Cancers. J. Oncol. 2012, 2012, 1–17. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The matrisome: In silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell Proteom. 2012, 11, M111.014647. [Google Scholar] [CrossRef]
- Naba, A.; Clauser, K.R.; Lamar, J.M.; Carr, S.A.; Hynes, R.O. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. Elife 2014, 3, e01308. [Google Scholar] [CrossRef]
- Tian, C.; Clauser, K.R.; Öhlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R.; Carr, S.A.; Tuveson, D.A.; Hynes, R.O. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19609–19618. [Google Scholar] [CrossRef]
- Tian, C.; Öhlund, D.; Rickelt, S.; Lidstrom, T.; Huang, Y.; Hao, L.; Zhao, R.T.; Franklin, O.; Bhatia, S.N.; Tuveson, D.A.; et al. Cancer Cell-Derived Matrisome Proteins Promote Metastasis in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2020, 80, 1461–1474. [Google Scholar] [CrossRef]
- Norton, J.; Foster, D.; Chinta, M.; Titan, A.; Longaker, M. Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment. Cancers 2020, 12, 1347. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, A.; Martin, I. Extracellular Matrices to Modulate the Innate Immune Response and Enhance Bone Healing. Front. Immunol. 2019, 10, 2256. [Google Scholar] [CrossRef] [PubMed]
- Jürgensen, H.J.; van Putten, S.; Norregaard, K.S.; Bugge, T.H.; Engelholm, L.H.; Behrendt, N.; Madsen, D.H. Cellular uptake of collagens and implications for immune cell regulation in disease. Cell Mol. Life Sci. 2020, 77, 3161–3176. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Liu, J.; Yang, G.; Li, Y. The tumor-stromal ratio as a strong prognosticator for advanced gastric cancer patients: Proposal of a new TSNM staging system. J. Gastroenterol. 2018, 53, 606–617. [Google Scholar] [CrossRef]
- Conti, J.; Thomas, G. The role of tumour stroma in colorectal cancer invasion and metastasis. Cancers 2011, 3, 2160–2168. [Google Scholar] [CrossRef]
- Lundgren, S.; Micke, P.; Elebro, J.; Heby, M.; Hrynchyk, I.; Nodin, B.; Leandersson, K.; Mezheyeuski, A.; Jirstrom, K. Topographical Distribution and Spatial Interactions of Innate and Semi-Innate Immune Cells in Pancreatic and Other Periampullary Adenocarcinoma. Front. Immunol. 2020, 11, 558169. [Google Scholar] [CrossRef]
- Lundgren, S.; Elebro, J.; Heby, M.; Nodin, B.; Leandersson, K.; Micke, P.; Jirstrom, K.; Mezheyeuski, A. Quantitative, qualitative and spatial analysis of lymphocyte infiltration in periampullary and pancreatic adenocarcinoma. Int. J. Cancer 2020, 146, 3461–3473. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases--elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.E.; Beatty, G.L.; Vonderheide, R.H. Immunosurveillance of pancreatic adenocarcinoma: Insights from genetically engineered mouse models of cancer. Cancer Lett. 2009, 279, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ireland, L.; Luckett, T.; Schmid, M.C.; Mielgo, A. Blockade of Stromal Gas6 Alters Cancer Cell Plasticity, Activates NK Cells, and Inhibits Pancreatic Cancer Metastasis. Front. Immunol. 2020, 11, 297. [Google Scholar] [CrossRef] [PubMed]
- Miyara, M.; Sakaguchi, S. Natural regulatory T cells: Mechanisms of suppression. Trends Mol. Med. 2007, 13, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Inman, K.S.; Francis, A.A.; Murray, N.R. Complex role for the immune system in initiation and progression of pancreatic cancer. World J. Gastroenterol. 2014, 20, 11160–11181. [Google Scholar] [CrossRef]
- Goswami, K.K.; Ghosh, T.; Ghosh, S.; Sarkar, M.; Bose, A.; Baral, R. Tumor promoting role of anti-tumor macrophages in tumor microenvironment. Cell Immunol. 2017, 316, 1–10. [Google Scholar] [CrossRef]
- Laviron, M.; Boissonnas, A. Ontogeny of Tumor-Associated Macrophages. Front. Immunol. 2019, 10, 1799. [Google Scholar] [CrossRef]
- Arseni, L.; Lombardi, A.; Orioli, D. From Structure to Phenotype: Impact of Collagen Alterations on Human Health. Int. J. Mol. Sci. 2018, 19, 1407. [Google Scholar] [CrossRef]
- Holmes, D.F.; Lu, Y.; Starborg, T.; Kadler, K.E. Collagen Fibril Assembly and Function. Curr. Top. Dev. Biol. 2018, 130, 107–142. [Google Scholar] [CrossRef]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect Biol. 2011, 3, a004978. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Raines, R.T. Collagen structure and stability. Annu. Rev. Biochem. 2009, 78, 929–958. [Google Scholar] [CrossRef] [PubMed]
- Delaine-Smith, R.M.; Green, N.H.; Matcher, S.J.; MacNeil, S.; Reilly, G.C. Monitoring fibrous scaffold guidance of three-dimensional collagen organisation using minimally-invasive second harmonic generation. PLoS ONE 2014, 9, e89761. [Google Scholar] [CrossRef] [PubMed]
- Trappmann, B.; Gautrot, J.E.; Connelly, J.T.; Strange, D.G.; Li, Y.; Oyen, M.L.; Cohen Stuart, M.A.; Boehm, H.; Li, B.; Vogel, V.; et al. Extracellular-matrix tethering regulates stem-cell fate. Nat. Mater. 2012, 11, 642–649. [Google Scholar] [CrossRef]
- Sorushanova, A.; Delgado, L.M.; Wu, Z.; Shologu, N.; Kshirsagar, A.; Raghunath, R.; Mullen, A.M.; Bayon, Y.; Pandit, A.; Raghunath, M.; et al. The Collagen Suprafamily: From Biosynthesis to Advanced Biomaterial Development. Adv. Mater. 2019, 31, e1801651. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Whittaker, C.A.; Carr, S.A.; Tanabe, K.K.; Hynes, R.O. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer 2014, 14, 518. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Pearce, O.M.T.; Del Rosario, A.; Ma, D.; Ding, H.; Rajeeve, V.; Cutillas, P.R.; Balkwill, F.R.; Hynes, R.O. Characterization of the Extracellular Matrix of Normal and Diseased Tissues Using Proteomics. J. Proteome Res. 2017, 16, 3083–3091. [Google Scholar] [CrossRef]
- Ohno, S.; Tachibana, M.; Fujii, T.; Ueda, S.; Kubota, H.; Nagasue, N. Role of stromal collagen in immunomodulation and prognosis of advanced gastric carcinoma. Int. J. Cancer 2002, 97, 770–774. [Google Scholar] [CrossRef]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef]
- Carstens, J.L.; Correa de Sampaio, P.; Yang, D.; Barua, S.; Wang, H.; Rao, A.; Allison, J.P.; LeBleu, V.S.; Kalluri, R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 2017, 8, 15095. [Google Scholar] [CrossRef]
- Li, X.; Gruosso, T.; Zuo, D.; Omeroglu, A.; Meterissian, S.; Guiot, M.C.; Salazar, A.; Park, M.; Levine, H. Infiltration of CD8(+) T cells into tumor cell clusters in triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3678–3687. [Google Scholar] [CrossRef]
- Kuczek, D.E.; Larsen, A.M.H.; Thorseth, M.L.; Carretta, M.; Kalvisa, A.; Siersbaek, M.S.; Simoes, A.M.C.; Roslind, A.; Engelholm, L.H.; Noessner, E.; et al. Collagen density regulates the activity of tumor-infiltrating T cells. J. Immunother. Cancer 2019, 7, 68. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Fujitani, Y.; Fujitani, S.; Wright, C.V.; Moses, H.L. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006, 20, 3147–3160. [Google Scholar] [CrossRef] [PubMed]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.L.; Rios, E.; Silva, A.C.; Neves, S.C.; Caires, H.R.; Pinto, A.T.; Duraes, C.; Carvalho, F.A.; Cardoso, A.P.; Santos, N.C.; et al. Decellularized human colorectal cancer matrices polarize macrophages towards an anti-inflammatory phenotype promoting cancer cell invasion via CCL18. Biomaterials 2017, 124, 211–224. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, W.; Dai, X.; Zhang, C.; Zhang, Q.; Lu, J. Identification of the collagen family as prognostic biomarkers and immune-associated targets in gastric cancer. Int. Immunopharmacol. 2020, 87, 106798. [Google Scholar] [CrossRef]
- Chen, J.; Hou, C.; Zheng, Z.; Lin, H.; Lv, G.; Zhou, D. Identification of Secreted Phosphoprotein 1 (SPP1) as a Prognostic Factor in Lower-Grade Gliomas. World Neurosurg. 2019, 130, e775–e785. [Google Scholar] [CrossRef]
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419. [Google Scholar] [CrossRef]
- Zardi, L.; Cecconi, C.; Barbieri, O.; Carnemolla, B.; Picca, M.; Santi, L. Concentration of fibronectin in plasma of tumor-bearing mice and synthesis by Ehrlich ascites tumor cells. Cancer Res. 1979, 39, 3774–3779. [Google Scholar]
- Mao, Y.; Schwarzbauer, J.E. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol. 2005, 24, 389–399. [Google Scholar] [CrossRef]
- To, W.S.; Midwood, K.S. Plasma and cellular fibronectin: Distinct and independent functions during tissue repair. Fibrogenesis Tissue Repair 2011, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Chen, Q.; Sivakumar, P. Dynamics of assembly and reorganization of extracellular matrix proteins. Curr. Top. Dev. Biol. 2006, 75, 1–24. [Google Scholar] [CrossRef]
- Digiacomo, G.; Tusa, I.; Bacci, M.; Cipolleschi, M.G.; Dello Sbarba, P.; Rovida, E. Fibronectin induces macrophage migration through a SFK-FAK/CSF-1R pathway. Cell Adhes. Migr. 2017, 11, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Abshire, M.Y.; Thomas, K.S.; Owen, K.A.; Bouton, A.H. Macrophage motility requires distinct alpha5beta1/FAK and alpha4beta1/paxillin signaling events. J. Leukoc. Biol. 2011, 89, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Kamoshida, G.; Matsuda, A.; Sekine, W.; Mizuno, H.; Oku, T.; Itoh, S.; Irimura, T.; Tsuji, T. Monocyte differentiation induced by co-culture with tumor cells involves RGD-dependent cell adhesion to extracellular matrix. Cancer Lett. 2012, 315, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Kamoshida, G.; Matsuda, A.; Miura, R.; Takashima, Y.; Katsura, A.; Tsuji, T. Potentiation of tumor cell invasion by co-culture with monocytes accompanying enhanced production of matrix metalloproteinase and fibronectin. Clin. Exp. Metastasis 2013, 30, 289–297. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef]
- Rybak, J.N.; Roesli, C.; Kaspar, M.; Villa, A.; Neri, D. The extra-domain A of fibronectin is a vascular marker of solid tumors and metastases. Cancer Res. 2007, 67, 10948–10957. [Google Scholar] [CrossRef]
- Rossnagl, S.; Altrock, E.; Sens, C.; Kraft, S.; Rau, K.; Milsom, M.D.; Giese, T.; Samstag, Y.; Nakchbandi, I.A. EDA-Fibronectin Originating from Osteoblasts Inhibits the Immune Response against Cancer. PLoS Biol. 2016, 14, e1002562. [Google Scholar] [CrossRef]
- Qin, Y.; Rodin, S.; Simonson, O.E.; Hollande, F. Laminins and cancer stem cells: Partners in crime? Semin. Cancer Biol. 2017, 45, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Timpl, R.; Rohde, H.; Robey, P.G.; Rennard, S.I.; Foidart, J.M.; Martin, G.R. Laminin—A glycoprotein from basement membranes. J. Biol. Chem. 1979, 254, 9933–9937. [Google Scholar] [PubMed]
- Aumailley, M. The laminin family. Cell Adhes. Migr. 2013, 7, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Sekiguchi, K. Molecular Basis of Laminin-Integrin Interactions. Curr. Top. Membr. 2015, 76, 197–229. [Google Scholar] [CrossRef]
- Rodin, S.; Domogatskaya, A.; Strom, S.; Hansson, E.M.; Chien, K.R.; Inzunza, J.; Hovatta, O.; Tryggvason, K. Long-term self-renewal of human pluripotent stem cells on human recombinant laminin-511. Nat. Biotechnol. 2010, 28, 611–615. [Google Scholar] [CrossRef]
- Miyazaki, T.; Futaki, S.; Hasegawa, K.; Kawasaki, M.; Sanzen, N.; Hayashi, M.; Kawase, E.; Sekiguchi, K.; Nakatsuji, N.; Suemori, H. Recombinant human laminin isoforms can support the undifferentiated growth of human embryonic stem cells. Biochem. Biophys. Res. Commun. 2008, 375, 27–32. [Google Scholar] [CrossRef]
- Simon, T.; Bromberg, J.S. Regulation of the Immune System by Laminins. Trends Immunol. 2017, 38, 858–871. [Google Scholar] [CrossRef]
- Fernandes, N.R.J.; Reilly, N.S.; Schrock, D.C.; Hocking, D.C.; Oakes, P.W.; Fowell, D.J. CD4(+) T Cell Interstitial Migration Controlled by Fibronectin in the Inflamed Skin. Front. Immunol. 2020, 11, 1501. [Google Scholar] [CrossRef]
- Sixt, M.; Engelhardt, B.; Pausch, F.; Hallmann, R.; Wendler, O.; Sorokin, L.M. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J. Cell Biol. 2001, 153, 933–946. [Google Scholar] [CrossRef]
- Warren, K.J.; Iwami, D.; Harris, D.G.; Bromberg, J.S.; Burrell, B.E. Laminins affect T cell trafficking and allograft fate. J. Clin. Investig. 2014, 124, 2204–2218. [Google Scholar] [CrossRef]
- Wu, C.; Ivars, F.; Anderson, P.; Hallmann, R.; Vestweber, D.; Nilsson, P.; Robenek, H.; Tryggvason, K.; Song, J.; Korpos, E.; et al. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat. Med. 2009, 15, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Rodriguez, L.; Martinez-Rey, D.; Fernandez-Acenero, M.J.; Gonzalez-Martin, A.; Paz-Cabezas, M.; Rodriguez-Rodriguez, N.; Perez-Villamil, B.; Saez, M.E.; Diaz-Rubio, E.; Mira, E.; et al. SOD3 induces a HIF-2alpha-dependent program in endothelial cells that provides a selective signal for tumor infiltration by T cells. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Kohl, K.; Schnautz, S.; Pesch, M.; Klein, E.; Aumailley, M.; Bieber, T.; Koch, S. Subpopulations of human dendritic cells display a distinct phenotype and bind differentially to proteins of the extracellular matrix. Eur. J. Cell Biol. 2007, 86, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.H. Laminins and their roles in mammals. Microsc. Res. Tech. 2008, 71, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Phillippi, B.; Singh, M.; Loftus, T.; Smith, H.; Muccioli, M.; Wright, J.; Pate, M.; Benencia, F. Effect of laminin environments and tumor factors on the biology of myeloid dendritic cells. Immunobiology 2020, 225, 151854. [Google Scholar] [CrossRef]
- Lin, J.H.; Huffman, A.P.; Wattenberg, M.M.; Walter, D.M.; Carpenter, E.L.; Feldser, D.M.; Beatty, G.L.; Furth, E.E.; Vonderheide, R.H. Type 1 conventional dendritic cells are systemically dysregulated early in pancreatic carcinogenesis. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Scandolera, A.; Odoul, L.; Salesse, S.; Guillot, A.; Blaise, S.; Kawecki, C.; Maurice, P.; El Btaouri, H.; Romier-Crouzet, B.; Martiny, L.; et al. The Elastin Receptor Complex: A Unique Matricellular Receptor with High Anti-tumoral Potential. Front. Pharmacol. 2016, 7, 32. [Google Scholar] [CrossRef]
- Vindin, H.; Mithieux, S.M.; Weiss, A.S. Elastin architecture. Matrix Biol. 2019, 84, 4–16. [Google Scholar] [CrossRef]
- Wise, S.G.; Weiss, A.S. Tropoelastin. Int. J. Biochem. Cell Biol. 2009, 41, 494–497. [Google Scholar] [CrossRef]
- Salesse, S.; Odoul, L.; Chazee, L.; Garbar, C.; Duca, L.; Martiny, L.; Mahmoudi, R.; Debelle, L. Elastin molecular aging promotes MDA-MB-231 breast cancer cell invasiveness. FEBS Open Bio 2018, 8, 1395–1404. [Google Scholar] [CrossRef]
- Li, J.; Xu, X.; Jiang, Y.; Hansbro, N.G.; Hansbro, P.M.; Xu, J.; Liu, G. Elastin is a key factor of tumor development in colorectal cancer. BMC Cancer 2020, 20, 217. [Google Scholar] [CrossRef] [PubMed]
- Houghton, A.M.; Quintero, P.A.; Perkins, D.L.; Kobayashi, D.K.; Kelley, D.G.; Marconcini, L.A.; Mecham, R.P.; Senior, R.M.; Shapiro, S.D. Elastin fragments drive disease progression in a murine model of emphysema. J. Clin. Investig. 2006, 116, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Murphy-Ullrich, J.E.; Sage, E.H. Revisiting the matricellular concept. Matrix Biol. 2014, 37, 1–14. [Google Scholar] [CrossRef]
- Sangaletti, S.; Di Carlo, E.; Gariboldi, S.; Miotti, S.; Cappetti, B.; Parenza, M.; Rumio, C.; Brekken, R.A.; Chiodoni, C.; Colombo, M.P. Macrophage-derived SPARC bridges tumor cell-extracellular matrix interactions toward metastasis. Cancer Res. 2008, 68, 9050–9059. [Google Scholar] [CrossRef] [PubMed]
- Viloria, K.; Hill, N.J. Embracing the complexity of matricellular proteins: The functional and clinical significance of splice variation. Biomol. Concepts 2016, 7, 117–132. [Google Scholar] [CrossRef]
- Sage, E.H.; Reed, M.; Funk, S.E.; Truong, T.; Steadele, M.; Puolakkainen, P.; Maurice, D.H.; Bassuk, J.A. Cleavage of the matricellular protein SPARC by matrix metalloproteinase 3 produces polypeptides that influence angiogenesis. J. Biol. Chem. 2003, 278, 37849–37857. [Google Scholar] [CrossRef]
- Kehlet, S.N.; Manon-Jensen, T.; Sun, S.; Brix, S.; Leeming, D.J.; Karsdal, M.A.; Willumsen, N. A fragment of SPARC reflecting increased collagen affinity shows pathological relevance in lung cancer—Implications of a new collagen chaperone function of SPARC. Cancer Biol. Ther. 2018, 19, 904–912. [Google Scholar] [CrossRef]
- Bradshaw, A.D.; Sage, E.H. SPARC, a matricellular protein that functions in cellular differentiation and tissue response to injury. J. Clin. Investig. 2001, 107, 1049–1054. [Google Scholar] [CrossRef]
- Bradshaw, A.D. The role of SPARC in extracellular matrix assembly. J. Cell Commun. Signal. 2009, 3, 239–246. [Google Scholar] [CrossRef]
- Gilles, C.; Bassuk, J.A.; Pulyaeva, H.; Sage, E.H.; Foidart, J.M.; Thompson, E.W. SPARC/osteonectin induces matrix metalloproteinase 2 activation in human breast cancer cell lines. Cancer Res. 1998, 58, 5529–5536. [Google Scholar]
- Weaver, M.S.; Workman, G.; Sage, E.H. The copper binding domain of SPARC mediates cell survival in vitro via interaction with integrin beta1 and activation of integrin-linked kinase. J. Biol. Chem. 2008, 283, 22826–22837. [Google Scholar] [CrossRef] [PubMed]
- Guweidhi, A.; Kleeff, J.; Adwan, H.; Giese, N.A.; Wente, M.N.; Giese, T.; Büchler, M.W.; Berger, M.R.; Friess, H. Osteonectin influences growth and invasion of pancreatic cancer cells. Ann. Surg. 2005, 242, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, A.D.; Puolakkainen, P.; Dasgupta, J.; Davidson, J.M.; Wight, T.N.; Helene Sage, E. SPARC-null mice display abnormalities in the dermis characterized by decreased collagen fibril diameter and reduced tensile strength. J. Investig. Dermatol. 2003, 120, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Sage, E.H. SPARC, a matricellular glycoprotein with important biological functions. J. Histochem. Cytochem. 1999, 47, 1495–1506. [Google Scholar] [CrossRef]
- Chen, J.; Wang, M.; Xi, B.; Xue, J.; He, D.; Zhang, J.; Zhao, Y. SPARC is a key regulator of proliferation, apoptosis and invasion in human ovarian cancer. PLoS ONE 2012, 7, e42413. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, Y.; Da, J.; Jia, Z.; Wu, H.; Gu, K. Downregulation of SPARC Expression Decreases Cell Migration and Invasion Involving Epithelial-Mesenchymal Transition through the p-FAK/p-ERK Pathway in Esophageal Squamous Cell Carcinoma. J. Cancer 2020, 11, 414–420. [Google Scholar] [CrossRef]
- Rempel, S.A.; Golembieski, W.A.; Fisher, J.L.; Maile, M.; Nakeff, A. SPARC modulates cell growth, attachment and migration of U87 glioma cells on brain extracellular matrix proteins. J. Neurooncol. 2001, 53, 149–160. [Google Scholar] [CrossRef]
- Schultz, C.; Lemke, N.; Ge, S.; Golembieski, W.A.; Rempel, S.A. Secreted protein acidic and rich in cysteine promotes glioma invasion and delays tumor growth in vivo. Cancer Res. 2002, 62, 6270–6277. [Google Scholar]
- Chin, D.; Boyle, G.M.; Williams, R.M.; Ferguson, K.; Pandeya, N.; Pedley, J.; Campbell, C.M.; Theile, D.R.; Parsons, P.G.; Coman, W.B. Novel markers for poor prognosis in head and neck cancer. Int. J. Cancer 2005, 113, 789–797. [Google Scholar] [CrossRef]
- Kato, Y.; Nagashima, Y.; Baba, Y.; Kawano, T.; Furukawa, M.; Kubota, A.; Yanoma, S.; Imagawa-Ishiguro, Y.; Satake, K.; Taguchi, T.; et al. Expression of SPARC in tongue carcinoma of stage II is associated with poor prognosis: An immunohistochemical study of 86 cases. Int. J. Mol. Med. 2005, 16, 263–268. [Google Scholar] [CrossRef]
- Kurtul, N.; Tasdemir, E.A.; Unal, D.; Izmirli, M.; Eroglu, C. SPARC: As a prognostic biomarker in rectal cancer patients treated with chemo-radiotherapy. Cancer Biomark. 2017, 18, 459–466. [Google Scholar] [CrossRef] [PubMed]
- John, B.; Naczki, C.; Patel, C.; Ghoneum, A.; Qasem, S.; Salih, Z.; Said, N. Regulation of the bi-directional cross-talk between ovarian cancer cells and adipocytes by SPARC. Oncogene 2019, 38, 4366–4383. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Matsubayashi, H.; Sato, N.; Tonascia, J.; Klein, A.P.; Riall, T.A.; Yeo, C.; Iacobuzio-Donahue, C.; Goggins, M. Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J. Clin. Oncol. 2007, 25, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Puolakkainen, P.A.; Brekken, R.A.; Muneer, S.; Sage, E.H. Enhanced growth of pancreatic tumors in SPARC-null mice is associated with decreased deposition of extracellular matrix and reduced tumor cell apoptosis. Mol. Cancer Res. 2004, 2, 215–224. [Google Scholar]
- Zhang, J.; Wang, P.; Zhu, J.; Wang, W.; Yin, J.; Zhang, C.; Chen, Z.; Sun, L.; Wan, Y.; Wang, X.; et al. SPARC expression is negatively correlated with clinicopathological factors of gastric cancer and inhibits malignancy of gastric cancer cells. Oncol. Rep. 2014, 31, 2312–2320. [Google Scholar] [CrossRef]
- Arnold, S.A.; Rivera, L.B.; Miller, A.F.; Carbon, J.G.; Dineen, S.P.; Xie, Y.; Castrillon, D.H.; Sage, E.H.; Puolakkainen, P.; Bradshaw, A.D.; et al. Lack of host SPARC enhances vascular function and tumor spread in an orthotopic murine model of pancreatic carcinoma. Dis. Model. Mech. 2010, 3, 57–72. [Google Scholar] [CrossRef]
- Kzhyshkowska, J.; Workman, G.; Cardo-Vila, M.; Arap, W.; Pasqualini, R.; Gratchev, A.; Krusell, L.; Goerdt, S.; Sage, E.H. Novel function of alternatively activated macrophages: Stabilin-1-mediated clearance of SPARC. J. Immunol. 2006, 176, 5825–5832. [Google Scholar] [CrossRef]
- Arnold, S.A.; Rivera, L.B.; Carbon, J.G.; Toombs, J.E.; Chang, C.L.; Bradshaw, A.D.; Brekken, R.A. Losartan slows pancreatic tumor progression and extends survival of SPARC-null mice by abrogating aberrant TGFbeta activation. PLoS ONE 2012, 7, e31384. [Google Scholar] [CrossRef]
- Rempel, S.A.; Hawley, R.C.; Gutierrez, J.A.; Mouzon, E.; Bobbitt, K.R.; Lemke, N.; Schultz, C.R.; Schultz, L.R.; Golembieski, W.; Koblinski, J.; et al. Splenic and immune alterations of the Sparc-null mouse accompany a lack of immune response. Genes Immun. 2007, 8, 262–274. [Google Scholar] [CrossRef]
- Hu, J.; Ma, Y.; Ma, J.; Chen, S.; Zhang, X.; Guo, S.; Huang, Z.; Yue, T.; Yang, Y.; Ning, Y.; et al. Macrophage-derived SPARC Attenuates M2-mediated Pro-tumour Phenotypes. J. Cancer 2020, 11, 2981–2992. [Google Scholar] [CrossRef]
- Said, N.; Frierson, H.F.; Sanchez-Carbayo, M.; Brekken, R.A.; Theodorescu, D. Loss of SPARC in bladder cancer enhances carcinogenesis and progression. J. Clin. Investig. 2013, 123, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Denhardt, D.T.; Guo, X. Osteopontin: A protein with diverse functions. FASEB J. 1993, 7, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Irby, R.B.; McCarthy, S.M.; Yeatman, T.J. Osteopontin regulates multiple functions contributing to human colon cancer development and progression. Clin. Exp. Metastasis 2004, 21, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Patarca, R.; Freeman, G.J.; Singh, R.P.; Wei, F.Y.; Durfee, T.; Blattner, F.; Regnier, D.C.; Kozak, C.A.; Mock, B.A.; Morse, H.C., 3rd; et al. Structural and functional studies of the early T lymphocyte activation 1 (Eta-1) gene. Definition of a novel T cell-dependent response associated with genetic resistance to bacterial infection. J. Exp. Med. 1989, 170, 145–161. [Google Scholar] [CrossRef]
- Weber, G.F.; Ashkar, S.; Glimcher, M.J.; Cantor, H. Receptor-ligand interaction between CD44 and osteopontin (Eta-1). Science 1996, 271, 509–512. [Google Scholar] [CrossRef]
- Icer, M.A.; Gezmen-Karadag, M. The multiple functions and mechanisms of osteopontin. Clin. Biochem. 2018, 59, 17–24. [Google Scholar] [CrossRef]
- Baaten, B.J.; Li, C.R.; Bradley, L.M. Multifaceted regulation of T cells by CD44. Commun. Integr. Biol. 2010, 3, 508–512. [Google Scholar] [CrossRef]
- Jefferies, C.A. Regulating IRFs in IFN Driven Disease. Front. Immunol. 2019, 10, 325. [Google Scholar] [CrossRef]
- McGough, J.M.; Yang, D.; Huang, S.; Georgi, D.; Hewitt, S.M.; Rocken, C.; Tanzer, M.; Ebert, M.P.; Liu, K. DNA methylation represses IFN-gamma-induced and signal transducer and activator of transcription 1-mediated IFN regulatory factor 8 activation in colon carcinoma cells. Mol. Cancer Res. 2008, 6, 1841–1851. [Google Scholar] [CrossRef]
- Klement, J.D.; Paschall, A.V.; Redd, P.S.; Ibrahim, M.L.; Lu, C.; Yang, D.; Celis, E.; Abrams, S.I.; Ozato, K.; Liu, K. An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J. Clin. Investig. 2018, 128, 5549–5560. [Google Scholar] [CrossRef]
- Lin, C.N.; Wang, C.J.; Chao, Y.J.; Lai, M.D.; Shan, Y.S. The significance of the co-existence of osteopontin and tumor-associated macrophages in gastric cancer progression. BMC Cancer 2015, 15, 128. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Lee, S.H.; Go du, M.; Kim, H.K.; Kwon, H.J.; Kim, D.Y. Osteopontin depletion decreases inflammation and gastric epithelial proliferation during Helicobacter pylori infection in mice. Lab. Investig. 2015, 95, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, W.; Chen, Z.; Xiang, C. Upregulation of PD-L1 by SPP1 mediates macrophage polarization and facilitates immune escape in lung adenocarcinoma. Exp. Cell Res. 2017, 359, 449–457. [Google Scholar] [CrossRef]
- Kudo, A. Introductory review: Periostin-gene and protein structure. Cell Mol. Life Sci. 2017, 74, 4259–4268. [Google Scholar] [CrossRef]
- Bonnet, N.; Garnero, P.; Ferrari, S. Periostin action in bone. Mol. Cell Endocrinol. 2016, 432, 75–82. [Google Scholar] [CrossRef]
- Canty, E.G.; Kadler, K.E. Procollagen trafficking, processing and fibrillogenesis. J. Cell Sci. 2005, 118, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Jin, R.; Norris, R.A.; Zhang, L.; Yu, S.; Wu, F.; Markwald, R.R.; Nanda, A.; Conway, S.J.; Smyth, S.S.; et al. Periostin mediates vascular smooth muscle cell migration through the integrins alphavbeta3 and alphavbeta5 and focal adhesion kinase (FAK) pathway. Atherosclerosis 2010, 208, 358–365. [Google Scholar] [CrossRef]
- Kii, I.; Ito, H. Periostin and its interacting proteins in the construction of extracellular architectures. Cell Mol. Life Sci. 2017, 74, 4269–4277. [Google Scholar] [CrossRef]
- Liu, Y.; Du, L. Role of pancreatic stellate cells and periostin in pancreatic cancer progression. Tumour Biol. 2015, 36, 3171–3177. [Google Scholar] [CrossRef]
- Liu, Y.; Li, F.; Gao, F.; Xing, L.; Qin, P.; Liang, X.; Zhang, J.; Qiao, X.; Lin, L.; Zhao, Q.; et al. Role of microenvironmental periostin in pancreatic cancer progression. Oncotarget 2017, 8, 89552–89565. [Google Scholar] [CrossRef]
- González-González, L.; Alonso, J. Periostin: A Matricellular Protein With Multiple Functions in Cancer Development and Progression. Front. Oncol. 2018, 8, 225. [Google Scholar] [CrossRef]
- Shi, C.; Washington, M.K.; Chaturvedi, R.; Drosos, Y.; Revetta, F.L.; Weaver, C.J.; Buzhardt, E.; Yull, F.E.; Blackwell, T.S.; Sosa-Pineda, B.; et al. Fibrogenesis in pancreatic cancer is a dynamic process regulated by macrophage-stellate cell interaction. Lab. Investig. 2014, 94, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.R.; Quaranta, V.; Linford, A.; Emeagi, P.; Rainer, C.; Santos, A.; Ireland, L.; Sakai, T.; Sakai, K.; Kim, Y.S.; et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat. Cell Biol. 2016, 18, 549–560. [Google Scholar] [CrossRef]
- Koh, S.J.; Kim, J.W.; Kim, B.G.; Lee, K.L.; Kim, D.W.; Kim, J.S. Matricellular protein periostin promotes colitis-associated colon tumorigenesis in mice. Carcinogenesis 2019, 40, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Han, Z.; Chandoo, A.; Huang, X.; Sun, X.; Ye, L.; Hu, C.; Xue, X.; Huang, Y.; Shen, X.; et al. Low periostin expression predicts poor survival in intestinal type gastric cancer patients. Cancer Manag. Res. 2019, 11, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Liu, B.; Bu, X.; Zhao, P. Cross-talk between ovarian cancer cells and macrophages through periostin promotes macrophage recruitment. Cancer Sci. 2018, 109, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Yokota, K.; Kobayakawa, K.; Saito, T.; Hara, M.; Kijima, K.; Ohkawa, Y.; Harada, A.; Okazaki, K.; Ishihara, K.; Yoshida, S.; et al. Periostin Promotes Scar Formation through the Interaction between Pericytes and Infiltrating Monocytes/Macrophages after Spinal Cord Injury. Am. J. Pathol. 2017, 187, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Ohno, F.; Nakahara, T.; Kido-Nakahara, M.; Ito, T.; Nunomura, S.; Izuhara, K.; Furue, M. Periostin Links Skin Inflammation to Melanoma Progression in Humans and Mice. Int. J. Mol. Sci. 2019, 20, 169. [Google Scholar] [CrossRef] [PubMed]
- Skonier, J.; Neubauer, M.; Madisen, L.; Bennett, K.; Plowman, G.D.; Purchio, A.F. cDNA cloning and sequence analysis of beta ig-h3, a novel gene induced in a human adenocarcinoma cell line after treatment with transforming growth factor-beta. DNA Cell Biol. 1992, 11, 511–522. [Google Scholar] [CrossRef]
- Skonier, J.; Bennett, K.; Rothwell, V.; Kosowski, S.; Plowman, G.; Wallace, P.; Edelhoff, S.; Disteche, C.; Neubauer, M.; Marquardt, H.; et al. beta ig-h3: A transforming growth factor-beta-responsive gene encoding a secreted protein that inhibits cell attachment in vitro and suppresses the growth of CHO cells in nude mice. DNA Cell Biol. 1994, 13, 571–584. [Google Scholar] [CrossRef]
- Bae, J.S.; Lee, S.H.; Kim, J.E.; Choi, J.Y.; Park, R.W.; Yong Park, J.; Park, H.S.; Sohn, Y.S.; Lee, D.S.; Bae Lee, E.; et al. Betaig-h3 supports keratinocyte adhesion, migration, and proliferation through alpha3beta1 integrin. Biochem. Biophys. Res. Commun. 2002, 294, 940–948. [Google Scholar] [CrossRef]
- Nam, J.O.; Kim, J.E.; Jeong, H.W.; Lee, S.J.; Lee, B.H.; Choi, J.Y.; Park, R.W.; Park, J.Y.; Kim, I.S. Identification of the alphavbeta3 integrin-interacting motif of betaig-h3 and its anti-angiogenic effect. J. Biol. Chem. 2003, 278, 25902–25909. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Noshiro, M.; Makihira, S.; Kawamoto, T.; Shen, M.; Yan, W.; Kawashima-Ohya, Y.; Fujimoto, K.; Tanne, K.; Kato, Y. RGD-CAP ((beta)ig-h3) enhances the spreading of chondrocytes and fibroblasts via integrin alpha(1)beta(1). Biochim. Biophys. Acta 1999, 1451, 196–205. [Google Scholar] [CrossRef]
- Ma, C.; Rong, Y.; Radiloff, D.R.; Datto, M.B.; Centeno, B.; Bao, S.; Cheng, A.W.; Lin, F.; Jiang, S.; Yeatman, T.J.; et al. Extracellular matrix protein betaig-h3/TGFBI promotes metastasis of colon cancer by enhancing cell extravasation. Genes Dev. 2008, 22, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Noshiro, M.; Ohno, S.; Kawamoto, T.; Satakeda, H.; Akagawa, Y.; Nakashima, K.; Okimura, A.; Ishida, H.; Okamoto, T.; et al. Characterization of a cartilage-derived 66-kDa protein (RGD-CAP/beta ig-h3) that binds to collagen. Biochim. Biophys. Acta 1997, 1355, 303–314. [Google Scholar] [CrossRef]
- Billings, P.C.; Whitbeck, J.C.; Adams, C.S.; Abrams, W.R.; Cohen, A.J.; Engelsberg, B.N.; Howard, P.S.; Rosenbloom, J. The transforming growth factor-beta-inducible matrix protein (beta)ig-h3 interacts with fibronectin. J. Biol. Chem. 2002, 277, 28003–28009. [Google Scholar] [CrossRef] [PubMed]
- Goehrig, D.; Nigri, J.; Samain, R.; Wu, Z.; Cappello, P.; Gabiane, G.; Zhang, X.; Zhao, Y.; Kim, I.S.; Chanal, M.; et al. Stromal protein betaig-h3 reprogrammes tumour microenvironment in pancreatic cancer. Gut 2019, 68, 693–707. [Google Scholar] [CrossRef] [PubMed]
- Costanza, B.; Rademaker, G.; Tiamiou, A.; De Tullio, P.; Leenders, J.; Blomme, A.; Bellier, J.; Bianchi, E.; Turtoi, A.; Delvenne, P.; et al. Transforming growth factor beta-induced, an extracellular matrix interacting protein, enhances glycolysis and promotes pancreatic cancer cell migration. Int. J. Cancer 2019, 145, 1570–1584. [Google Scholar] [CrossRef]
- Patry, M.; Teinturier, R.; Goehrig, D.; Zetu, C.; Ripoche, D.; Kim, I.S.; Bertolino, P.; Hennino, A. betaig-h3 Represses T-Cell Activation in Type 1 Diabetes. Diabetes 2015, 64, 4212–4219. [Google Scholar] [CrossRef]
- Fico, F.; Santamaria-Martinez, A. TGFBI modulates tumour hypoxia and promotes breast cancer metastasis. Mol. Oncol. 2020, 14, 3198–3210. [Google Scholar] [CrossRef]
- Nacu, N.; Luzina, I.G.; Highsmith, K.; Lockatell, V.; Pochetuhen, K.; Cooper, Z.A.; Gillmeister, M.P.; Todd, N.W.; Atamas, S.P. Macrophages produce TGF-beta-induced (beta-ig-h3) following ingestion of apoptotic cells and regulate MMP14 levels and collagen turnover in fibroblasts. J. Immunol. 2008, 180, 5036–5044. [Google Scholar] [CrossRef] [PubMed]
- Steitz, A.M.; Steffes, A.; Finkernagel, F.; Unger, A.; Sommerfeld, L.; Jansen, J.M.; Wagner, U.; Graumann, J.; Muller, R.; Reinartz, S. Tumor-associated macrophages promote ovarian cancer cell migration by secreting transforming growth factor beta induced (TGFBI) and tenascin C. Cell Death Dis. 2020, 11, 249. [Google Scholar] [CrossRef] [PubMed]
- Wight, T.N.; Kang, I.; Evanko, S.P.; Harten, I.A.; Chang, M.Y.; Pearce, O.M.T.; Allen, C.E.; Frevert, C.W. Versican-A Critical Extracellular Matrix Regulator of Immunity and Inflammation. Front. Immunol. 2020, 11, 512. [Google Scholar] [CrossRef] [PubMed]
- Wight, T.N. Versican: A versatile extracellular matrix proteoglycan in cell biology. Curr. Opin. Cell Biol. 2002, 14, 617–623. [Google Scholar] [CrossRef]
- Bögels, M.; Braster, R.; Nijland, P.G.; Gül, N.; van de Luijtgaarden, W.; Fijneman, R.J.; Meijer, G.A.; Jimenez, C.R.; Beelen, R.H.; van Egmond, M. Carcinoma origin dictates differential skewing of monocyte function. Oncoimmunology 2012, 1, 798–809. [Google Scholar] [CrossRef]
- Kim, S.; Takahashi, H.; Lin, W.W.; Descargues, P.; Grivennikov, S.; Kim, Y.; Luo, J.L.; Karin, M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009, 457, 102–106. [Google Scholar] [CrossRef]
- Hope, C.; Foulcer, S.; Jagodinsky, J.; Chen, S.X.; Jensen, J.L.; Patel, S.; Leith, C.; Maroulakou, I.; Callander, N.; Miyamoto, S.; et al. Immunoregulatory roles of versican proteolysis in the myeloma microenvironment. Blood 2016, 128, 680–685. [Google Scholar] [CrossRef]
- Hope, C.; Emmerich, P.B.; Papadas, A.; Pagenkopf, A.; Matkowskyj, K.A.; Van De Hey, D.R.; Payne, S.N.; Clipson, L.; Callander, N.S.; Hematti, P.; et al. Versican-Derived Matrikines Regulate Batf3-Dendritic Cell Differentiation and Promote T Cell Infiltration in Colorectal Cancer. J. Immunol. 2017, 199, 1933–1941. [Google Scholar] [CrossRef]
- Li, W.; Han, F.; Fu, M.; Wang, Z. High expression of VCAN is an independent predictor of poor prognosis in gastric cancer. J. Int. Med. Res. 2020, 48, 300060519891271. [Google Scholar] [CrossRef]
- Cheng, Y.; Sun, H.; Wu, L.; Wu, F.; Tang, W.; Wang, X.; Lv, C. VUp-Regulation of VCAN Promotes the Proliferation, Invasion and Migration and Serves as a Biomarker in Gastric Cancer. Onco Targets Ther. 2020, 13, 8665–8675. [Google Scholar] [CrossRef]
- Gorter, A.; Zijlmans, H.J.; van Gent, H.; Trimbos, J.B.; Fleuren, G.J.; Jordanova, E.S. Versican expression is associated with tumor-infiltrating CD8-positive T cells and infiltration depth in cervical cancer. Mod. Pathol. 2010, 23, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Pappas, A.G.; Magkouta, S.; Pateras, I.S.; Skianis, I.; Moschos, C.; Vazakidou, M.E.; Psarra, K.; Gorgoulis, V.G.; Kalomenidis, I. Versican modulates tumor-associated macrophage properties to stimulate mesothelioma growth. Oncoimmunology 2019, 8, e1537427. [Google Scholar] [CrossRef] [PubMed]
- Hartley, G.; Regan, D.; Guth, A.; Dow, S. Regulation of PD-L1 expression on murine tumor-associated monocytes and macrophages by locally produced TNF-alpha. Cancer Immunol. Immunother 2017, 66, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Franklin, O.; Lundberg, E.; Lundin, C.; Sund, M. Type IV collagen stimulates pancreatic cancer cell proliferation, migration, and inhibits apoptosis through an autocrine loop. BMC Cancer 2013, 13, 154. [Google Scholar] [CrossRef] [PubMed]
- Amrutkar, M.; Aasrum, M.; Verbeke, C.S.; Gladhaug, I.P. Secretion of fibronectin by human pancreatic stellate cells promotes chemoresistance to gemcitabine in pancreatic cancer cells. BMC Cancer 2019, 19, 596. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, C.; Ye, Y.; Wang, Z.; He, Y.; Li, Y.; Mao, H. High expression of fibronectin 1 indicates poor prognosis in gastric cancer. Oncol. Lett. 2020, 19, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Liu, C.; Zhang, T.N.; Zhu, Y.W.; Dong, X.; Xue, P. Down-regulation of FN1 inhibits colorectal carcinogenesis by suppressing proliferation, migration, and invasion. J. Cell Biochem. 2018, 119, 4717–4728. [Google Scholar] [CrossRef]
- Lin, T.C.; Yang, C.H.; Cheng, L.H.; Chang, W.T.; Lin, Y.R.; Cheng, H.C. Fibronectin in Cancer: Friend or Foe. Cells 2019, 9, 27. [Google Scholar] [CrossRef]
- Zheng, B.; Qu, J.; Ohuchida, K.; Feng, H.; Chong, S.J.F.; Yan, Z.; Piao, Y.; Liu, P.; Sheng, N.; Eguchi, D.; et al. LAMA4 upregulation is associated with high liver metastasis potential and poor survival outcome of Pancreatic Cancer. Theranostics 2020, 10, 10274–10289. [Google Scholar] [CrossRef]
- Lin, Y.; Ge, X.; Zhang, X.; Wu, Z.; Liu, K.; Lin, F.; Dai, C.; Guo, W.; Li, J. Protocadherin-8 promotes invasion and metastasis via laminin subunit gamma2 in gastric cancer. Cancer Sci. 2018, 109, 732–740. [Google Scholar] [CrossRef]
- Galatenko, V.V.; Maltseva, D.V.; Galatenko, A.V.; Rodin, S.; Tonevitsky, A.G. Cumulative prognostic power of laminin genes in colorectal cancer. BMC Med. Genom. 2018, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, T.; Rybarczyk, P.; Bretaudeau, C.; Vanlaeys, A.; Cousin, R.; Brassart-Pasco, S.; Chatelain, D.; Dhennin-Duthille, I.; Ouadid-Ahidouch, H.; Brassart, B.; et al. TRPM7/RPSA Complex Regulates Pancreatic Cancer Cell Migration. Front. Cell Dev. Biol. 2020, 8, 549. [Google Scholar] [CrossRef] [PubMed]
- Maeng, H.Y.; Song, S.B.; Choi, D.K.; Kim, K.E.; Jeong, H.Y.; Sakaki, Y.; Furihata, C. Osteonectin-expressing cells in human stomach cancer and their possible clinical significance. Cancer Lett. 2002, 184, 117–121. [Google Scholar] [CrossRef]
- Drev, D.; Harpain, F.; Beer, A.; Stift, A.; Gruber, E.S.; Klimpfinger, M.; Thalhammer, S.; Reti, A.; Kenner, L.; Bergmann, M.; et al. Impact of Fibroblast-Derived SPARC on Invasiveness of Colorectal Cancer Cells. Cancers 2019, 11, 1421. [Google Scholar] [CrossRef]
- Khetan, K.; Baloda, V.; Sahoo, R.K.; Vishnubhathla, S.; Yadav, R.; Saraya, A.; Sharma, A.; Gupta, S.D.; Das, P. SPARC expression in desmoplastic and non desmoplastic pancreatic carcinoma and cholangiocarcinoma. Pathol. Res. Pract. 2019, 215, 152685. [Google Scholar] [CrossRef] [PubMed]
- Zhivkova-Galunska, M.; Adwan, H.; Eyol, E.; Kleeff, J.; Kolb, A.; Bergmann, F.; Berger, M.R. Osteopontin but not osteonectin favors the metastatic growth of pancreatic cancer cell lines. Cancer Biol. Ther. 2010, 10, 54–64. [Google Scholar] [CrossRef][Green Version]
- Gu, X.; Gao, X.S.; Ma, M.; Qin, S.; Qi, X.; Li, X.; Sun, S.; Yu, H.; Wang, W.; Zhou, D. Prognostic significance of osteopontin expression in gastric cancer: A meta-analysis. Oncotarget 2016, 7, 69666–69673. [Google Scholar] [CrossRef]
- Wei, R.; Wong, J.P.C.; Lyu, P.; Xi, X.; Tong, O.; Zhang, S.D.; Yuen, H.F.; Shirasawa, S.; Kwok, H.F. In vitro and clinical data analysis of Osteopontin as a prognostic indicator in colorectal cancer. J. Cell Mol. Med. 2018, 22, 4097–4105. [Google Scholar] [CrossRef]
- Likui, W.; Hong, W.; Shuwen, Z. Clinical significance of the upregulated osteopontin mRNA expression in human colorectal cancer. J. Gastrointest. Surg. 2010, 14, 74–81. [Google Scholar] [CrossRef]
- Sedivy, R.; Peters, K.; Kloppel, G. Osteopontin expression in ductal adenocarcinomas and undifferentiated carcinomas of the pancreas. Virchows Arch. 2005, 446, 41–45. [Google Scholar] [CrossRef]
- Liu, Y.; Li, F.; Gao, F.; Xing, L.; Qin, P.; Liang, X.; Zhang, J.; Qiao, X.; Lin, L.; Zhao, Q.; et al. Periostin promotes tumor angiogenesis in pancreatic cancer via Erk/VEGF signaling. Oncotarget 2016, 7, 40148–40159. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Yokobori, T.; Gombodorj, N.; Yashiro, M.; Turtoi, A.; Handa, T.; Ogata, K.; Oyama, T.; Shirabe, K.; Kuwano, H. High stromal transforming growth factor beta-induced expression is a novel marker of progression and poor prognosis in gastric cancer. J. Surg. Oncol. 2018, 118, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Skandalis, S.S.; Kletsas, D.; Kyriakopoulou, D.; Stavropoulos, M.; Theocharis, D.A. The greatly increased amounts of accumulated versican and decorin with specific post-translational modifications may be closely associated with the malignant phenotype of pancreatic cancer. Biochim. Biophys. Acta 2006, 1760, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.H.; Lin, W.R.; Xu, M.D.; Qi, P.; Dong, L.; Zhang, Q.Y.; Ni, S.J.; Weng, W.W.; Tan, C.; Huang, D.; et al. Prognostic significance of Versican expression in gastric adenocarcinoma. Oncogenesis 2015, 4, e178. [Google Scholar] [CrossRef] [PubMed]
- De Wit, M.; Carvalho, B.; Delis-van Diemen, P.M.; van Alphen, C.; Beliën, J.A.M.; Meijer, G.A.; Fijneman, R.J.A. Lumican and versican protein expression are associated with colorectal adenoma-to-carcinoma progression. PLoS ONE 2017, 12, e0174768. [Google Scholar] [CrossRef]
- Papadas, A.; Arauz, G.; Cicala, A.; Wiesner, J.; Asimakopoulos, F. Versican and Versican-matrikines in Cancer Progression, Inflammation, and Immunity. J. Histochem. Cytochem. 2020. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef]
- Goldman, J.; Eckhardt, S.G.; Borad, M.J.; Curtis, K.K.; Hidalgo, M.; Calvo, E.; Ryan, D.P.; Wirth, L.J.; Parikh, A.; Partyka, J.; et al. Phase I dose-escalation trial of the oral investigational Hedgehog signaling pathway inhibitor TAK-441 in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 1002–1009. [Google Scholar] [CrossRef]
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kate Kelley, R.; Lewis, S.; Chang, W.C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370–375. [Google Scholar] [CrossRef]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- De Jesus-Acosta, A.; Sugar, E.A.; O’Dwyer, P.J.; Ramanathan, R.K.; Von Hoff, D.D.; Rasheed, Z.; Zheng, L.; Begum, A.; Anders, R.; Maitra, A.; et al. Phase 2 study of vismodegib, a hedgehog inhibitor, combined with gemcitabine and nab-paclitaxel in patients with untreated metastatic pancreatic adenocarcinoma. Br. J. Cancer 2020, 122, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef]
- Cheng, X.B.; Sato, N.; Kohi, S.; Yamaguchi, K. Prognostic impact of hyaluronan and its regulators in pancreatic ductal adenocarcinoma. PLoS ONE 2013, 8, e80765. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Hingorani, S.R. Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br. J. Cancer 2013, 108, 1–8. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; LoBello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef]
- Sironen, R.K.; Tammi, M.; Tammi, R.; Auvinen, P.K.; Anttila, M.; Kosma, V.M. Hyaluronan in human malignancies. Exp. Cell Res. 2011, 317, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Caon, I.; Bartolini, B.; Parnigoni, A.; Carava, E.; Moretto, P.; Viola, M.; Karousou, E.; Vigetti, D.; Passi, A. Revisiting the hallmarks of cancer: The role of hyaluronan. Semin. Cancer Biol. 2020, 62, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Elahi-Gedwillo, K.Y.; Carlson, M.; Zettervall, J.; Provenzano, P.P. Antifibrotic Therapy Disrupts Stromal Barriers and Modulates the Immune Landscape in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2019, 79, 372–386. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Whatcott, C.J.; Han, H.; Posner, R.G.; Hostetter, G.; Von Hoff, D.D. Targeting the tumor microenvironment in cancer: Why hyaluronidase deserves a second look. Cancer Discov. 2011, 1, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Blair, A.B.; Kim, V.M.; Muth, S.T.; Saung, M.T.; Lokker, N.; Blouw, B.; Armstrong, T.D.; Jaffee, E.M.; Tsujikawa, T.; Coussens, L.M.; et al. Dissecting the Stromal Signaling and Regulation of Myeloid Cells and Memory Effector T Cells in Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 5351–5363. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa With Nab-Paclitaxel Plus Gemcitabine for Patients With Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar] [CrossRef]
- Wagner, K.; Schulz, P.; Scholz, A.; Wiedenmann, B.; Menrad, A. The targeted immunocytokine L19-IL2 efficiently inhibits the growth of orthotopic pancreatic cancer. Clin. Cancer Res. 2008, 14, 4951–4960. [Google Scholar] [CrossRef]
- Hosoi, H.; Ikeda, H.; Imai, N.; Amaike, C.; Wang, L.; Orito, Y.; Yamane, M.; Ueno, H.; Ideno, M.; Nukaya, I.; et al. Stimulation through very late antigen-4 and -5 improves the multifunctionality and memory formation of CD8(+) T cells. Eur. J. Immunol. 2014, 44, 1747–1758. [Google Scholar] [CrossRef]
- Ishikawa, T.; Kokura, S.; Enoki, T.; Sakamoto, N.; Okayama, T.; Ideno, M.; Mineno, J.; Uno, K.; Yoshida, N.; Kamada, K.; et al. Phase I clinical trial of fibronectin CH296-stimulated T cell therapy in patients with advanced cancer. PLoS ONE 2014, 9, e83786. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef]
- Zhao, J.; Guan, J.L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009, 28, 35–49. [Google Scholar] [CrossRef]
- Zaghdoudi, S.; Decaup, E.; Belhabib, I.; Samain, R.; Cassant-Sourdy, S.; Rochotte, J.; Brunel, A.; Schlaepfer, D.; Cros, J.; Neuzillet, C.; et al. FAK activity in cancer-associated fibroblasts is a prognostic marker and a druggable key metastatic player in pancreatic cancer. EMBO Mol. Med. 2020, 12, e12010. [Google Scholar] [CrossRef]
- Kapoor, C.; Vaidya, S.; Wadhwan, V.; Hitesh; Kaur, G.; Pathak, A. Seesaw of matrix metalloproteinases (MMPs). J. Cancer Res. Ther. 2016, 12, 28–35. [Google Scholar] [CrossRef]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Dreymueller, D.; Theodorou, K.; Donners, M.; Ludwig, A. Fine Tuning Cell Migration by a Disintegrin and Metalloproteinases. Mediat. Inflamm. 2017, 2017, 9621724. [Google Scholar] [CrossRef] [PubMed]
- Fields, G.B. The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma. Cells 2019, 8, 984. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J.; Tjulandin, S.; Davidson, N.; Shaw, H.; Desai, N.; Bhar, P.; Hawkins, M.; O’Shaughnessy, J. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J. Clin. Oncol. 2005, 23, 7794–7803. [Google Scholar] [CrossRef] [PubMed]
- Watkins, G.; Douglas-Jones, A.; Bryce, R.; Mansel, R.E.; Jiang, W.G. Increased levels of SPARC (osteonectin) in human breast cancer tissues and its association with clinical outcomes. Prostaglandins Leukot. Essent. Fat. Acids 2005, 72, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Manikhas, G.M.; Stroyakovsky, D.L.; Makhson, A.N.; Cheporov, S.V.; Orlov, S.V.; Yablonsky, P.K.; Bhar, P.; Iglesias, J. A dose finding study of weekly and every-3-week nab-Paclitaxel followed by carboplatin as first-line therapy in patients with advanced non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Giatromanolaki, A.; Brekken, R.A.; Sivridis, E.; Gatter, K.C.; Harris, A.L.; Sage, E.H. Enhanced expression of SPARC/osteonectin in the tumor-associated stroma of non-small cell lung cancer is correlated with markers of hypoxia/acidity and with poor prognosis of patients. Cancer Res. 2003, 63, 5376–5380. [Google Scholar]
- Hersh, E.M.; O’Day, S.J.; Ribas, A.; Samlowski, W.E.; Gordon, M.S.; Shechter, D.E.; Clawson, A.A.; Gonzalez, R. A phase 2 clinical trial of nab-paclitaxel in previously treated and chemotherapy-naive patients with metastatic melanoma. Cancer 2010, 116, 155–163. [Google Scholar] [CrossRef]
- Massi, D.; Franchi, A.; Borgognoni, L.; Reali, U.M.; Santucci, M. Osteonectin expression correlates with clinical outcome in thin cutaneous malignant melanomas. Hum. Pathol. 1999, 30, 339–344. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ramanathan, R.K.; Borad, M.J.; Laheru, D.A.; Smith, L.S.; Wood, T.E.; Korn, R.L.; Desai, N.; Trieu, V.; Iglesias, J.L.; et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: A phase I/II trial. J. Clin. Oncol. 2011, 29, 4548–4554. [Google Scholar] [CrossRef]
- Hidalgo, M.; Plaza, C.; Musteanu, M.; Illei, P.; Brachmann, C.B.; Heise, C.; Pierce, D.; Lopez-Casas, P.P.; Menendez, C.; Tabernero, J.; et al. SPARC Expression Did Not Predict Efficacy of nab-Paclitaxel plus Gemcitabine or Gemcitabine Alone for Metastatic Pancreatic Cancer in an Exploratory Analysis of the Phase III MPACT Trial. Clin. Cancer Res. 2015, 21, 4811–4818. [Google Scholar] [CrossRef]


{kind=link}
| Matrisome Category | Name | Digestive Cancer | Immune Modulatory Function | Secreted By |
|---|---|---|---|---|
| Collagens | PC [31] GC [58] CRC [66] | M2 macrophage polarization [66] T cell exclusion [58,59] T cell suppression [62] | Stromal fibroblasts [31,185] Tumor cells [31,185] | |
| Fibrous and non-fibrous glycoproteins | Fibronectin | PC [186] GC [187] CRC [188] | Monocyte recruitment [74,78] Monocyte differentiation [76,77] | Stromal fibroblasts [186] Monocytes/Macrophages [189] Tumor cells [189] |
| Laminin | PC [190] GC [191] CRC [192] | T cell infiltration/exclusion [93] DC priming [96] | Tumor basement membrane [82,83] | |
| Elastin | PC [193] CRC [102] | Macrophage activation [102] | Stromal fibroblasts [100] | |
| Matricellular glycoproteins | SPARC/ Osteonectin | PC [124] GC [194] CRC [195] | Macrophage recruitment/location [125] M2 macrophage suppression [127] TGF-β suppression [129] T reg cell suppression [129] | Stromal fibroblasts [124,195] Macrophages/macrophages [131] Tumor cells [196] |
| Osteopontin | PC [197] GC [198] CRC [199] | CD8+ T cell suppression [141] Macrophage recruitment/differentiation [142] | Tumor cells [200,201] Macrophages [201] | |
| Periostin | PC [202] GC [156] CRC [155] | Macrophage recruitment [155] M2 macrophage polarization [156] | Stromal fibroblasts [150,151,152] Tumor cells [152] | |
| βig-H3/ TGFβi | PC [168,169] GC [203] CRC [165] | T cell suppression [168,170] | Stromal fibroblasts [168,203] Tumor cells [168,203] Macrophages [173] | |
| Proteoglycans | Versican | PC [204] GC [205] CRC [176,206] | Macrophage differentiation [176] Macrophages- and DC-mediated T cell activation [178,179] T reg cell recruitment [181] | Stromal fibroblasts [174,207] Tumor cells [174,207] Infiltrating leukocytes [174,207] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gamradt, P.; De La Fouchardière, C.; Hennino, A. Stromal Protein-Mediated Immune Regulation in Digestive Cancers. Cancers 2021, 13, 146. https://doi.org/10.3390/cancers13010146
Gamradt P, De La Fouchardière C, Hennino A. Stromal Protein-Mediated Immune Regulation in Digestive Cancers. Cancers. 2021; 13(1):146. https://doi.org/10.3390/cancers13010146
Chicago/Turabian StyleGamradt, Pia, Christelle De La Fouchardière, and Ana Hennino. 2021. "Stromal Protein-Mediated Immune Regulation in Digestive Cancers" Cancers 13, no. 1: 146. https://doi.org/10.3390/cancers13010146
APA StyleGamradt, P., De La Fouchardière, C., & Hennino, A. (2021). Stromal Protein-Mediated Immune Regulation in Digestive Cancers. Cancers, 13(1), 146. https://doi.org/10.3390/cancers13010146