Simple Summary
A new class of drugs, termed Immune Checkpoint Inhibitors, has revolutionized cancer therapy during the last few years. Unfortunately, these drugs are only effective for a subset of patients and cancer types. Recent work has suggested that how well cancer cells present some of their molecules to the immune system is critical for patient responses to immunotherapy with immune checkpoint inhibitors. Here, we review the role of the biochemical pathway of antigen presentation in cancer and discuss how it can be modulated to enhance the efficacy of cancer immunotherapy.
Abstract
Recent clinical successes of cancer immunotherapy using immune checkpoint inhibitors (ICIs) are rapidly changing the landscape of cancer treatment. Regardless of initial impressive clinical results though, the therapeutic benefit of ICIs appears to be limited to a subset of patients and tumor types. Recent analyses have revealed that the potency of ICI therapies depends on the efficient presentation of tumor-specific antigens by cancer cells and professional antigen presenting cells. Here, we review current knowledge on the role of antigen presentation in cancer. We focus on intracellular antigen processing and presentation by Major Histocompatibility class I (MHCI) molecules and how it can affect cancer immune evasion. Finally, we discuss the pharmacological tractability of manipulating intracellular antigen processing as a complementary approach to enhance tumor immunogenicity and the effectiveness of ICI immunotherapy.
1. The Immune System and Cancer
The interplay between the immune system and cancer termed “cancer immunoediting” is a dynamic and continuously evolving process in which immune responses can eradicate tumor cells but also promote tumor progression through selective pressures [1,2]. The temporal evolution of the immune system–cancer interaction is usually considered to consist of at least three phases termed elimination, equilibrium, and escape [3]. During the elimination phase, often at the initial stages of carcinogenesis, the immune system aggressively destroys newly formed cancer cells. If this attack is successful in eliminating all pre-cancerous and cancerous cells, no clinically detectable tumors are formed. Failure to eliminate all cancer cells however can result in establishment of an equilibrium phase in which the immune system controls tumor growth but cannot fully eliminate it [4]. This phase is considered to include some degree of immune evasion and can lead to a strong selective pressure on cancer cells to mutate in ways to further avoid the immune surveillance either by becoming less immunogenic or by inducing a localized immunosuppressive state. Success in these processes leads to the escape phase that allows out-of-control cancer cell growth and the appearance of clinically visible tumors that are characterized by different mechanisms and magnitudes of immune evasion and suppression [5].
2. Mechanisms of Cancer Immune Evasion and the Role of Immune Checkpoints
Tumors can attempt to evade cellular immune responses either by excluding T cells from the tumor microenvironment (TME) or by establishing equilibrium with T cells that successfully migrate to the tumor [6]. The former mechanism, termed innate evasion, includes accumulation of defects in T cell priming and reduced intratumoral trafficking through aberrant cell-intrinsic signaling events. Such events include activation of the Wnt/β-catenin pathway [7], loss of function of PTEN [8], c-Myc signaling dependent activation [9], and loss of LKB1 signaling [10]. The latter mechanism, termed adaptive immune evasion can emerge from the selection of tumor cell clones that have progressively reduced their immunogenicity through the loss of expression of key tumor-specific antigens and/or the accumulation of mutations in genes involved in immune recognition [11,12,13]. Notably, loss of MHCI expression is a common mechanism utilized by tumors attempting to evade T cell cytotoxic responses [14,15]. Loss of immune signaling can also synergize with loss of antigenicity by interfering with interferon generation and function [16,17]. In general, synergism between innate and adaptive immune evasion can result to a major therapeutic challenge that may only be overcome by combining separate approaches that, in tandem, address problems in both the TME as well as tumor immunogenicity.
T-cell-mediated immunity is regulated by a balance between stimulatory and inhibitory signals [18]. After encountering their cognate antigen, T cells, via their CD28 receptor, are activated by stimulatory signals in the context of antigen presenting cells (APCs) in order to attack and eliminate cancerous cells. However, their inflammatory activity must then be diminished to preserve immune homeostasis. The inhibitory signals are provided by molecules called immune checkpoints (ICs) that, when activated, suppress T cell activity [19]. These molecules are receptors located on the surface of T lymphocytes that regulate the extent and duration of physiological immune responses and therefore limit tissue damage and maintain self-tolerance. Several inhibitory checkpoint molecules have been discovered to date, such as CTLA-4 (cytotoxic T lymphocyte-associated protein 4), PD-1 (programmed cell death protein 1), LAG-3 (lymphocyte activation Gene-3), TIM-3 (T-cell immunoglobulin and mucin-domain containing 3), TIGIT (T cell immunoglobulin and ITIM domain), VISTA (V-Domain Ig Suppressor of T-Cell Activation), B7-H3, BTLA (B and T lymphocyte attenuator 4), and Siglec-15 [20]. CTLA-4 and PD-1 are the most well studied and play central roles in state-of-the-art immunotherapy strategies. CTLA-4 is upregulated immediately after TCR engagement and through its competition with the co-stimulatory molecule CD28 for the B7 ligands (CD80/B7.1 and CD86/B7.2) of the APCs, it limits autoreactive T cells early at their activation stage leading to immune tolerance and prevention of autoimmunity [21,22]. Besides its surface expression being upregulated, additional CTLA-4 is recruited to the immunologic synapse via intracellular vesicles to further dampen T cell receptor (TCR) signaling [23]. Through the recruitment of phosphatases, CTLA-4 interferes with the TCR-induced stop signal for stable immune conjugate formation, leading to fewer contact periods between T cells and APCs and finally to decreased T cell priming and proliferation. The CTLA-4 suppressive functions can be also mediated by regulatory T cells (Tregs), as it is expressed on their surface [24,25]. Recently, it was shown that CTLA-4 can deplete, through trans-endocytosis, available CD80 and CD86 ligands from the membranes of neighboring APCs to prevent their interaction with CD28 on T cells [26]. PD-1 is also expressed on activated T cells but acts at later stages of an immune response and interferes with previously activated T cells. If the stimulating antigen is cleared, PD-1 expression levels decrease on responding T cells whilst in the opposite case, its expression remains elevated. PD-1 has two tyrosine motifs in its cytoplasmic tail. Through its interaction with its ligands, PD-L1 and PD-L2, PD-1 is phosphorylated at these tyrosine residues, which leads to phosphatase recruitment. These phosphatases can then dephosphorylate downstream kinases and antagonize positive signals that take place through TCR and CD28, affecting TCR-mediated downstream signaling. The final outcome is impaired T cell activation, survival, cytokine production, and altered metabolism [27]. Sustained expression of PD-1 is considered to render T cells exhausted. PD-L1 expression is induced in response to inflammatory cytokines, such as IFNγ, and thus PD-1 regulation of T cell activity occurs in response to cytolytic and effector T cell function [28].
Signaling through PD-1 is a common mechanism that tumors utilize in order to put T cells in check and escape immunosurveillance. This can be achieved by upregulating PD-L1 expression on tumor cells themselves or on stromal and immune cells in the TME [25,29]. In mouse tumor cells, the upregulated expression of PD-L1 has been associated with impaired T cell mediated antitumor responses [30,31,32]. The combination of these findings with the recognition of ICs as negative regulators of T cell activation, gave rise to the idea that blocking the inhibitory action of ICs on T cells by using specific monoclonal antibodies, could improve T cell functions and enhance immune responses against cancer [33]. These pioneering cancer therapy approaches have now shifted the focus from attacking the tumor to assisting the host’s immune system to attack cancer cells. The presence of pre-existing cancer-specific T cells capable of recognizing tumor-specific antigens and neoantigens has been considered a necessary premise for this therapeutic approach [34]. However, although for many years the main mechanism of action of ICIs has been considered to be the re-activation of primed T cells, recently the role of novel T cells that are primed and recruited to the tumors after the initiation of immunotherapy has been emerging [35,36]. Regardless of the exact mechanism, the main advantage of ICI therapy is that it can induce durable responses representative of tumor-specific immunological memory formation [37]. Several antibody ICIs have already been FDA-approved since 2011 and have shown clinical efficacy in many cancer types (Table 1) [38].

Table 1.
FDA-approved immune checkpoint inhibitors.
CTLA-4 blockade aims to induce robust activation of tumor reactive T cells. By sterically hindering the interaction of CTLA-4 receptor with B7 ligands, it leads to unrestrained CD28-mediated positive co-stimulation of T lymphocytes. The co-crystal structure of the first approved antibody against CTLA-4, ipilimumab, in complex with CTLA-4 revealed that the epitope recognized by ipilimumab overlaps with the B7 interaction domain [39]. Additionally, CTLA-4 blocking promotes antitumor responses through the deletion of Tregs via antibody mediated cytotoxicity, as demonstrated in murine cancer models [40,41]. CTLA-4 inhibition is also able to broaden and remodel the peripheral TCR repertoire, as it was observed in cancer patients undergoing ipilimumab treatment [42]. Loss of CTLA-4 may lower the threshold for TCR ligation required for effective T cell activation since CTLA-4 normally acts to dampen TCR signal strength [25]. Blockade with either a-PD-1 or a-PD-L1 antibodies abrogates inhibition of TCR signaling and removes the brakes from T cells, unleashing their effector properties, while it also appears to be able to restore the function of exhausted T cells [43,44]. PD-1 blockade seems to be more effective in tumors infiltrated by tumor antigen-specific T cells that express PD-1 receptor but were kept in an inactive state due to the interaction of PD-1 with its ligands expressed by tumor cells and stromal cells within the TME [45].
3. ICI Therapy Failure and Tumor Immunogenicity
Although immunotherapy with ICIs holds much promise for durable outcomes in cancer regression and in some cases even cure, the majority of patients do not benefit by this course of treatment and either do not respond (innate/primary resistance) or relapse after an initial period of response (acquired/adaptive resistance) [46]. Emerging evidence from studies with patients treated with cancer immunotherapies indicates that the mechanisms of resistance broadly overlap with those normally used by cancers as they undergo immunoediting [47]. Several tumor cell-intrinsic and cell-extrinsic factors contribute to the resistance to therapy, leading to three different outcomes: i) insufficient generation of antitumor T cells, ii) inadequate function of tumor-specific T cells, and iii) impaired formation of T cell memory [46,48]. Amongst them, the immunogenicity of a tumor is considered to be a critical determinant of response to ICI therapy, as tumors devoid of tumor-specific antigens can never be recognized as foreign [49]. Anagnostou and colleagues examined the evolving landscape of tumor neoantigens during the acquisition of resistance to ICIs in non-small cell lung cancer (NSCLC) patients, and attributed this resistance to loss of mutations encoding cancer-specific neoantigens [50]. However, even a high mutational and neoantigen burden cannot lead to efficacious response if the patients’ cells lack a functional machinery for tumor antigen processing and presentation [46,51] since generation of reactive CD8+ T cells requires successful antigen processing and presentation of tumor-specific antigens [48].
4. Antigen Processing and Presentation in Cancer
For T cells to recognize malignant cells and attack them, two conditions are essential. First, the tumor cells have to report their intracellular changes on their surface and second, these changes must be sensed by T lymphocytes. The cellular mechanism that determines this direct interaction between the cancer and the adaptive immune system is the antigen processing and presentation pathway (APP) [52]. CD8+ T cells, via their TCR, can only detect aberrant cells in the context of peptide-MHC class I complexes. MHCI complexes are expressed on professional APCs that can activate naïve T cells, but also on all nucleated healthy and infected or transformed cells [53].
For a peptide to serve as an epitope and therefore be capable of inducing an effective adaptive response, it has to be first processed by the cellular antigen processing machinery (APM) and then loaded onto an MHCI molecule (Figure 1). The processing and presentation pathway is a multi-step process in the context of the normal turnover of cellular proteins and often starts in the cytoplasm. There, intracellular proteins are ubiquitinated and fragmented into smaller pieces by the proteasome. An alternative pathway includes the proteasomal degradation of aberrant or misfolded proteins termed Defective Ribosomal Products (DRiPs) [54]. The constitutive proteasome is a barrel-shaped structure consisting of a catalytic 20S four-stacked ring core with chymotrypsin, trypsin, and caspase-like activities that is capped at each end by a regulatory 19S cap complex responsible for de-ubiquitination and unfolding of the trapped proteins that enter the main catalytic core. After exposure of the cells to inflammatory cytokines that generally enhance antigen presentation, new catalytic subunits named LMP2, LPM7, and LMP10 are produced and substitute these of the 20S proteasome to generate the immunoproteasome. This transition has been linked to changes in cleavage specificity, efficiency of MHCI ligand generation, and MHCI repertoire quantity [55]. Proteasomal cleavage generates peptides 2–26 residues long with a C-terminus anchor residue compatible with MHCI binding groove, but often extended at their N-terminus [55]. Peptides are then released in the cytosol and if they survive further degradation by cytosolic peptidases, are transferred into the endoplasmic reticulum (ER) by the Transporter associated with Antigen Processing, TAP. The TAP heterodimer, consisting from TAP1 and TAP2 subunits, forms a transmembrane pore in the ER membrane and preferentially transfers peptides 9–16 residues long, although longer peptides can be also transferred with much lower efficiency [56]. After a peptide enters the ER, its final assembly onto a nascent MHCI molecule is mainly orchestrated by a multi-subunit complex, called the Peptide Loading Complex (PLC) [57]. TAP constitutes an integral part of PLC, where it acts as a docking site for the MHC class I dedicated chaperone tapasin, and three other ER chaperones, the lectin calreticulin, calnexin, and the disulfide isomerase ERp57. Calnexin is important for early folding and oxidation of newly synthesized MHC heavy chain [56]. MHCI molecules are heterodimeric glycoproteins consisting of a polymorphic heavy chain (in humans encoded by the Human Leukocyte Antigen-HLA A, B, and C genes) and an invariable light chain, β2 microglobulin (β2m). MHCI molecules have a groove that can preferentially bind 8-11mer peptides. The exposed surface of this groove where the antigenic epitope is bound is the part of the MHCI complex that is recognized by the TCR [58]. Peptides that enter the ER and are too long to fit into MHCI are trimmed by the concerted action of two ER-resident aminopeptidases, ERAP1 and ERAP2 [59]. Calreticulin in combination with ERp57 assist with the folding and stabilization of the newly synthesized empty MHCI molecules. Tapasin mediates the recruitment of MHCI to the PLC and enables peptide loading and exchange, facilitating the formation of MHCI molecules with high affinity peptides. However, even after a peptide is loaded onto an MHCI molecule, an additional chaperone, TAP binding protein related protein, TAPBPR, assists with quality control to ensure peptide stable binding [60]. Having acquired a suitable peptide, the MHC class I molecule traffics to the cell surface through the Golgi network for presentation to T cells [56].
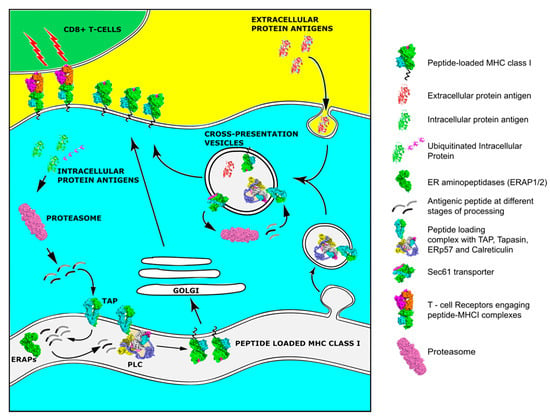
Figure 1.
Overview of the Major Histocompatibility Class (MHC) class I pathway of antigen processing and presentation and the alternative pathway of cross-presentation.
Tumors are particularly immunogenic and presentation of their specific antigens to T cells in an MHCI-restricted manner would lead to their eradication. Defects and alterations in the components of the APM are often found in tumors as cancer progression requires tumor cells to acquire the ability to avoid immune recognition [61]. Alterations in tumor APM can result not only in the downregulation of cell-surface expression MHCI molecules but can also alter the repertoire of antigenic peptides presented to the T lymphocytes. Since successful treatment with ICIs relies on re-activation of T cells, alterations in antigen processing and presentation of antigens can result to impaired antitumor responses and therapy resistance [16,62,63].
Alterations in antigen processing and presentation pathway may occur at any step of synthesis, assembly, transport, and surface expression of MHCI molecules or at any step of antigen editing (Figure 2). Truncating alterations, loss of heterozygosity, frameshift, and loss-of-function mutations affecting the β2m protein in human tumor cells that lead to instability of MHCI complexes, impaired folding, and diminished transport to the cell surface, have been associated with resistance to ICI therapy. In lung cancer patients, disruption of MHCI-mediated antigen presentation due to β2m loss of heterozygosity conferred resistance to PD-1 blockade therapy [64]. In melanoma metastatic patients treated with checkpoint inhibitors, point mutations, deletions, truncations, and loss of heterozygosity in β2m have been associated with resistance to ICI immunotherapy [16,62]. Furthermore, loss of expression of thiol reductase ERp57 has been demonstrated in several tumor types to correlate with poor prognosis [65,66,67]. Downregulation of calreticulin expression has been observed in colorectal and bladder cancers as well as in myeloproliferative neoplasms and has been associated with impaired antigen processing and presentation [68,69,70]. Defects have also been found in the IFNγ-inducible proteasome components [66,71,72]. Loss or downregulation of the transporter TAP have also been recorded in many cancer cell lines and primary tumors [72,73,74]. In all these cases, patients had a poor disease prognosis and diminished MHCI surface expression on tumor cells that correlated with changes in their antigenic peptide repertoire. In melanoma cells, micro-RNA downregulation of TAP expression led to reduction of MHCI surface expression and decreased T cell recognition [75]. In addition, the expression of Tapasin, another important APM component, has been found altered in several types of cancer [76,77,78]. MHCI surface expression was significantly decreased in all these patients and correlated with tumor progression. Impaired tapasin function led to MHCI molecules loaded with low-affinity, suboptimal antigenic epitopes. Furthermore, it reduced antigen presentation of tumor-specific antigens and blocked the presentation of certain immunodominant epitopes [77,78,79]. Mutations in tapasin and structural defects in IFNγ-related genes were found in recurrent metastatic melanoma with disease progression after active immunotherapy [80]. Furthermore, endoplasmic reticulum aminopeptidases, ERAP1 and ERAP2, exhibit variable expression levels in different cancer types [81]. Although mutations in these enzymes are rare, their expression in cancer is often either downregulated or upregulated while SNPs affecting their enzymatic activity can influence the immunopeptidome presented by MHCI molecules [82,83,84,85,86,87]. As these enzymes can both trim and destroy epitopes destined for binding onto MHCI molecules, their expression levels and activity strongly influence the peptide pool available for loading onto MHCI and can thus affect the immunogenicity of tumors [88,89]. In some cancers, ERAP1 overexpression leads to destruction of tumor-specific immunodominant epitopes and induction of anti-tumor CD8+ responses, linking antigen destruction with tumor escape [90,91,92]. In other cases, ERAP1 downregulation can lead to cancer rejection through Natural Killer cell mediated cytotoxicity [93,94]. ERAP2 overexpression in patients with oral cavity squamous cell carcinoma has been associated with metastasis from the primary tumor and poor prognosis [95], while the absence of ERAP2 in choriocarcinoma cells reduced their ability to activate T lymphocytes [96]. Downregulation of the mouse homologue, ERAAP, in mouse tumors increased the efficacy of a-PD1 blockade therapy [97]. In bladder cancer patients receiving a-PD1 therapy, expression quantitative trait loci affecting the expression of both ERAP1 and ERAP2 were found to associate with favored response to therapy and prolonged survival, probably due to alterations in the repertoire of peptides available for presentation to T cells [98]. Recently, functional ERAP1 allotypes have been correlated with tumor-infiltration by CD8+ T cells in cervical and oropharyngeal squamous cell carcinomas due to changes in processing of particular antigenic epitopes [99]. Overall, intracellular antigen processing is emerging as a master regulator of the immunogenicity of cancer [100,101].
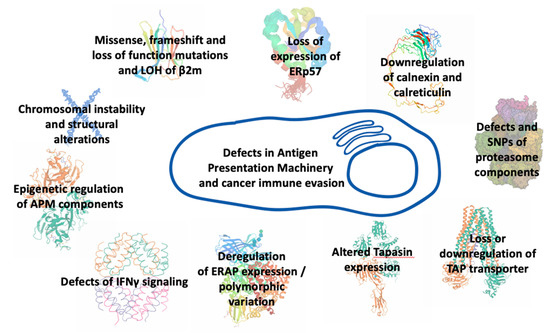
Figure 2.
Defects in different components of the antigen processing and presentation machinery that can underlie immune evasion by cancer.
As most components of the APP machinery are IFNγ inducible, defects in IFNγ signaling cascade can limit MHCI surface expression. The main proteins that interfere with this pathway are the transcription factors IFN-regulatory factor 1 (IRF-1) and STAT-1, and the kinases Janus-associated kinase JAK-1 and JAK-2. Tumor cells with activated IFNγ pathways can respond to cytokine secretion by immune cells located into the TME and become visible to T cells. Multiple studies have linked defects in IFNγ signaling with resistance to ICI therapy [16,17,102,103]. Genetic analysis of tumors from patients with melanoma and colon cancer who did not respond to PD1 blockade therapy despite their high mutation burden and high percentage of pre-existing tumor specific T cells, were identified to acquire loss of function mutations in JAK1/2 kinases and decreased MHCI surface expression [102,104]. Although indirectly associated to the IFNγ pathway, loss of the protein tyrosine phosphatase Ptpn2 was correlated to enhanced levels of antigen-loaded MHCI molecules on the surface of tumors and to sensitivity of tumor cells to immunotherapy [97].
Epigenetic events in cancer cells can regulate the expression of immune-related genes, resulting in changes in antigen processing and presentation that impair tumor recognition [105]. DNA methylation and histone modifications of MHCI heavy chain gene promoters leads to transcriptional silencing and decreased MHCI surface expression, causing impaired antigen presentation and immune evasion [106,107]. Additional components of the APM machinery have been found to be epigenetically regulated in many cancer types [107,108]. In melanoma, increased histone methyltransferase Ezh2 expression during a-CTLA-4 immunotherapy, decreased the antigen presentation ability of cancer cells while its inactivation reversed the resistance to therapy and synergistically suppressed tumor growth [109]. In prostate cancer, epigenetic silencing of the crucial component JAK1 kinase of the IFNγ signaling pathway led to IFNγ-insensitivity-mediated tumor evasion and resistance to immunotherapy [110]. Moreover, methylation of the NLRC5 MHCI trans-activator caused suppression of MHCI molecules and other components of the APM machinery in mice and an impaired ability to induce CD8+ T cell activation in cells [111]. In addition, Merkel cell carcinoma patients with low expression of APM components that was mediated by histone deacetylation, were resistant to a-PD1 therapy [112,113]. Antigen presentation efficiency is also diminished in human tumors characterized by large chromosomal instability and structural alterations. Although these tumors initially show induction of MHCI-restricted antigen presentation due to activation of cGAS/STING cytosolic DNA sensing pathway that detects tumor derived DNA and other pro-inflammatory signaling pathways, as they evolve under immune pressure, they suppress their antigen presentation machinery and adopt an immunologically poor phenotype. An experimental model of such tumor aneuploidy revealed that the suppression of antigen processing and presentation genes can be at least partly attributed to DNA hypermethylation of the corresponding genes, while the expression level of DNA methylotransferases was found significantly elevated [114].
5. MHCI Expression in Cancers
Downregulation of MHCI favors escape of tumor cells from immune surveillance [115]. Many studies in tumor cell lines and biopsies from patients reported total or partial loss of MHCI surface expression as a frequent event in cancer [72,116,117,118]. According to Garrido and colleagues, the loss or downregulation of surface MHCI expression is an active process that takes place gradually as tumors develop [119]. As such, at the early phase of tumor development, cancer cells are mostly MHCI positive. This induces T cell infiltration at the tumor microenvironment that recognize and kill cancer cells capable of presenting tumor-specific antigens on their MHCI molecules. Gradually, a vast diversity of tumor clones with variable MHCI surface expression levels is generated. A Darwinian type T cell-mediated selective pressure leads to tumors characterized by the presence of only MHCI negative cancer cells. This phase is accompanied by dramatic changes of the tumor tissue architecture that prevents T cells from entering the cancer niche as they are retained in the surrounding stroma [119,120]. This immune selection of MHCI-negative tumor cells has been demonstrated after immunotherapy in cancer patients and in experimental cancer models as the therapeutic application of checkpoint blockade increases the selective pressure towards tumor cells [121].
Apart from mutations, epigenetic modifications and structural alterations, tumors can adopt additional mechanisms to decrease their MHCI surface expression. A phenomenon often observed in tumors is the surface expression of non-classical MHCI molecules, such as HLA-G, HLA-E, and HLA-F. Although these molecules can present antigenic peptides in the context of antiviral defense, it is not clear whether their role in cancer is related to antigen presentation or they function as inhibitory ligands through their interaction with receptors on effector cells [122,123]. MHCI molecules can also be downregulated by other regulatory mechanisms, that involve signal transduction cascades, oncogenes, and tumor suppressor genes. Mutations in the BRAF oncogene (such as the V600E) lead to internalization of MHCI molecules from the cell surface of melanoma tumor cells and its sequestration within endocytic compartments, resulting in impaired recognition by the adaptive immune system [124]. In addition, autophagy, a conserved nutrient sensing system that induces the degradation of cytoplasmic proteins and damaged organelles by lysosomes can interfere with MHCI surface expression. In pancreatic ductal adenocarcinoma, Yamamoto and colleagues showed that MHCI molecules are selectively targeted for lysosomal degradation by an autophagy-dependent manner leading to alterations of immunogenicity of the tumor and impaired antigen presentation while its inhibition acts in synergism with ICI therapy and results in enhanced antitumor responses [125]. The SND1 oncoprotein, highly expressed in various cancers, prevents normal assembly of MHCI molecules by leading nascent synthesized MHCI heavy chain to ER-associated degradation (ERAD). Deletion of SND1 in tumor mouse models restores tumor antigen presentation to T cells both in vitro and in vivo and enhances T cell infiltration into the tumors [126]. Moreover, additional tumor suppressor genes (such as Fhit and p53) and oncogenes (such as Her2), interfere with MHCI expression in cancer cells [112,127,128]. In many human cancers, MHCI downregulation also associates with impaired signaling by transcription factors, such as NFkB and IRF2 that regulate activation of transcription of the MHCI heavy chain [112]. Additionally, IRF2 loss is associated with impaired peptide transport from the cytosol to the ER and peptide trimming [129]. Recently, it was demonstrated that the RNA binding protein MEX3B is linked with resistance to cancer immunotherapy in melanoma patients, by binding and destabilizing the HLA-A mRNA resulting in decreased HLA-A expression on the surface of tumor cells and thereby protecting the tumor cells by T cell-mediated recognition and elimination [130]. Finally, several long non-coding RNAs and miRNAs have been shown to modulate MHCI expression levels in several cancers [131,132,133,134].
In a recently published study, Chowell and colleagues analyzed the impact of individual’s specific MHC class I germline alleles on the clinical outcome of ICI therapy. By carrying out high-resolution MHCI genotyping of two patient cohorts with advanced melanoma and NSCLC that had received treatment with IC molecules, the authors observed that homozygosity in at least one human MHCI locus was linked to reduced survival periods, independently of mutational load, age, tumor stage, or type of therapy. Antigen presenting MHCI molecules are highly polymorphic, especially at their peptide binding grooves, and therefore each allele can bind and present a restricted set of antigenic epitopes. As result/Consequently, individuals homozygous in at least one MHCI locus may present a smaller, less diverse repertoire of tumor antigens to CD8+ T cells and thus may be less likely to present potent epitopes that induce highly effective antitumor responses that can be enhanced by ICI therapy [135,136]. Given that only a small percentage of presented tumor antigens in cancer patients are immunogenic, it seems that even small differences in MHCI molecules can significantly affect the adaptive responses and the efficacy of immunotherapy. This may also explain why MHCI homozygous patients with tumors bearing low neoantigen load show decreased survival and fail to respond to ICI therapeutic strategies compared to heterozygous patients and why specific HLA supertypes are associated with increased immune responsiveness. Moreover, Chowell and colleagues provided an additional link between MHCI heterozygosity and the presentation of a greater variety of tumor-specific antigens. By deep-sequencing of TCRs from tumor samples collected on-therapy, the authors observed enhanced clonality in heterozygous patients, concluding that the diversity of MHCI molecules modulates the selection and the resulting clonal expansion of T cells reactive against neoantigens and tumor-specific antigens after treatment with ICIs [137]. Accordingly, Marty and coworkers demonstrated that an individual’s MHCI genotype can predict cancer susceptibility as oncogenic mutations found in a tumor were linked to this genotype. Their study suggests that MHCI genotypes can act as a barrier that constrains the possible mutations that a developing tumor can accumulate [138].
6. Dendritic Cells and Cross-Presentation in Cancer
Although tumor cells often express MHCI molecules on their cell surface they tend to be poor antigen presenters and immune stimulators since they often lack costimulatory molecules and thus cannot effectively stimulate naïve T lymphocytes [139]. De novo generation and initiation of adaptive immune responses specific to tumor antigens, requires the cross-presentation capability of professional APCs that capture exogenous derived antigens, process and present them in order to prime naïve T cells [140]. The most potent known APCs are the dendritic cells (DCs), that constitute a heterogeneous cell population subdivided to several different subtypes [141]. DCs differentiate from bone marrow progenitors and reside in lymphoid and peripheral tissues where they act as sentinels of the immune system. Under steady-state conditions, differentiated DCs are found in their immature form. Immature DCs show a high endocytic potential and capture antigens but express low levels of MHCI and costimulatory molecules and as a result they do not prime T cells, but rather induce immune tolerance [142]. In order to be able to prime naïve T cells, DCs must first be activated and shift to their mature form. Their maturation is characterized by movement of MHCI to the cell surface, upregulation of the costimulatory molecules CD80 and CD86, higher expression of the C-C chemokine receptor 7 (CCR7), enhanced migration to lymph nodes (LNs), and increased cytokine production that drive T cell stimulation and clonal expansion [143,144,145]. DC activation is normally considered to result from detection of pathogen or damage associated molecular patterns (PAMPs/DAMPs) recognized by specific receptors. Within tumors, several of these receptors recognize endogenous DAMPs released or expressed on the surface of dead or dying cells. Immunogenic death of cancer cells, either spontaneously or due to therapeutic interventions, is an active process that releases alarmins and chemotactic factors that attract and activate DCs [146].
The inflammatory environment of a tumor, which includes cytokines, chemokines, and growth factors, fosters infiltration by DCs [147]. In tumors, DCs have access to large amounts of tumor antigens. After capturing and processing them through either the cytosolic or the vacuolar pathway, DCs migrate to the draining LNs to present these antigens and prime tumor-specific T cells. Memory and effector T cells return to the tumor site to perform surveillance and killing activities [148]. Studies with DCs isolated from tumor-bearing mice confirm their ability to cross-present tumor antigens and induce adaptive immune responses [149,150]. Apart from migratory DCs, non-migratory DCs that remain in the tumor may interact with T cells and prime them [147]. Additionally, by secreting IL-12 and other cytokines, non-migratory DCs can maintain and regulate antitumor responses. Antigen experienced T cells require cognate interactions with tissue DCs presenting antigens at a sufficient dose and duration to expand in situ and achieve their full effector activity [151]. Additionally, DCs in tumors can also be involved in priming of T cells, when found in ectopic or tertiary lymphoid structures in the immediate proximity of the tumor mass [152,153]. This phenomenon is especially important for the response against neoantigens that develop as tumor progresses [145]. Infiltration of DCs into tumor sites is associated with prolonged survival and reduced incidence of metastasis in patients with various types of solid tumors [154].
Cancer often develops evasion mechanisms that interfere with proper DC function [155,156,157]. In cancer patients, defective DC function is highly associated with impaired immune responses against antigens expressed by tumors [158]. The inherent plasticity of DCs and the balance between stimulatory and suppressive signals within the TME dictate whether DCs can induce and maintain a T cell response or not. In many cases, their number, distribution, phenotype, and function can change as the tumor progresses. Studies have shown that the number of DCs in peripheral blood of patients with head and neck squamous carcinoma is different from that of healthy individuals [159]. In a model of spontaneous ovarian cancer, Scarlett and colleagues observed a functional switch in DCs from an immunostimulatory to an immunosuppressive phenotype. Moreover, the depletion of DCs at early stages correlates to tumor growth while the depletion in later stages results in tumor regression. Finally, tumor DCs progressively upregulate PD-1 and PD-L1 and this phenomenon has been associated with T cell suppression and loss of Tumor infiltrating Lymphocytes (TILs) [160]. In the TME, DCs have either inefficient or totally absent antigen presenting capability or are polarized into immunosuppressive/tolerogenic regulatory DCs that suppress T cell activity [147,161,162]. The TME constitutes a challenging environment with limited availability of oxygen due to poor vascularization and nutrients as well as increased concentration of metabolic products, which interfere with DC function, attenuating DC efficiency of cross priming [163]. A prerequisite for DC activation is their metabolic reprogramming to meet increased demands for protein synthesis and secretion of chemokines and cytokines that is accompanied by an increase in glucose uptake and enhanced levels of glycolysis. Competition for glucose uptake with other cells in the TME can render DCs unable to function properly [147]. The unique nature of the TME has also been highlighted by two separate recent studies that associated the impaired antigen presentation capacity of DCs with defects in trafficking of MHCI to the cell surface due to incorporation of tumor-derived oxidized lipids into DC lipid bodies. In this case, MHCI rather accumulate inside late endosomes [157,164]. Activation of the β-catenin pathway is another mechanism that cancer utilizes in order to inhibit cross-priming as activation of this pathway induces a tolerogenic state in DCs. Wnt ligands and other molecules, both in tumor cells and inside DCs, mediate DC exclusion from TME and inhibition of their antitumor activity, respectively. The DC intrinsic signaling route is also active in tumor infiltrating DCs in order to disrupt cross-presentation and reprogram DCs to induce tolerance [161]. In addition, many other factors (VEGF, IL-10, IL-6, colony stimulating factor CSF-1) inhibit maturation of bone marrow progenitors or monocytes into DCs, and instead drive monocytes toward a suppressive phenotype as they promote development of MDSCs and TAMs [165].
The clinical success of immunotherapy with checkpoint inhibitors relies significantly on effective processing and cross-presentation of tumor-specific antigens captured by DCs [158]. The blockade of inhibitory receptors on the cell surface of T cells by monoclonal antibodies, can intensify antitumor responses initially primed by DCs [166]. Tumor-bearing mice with impaired cross-presentation pathways showed resistance to therapy with antibodies targeting ICs [167,168]. During the last few years, new strategies have been emerging that aim to strengthen the therapeutic efficacy of checkpoint blockade treatment with DC-based vaccination, i.e., DCs loaded with tumor (neo)antigens for presentation to the immune system, as available preclinical and clinical data have demonstrated that DC-vaccination synergizes with ICIs for improved therapeutic outcomes [169,170]. In intracranial glioma tumor-bearing mice, the combined administration of an a-PD1 monoclonal antibody with a DC vaccine, led to long-term survival that was dependent on CD8+ T cells that infiltrated the tumor [170], while in a murine lung cancer model the combination of DC vaccination with ICIs led to 80% tumor eradication. The treated mice developed immunological memory that fostered cancer recurrence-free survival. In the same mice, monotherapy using either agent did not result in eradication of the tumor [171,172]. In human patients with active myeloma, the synergistic effect of the two therapies led to enhanced T cell responses against myeloma targets [173]. Ge and colleagues demonstrated that blocking the PD1/PD-L1 pathway with monoclonal antibodies, induces DC maturation and proliferation and that suppressing IC molecules during DC vaccination prolonged survival in a breast tumor-bearing mouse model [174]. Okada and colleagues used different MHCI-restricted tumor-associated neoantigens simultaneously with mature DCs and proposed that using this type of therapy at early stages of cancer can lead to generation of clinically useful neoantigen-specific T cells [175]. According to Linette and colleagues, vaccination using DCs appears to be necessary as an adjuvant to ICI therapy since most T cell clones specific for tumor neoantigens have been demonstrated to be naïve and below the limit of detection in patients with melanoma. In other words, a combinational therapy enhances both direct and cross-presentation and has the potential to boost the frequency and diversity of tumor-specific T cells and thus strengthen immune responses [176]. Finally, numerous studies have explored whether modulation of intratumoral APCs could increase the response to ICI therapies. These studies demonstrated that intratumoral DCs that sustain the potential to re-stimulate immune cells in the context of tumor microenvironment, are required for efficacious therapy outcomes, while their paucity limits the efficacy of ICIs [177,178,179].
7. The Immunopeptidome and Cancer
The sum of peptides bound and presented by MHCI on the surface of cells is increasingly referred to as the cellular immunopeptidome [180]. Under malignant conditions, the iummunopeptidome has been found altered both quantitatively and qualitatively. These altered tumor antigenic peptides may be recognized by the adaptive immune system as foreign and therefore induce immune responses. Indeed, in 2014, Gubin and colleagues first demonstrated that cancer immunotherapy treatments that boost T cell activity, overcoming tumor suppression induced by the tumor themselves, depend on T cell recognition of tumor-specific antigens [49]. Therefore, a detailed knowledge of the immunopeptidome constitution and deeper understanding of the characteristics of suitable tumor-associated rejection antigens can improve the current therapeutic immunotherapy interventions and offer new opportunities towards the development of personalized treatments. Several research efforts have already aimed at the identification of naturally presented antigens in different types of hematological and solid tumors while new tools and techniques (i.e., mass spectrometry-based and in silico, proteogenomic techniques etc.) are being developed and integrated, aiming for a more precise characterization and validation of the cancer-specific immunopeptidomes [181,182,183,184,185,186,187,188].
8. Tumor Antigens and Tumor-Associated Antigenic Peptides
A significant challenge in the field of immunotherapy is the identification of MHCI-presented peptides that are able to mediate T cell-based tumor rejection. Long-term clinical benefits of cancer immunotherapy treatments rely on T lymphocytes that recognize tumor antigens [189]. The major factors that determine whether an antigen is a good immunotherapy target are: (i) its immunogenicity, i.e., its ability to provoke an immune response after (re)-activation of T cells induced by ICIs, (ii) its tumor specificity, (iii) its prevalence and expression level on tumor cells, and (iv) its role in the oncogenic process [190].
Tumor antigens are classified into antigens of high tumoral and low tumoral specificity [189]. The first category includes antigens that are strictly tumor-specific, such as viral antigens generated in cancers of viral etiology and antigens derived from mutations or rearrangements in coding sequences and chromosome translocation events. The second group encompasses differentiation antigens, i.e., antigens expressed in both tumor and the corresponding healthy tissue but over-expressed in tumors [189]. A special category of tumor-associated antigens are RNA-editing derived epitopes. RNA editing is a posttranscriptional mechanism that generates sequence variations in proteins by enzymatic modification of nucleotides in mRNA sequences. This mechanism has been found dysregulated in cancers and peptides generated this way are presented to the immune system and elicit immune responses. In a recent study, Zhang and collaborators identified over-edited peptides from tumor tissues and provided evidence that effector CD8+ T cells specific for these peptides can be found in human tumors [191]. Moreover, potential tumor antigens can emerge after posttranslational modifications that occur on the antigens themselves and influence their binding affinity for MHCI [192,193]. Another class of antigenic peptides demonstrated to provoke immune responses and may constitute tumor rejection antigens, are the proteasome-generated spliced peptides [194]. In this case, distinct peptidic fragments (from the same protein or from different proteins) produced by the proteasome, are ligated in situ, producing sequences that are non-contiguous in the genome and are not found in proteins in the cell. In a recent study, Liepe and coworkers provided evidence that up to 30% of peptides bound to MHCI molecules can be spliced peptides [195]. However, this high prevalence of spliced peptides has been controversial and reanalysis of the original results by Mylonas and colleagues using multiple computational and verification tools estimated spliced peptides percentage to be much lower, in the range of 2–6% [196]. Finally, tumor antigens, named cryptic antigens, may also be derived from non-canonical translation of protein-coding genes or from translation of non-coding sequences. It has been proposed that up to 10% of the MHCI bound peptides can originate from non-coding genomic regions, untranslated regions and exonic out-of-frame translation. Additional sources of cryptic antigens can be long non-coding RNAs, altered mRNA splicing events, small nucleolar RNAs, and proteins encoded in ribosomal DNA [197]. If one takes into account that 99% of tumor-specific mutations are located in non-coding regions, these cryptic MHCI antigens can be a very rich source of tumor-specific antigens [197,198].
9. Epigenetic Control of Tumor Antigen Expression and Presentation
Tumor cells frequently exhibit epigenetic aberrations that significantly impact the repertoire of expressed proteins and therefore presented peptides, affecting recognition by immune cells. A class of tumor antigens that is epigenetically regulated and re-expressed in tumors is cancer testis antigens (CTAs). In healthy adults, CTAs are expressed only in male germ and trophoblastic cells [199]. However, ectopic expression has been observed in tumor cells of different histology—possibly indicating a role in oncogenesis and tumor growth—and is associated with global and promoter-specific DNA demethylation and histone modifications [115,199]. CTAs expressed by cancer cells are considered as tumor-specific antigens due to the fact that germ cells do not normally express MHCI molecules on their surface and additionally, due to the highly immunogenic capacity of CTAs. Indeed, potent cellular and humoral responses against these antigens, especially melanoma-associated MAGE and PRAME families and NY-ESO-1, have been observed in patients, while the use of demethylating agents in tumor cell lines increased their expression leading to recognition and destruction of the cancer cells by antigen-specific T cells [200,201].
Combination of ICI treatment with CTA vaccines has been demonstrated to have a synergistic positive effect. In melanoma patients, utilization of such a combinational treatment led to higher treatment response rates [202,203]. Immunological analysis showed that treatment with CTLA-4 immune-checkpoint antibody ipilimumab in metastatic melanoma patients enhanced NY-ESO-1 specific T cell responses and provided durable clinical benefits [204]. However, tumors can still find mechanisms to evade CTA-specific immune recognition and CTAs have been found downregulated in many cancers [202]. Moreover, dedifferentiated liposarcoma, leiomyosarcoma, and synovial sarcoma tumors with positive expression of PRAME cancer testis antigens have been demonstrated to reduce the expression levels of many components of the APP (such as MHC molecules, β2m, TAP2 and LMPs) in order to avoid immune recognition [205].
Apart from cancer testis antigens, other categories of antigens that could serve as tumor rejection antigens also appear to be under epigenetic regulation. Studies using DNA methyltransferase inhibitors demonstrated that these agents can induce the expression of transposable elements including mainly endogenous retroviruses (ERVs) [206]. ERVs are the most abundant viral elements in the human virome that are silenced due to DNA methylation in somatic cells [207]. Their activation in tumors results in a state of viral mimicry that can lead to generation of neoantigens in treated cancer cells. Their induced expression mimics exogenous retroviral infection and turns on viral defense genes resulting in innate immune responses, attraction of cytotoxic T cells in the TME, and IFNγ release that in turn induces transcription of APM components [207]. Indeed, a-PD-1 responsiveness has been positively correlated with ERVs expression in cancers [208]. Moreover, high molecular weight melanoma-associated antigens (HMW-MAAs) have been demonstrated to undergo demethylation at their gene promoter in melanoma lesions and cell lines, resulting in their re-expression, but whether these antigens can provoke immune responses remains elusive [209]. In a recent study, neoantigen expression levels were affected by promoter hypermethylation of genes harboring neoantigenic mutations in 23% of cases studied [11]. Qamra and colleagues analyzed chromatin profiles and the epigenomic promoter landscape in gastric adenocarcinoma and observed that epigenetically activated alternative tumor-specific promoters can favor immune evasion through depletion of immunogenic peptides and reduction of tumor antigenicity [210].
10. Neoantigens
Tumorigenesis and cancer outgrowth are closely related to genetic diversity and accumulation of non-synonymous somatic alterations. These alterations can be missense mutations, silent mutations, insertions, and deletions as well as copy number gains and losses that result in new peptide sequences which are strictly tumor-specific. A single alteration in amino acid sequence can interfere with T cell recognition in three different ways: (i) by creating an anchor residue that changes the binding affinity of the new peptide with the MHCI molecule; (ii) by changing the TCR binding properties resulting in a conformationally altered peptide-MHCI complex that can be recognized by different T cell populations and (iii) by altering processing of the protein by the cellular APM that could result to presentation of an epitope that normally would be degraded [211,212]. The number of mutations within a tumor genome is defined as tumor mutation burden (TMB). A high level of TMB raises the possibilities of generation of neoantigens and the emergence of neoantigens diverges cancer cells from normal, healthy cells. Cancer cells can now be recognized as foreign by the immune system as high levels of mutational load is believed to enhance antigen presentation to T cells and increase the chances of tumors being identified by widening the T cell killing repertoire [213,214]. The success of ICI therapy relies on reinvigoration of pre-existing T cells that although are kept under tight control by modulatory mechanisms, have the ability to recognize cancer cells and attack them when this control is unleashed [215]. Indeed, studies have demonstrated that neoantigens can elicit responses after immunotherapy treatment and T cells recognizing tumor-related neoepitopes have been identified in different cancers [216,217,218]. There is extensive published literature that correlates high mutation burden and neoantigen frequency with durable survival and regression benefit from/after ICI therapy in several types of tumors. Neoantigens have been proposed to be good predictive and prognostic markers of better clinical outcomes, although tumors with low mutational load can still respond to checkpoint blockade, indicating a non-linear correlation and the involvement of additional factors [219,220,221,222,223,224,225,226,227,228,229,230,231,232,233,234,235,236].
An important source of neoantigens comes from the accumulation of mutations that occur in the genome when the DNA repair mechanisms of the cell are deregulated. During the cell cycle, cells progress through a series of checkpoints before mitotic division to ensure replication fidelity. Cells are well equipped with mechanisms that recognize and correct DNA damages, such as proofreading polymerases, mismatch repair pathways, base and nucleotide excision pathways, and homologous repair mechanisms. However, tumors often develop defects in these mechanisms and, as a result, DNA replication errors accumulate leading to a large number of mutations that induce genomic instability, which in turn promote cancer growth. The major causes that drive repair deficiencies in cancer correlate with inherited and de novo germline and somatic alterations, at the DNA sequence level, in genes that constitute components of the repair machinery, as well as epigenetic alterations (DNA methylation, histone modifications, nucleosome remodeling, and RNA-mediated targeting) that lead to transcriptional silencing of the associated genes or changes in chromatin dynamics required for DNA repair [237].
Many studies have demonstrated the strong correlation between inactivation of DNA repair pathways and genomic instability with significant higher mutational burden, tumor neoantigen load, and immune cell infiltration [238,239,240,241,242]. Rospo and colleagues used a colorectal cancer model system and found that alterations in DNA repair genes facilitate the acquisition of dynamic neoantigen profiles that fluctuate over time [243] while similar results were also observed in lung squamous cell carcinoma [240]. A CRISPR/Cas9-mediated targeting of the mismatch repair (MMR) component Mlh1 in murine breast, colon, and pancreatic ductal adenocarcinomas, revealed that MMR deficiency is associated with high mutational burden, TCR diversity, and significantly elevated neoantigen production. Furthermore, neoantigen production had continuous renewal potential compared to MMR-proficient cells that exhibited stable mutational load and neoantigen profiles [244]. The hyper-mutated phenotype that characterizes these types of tumors has been demonstrated to associate with higher rates of response to ICI therapy and durable clinical benefit [245,246,247,248]. In a study evaluating clinical data in patients with 12 different types of MMR-deficient tumors treated with an a-PD-1 agent, Le and colleagues observed rapid in vivo expansion of neoantigen-specific T cell clones reactive to mutant neoantigenic peptides found in the tumor. Such peptides may constitute a cohort of neoantigens useful for evaluating responses to IC treatment [247]. Collectively, it appears that there is growing evidence that the MMR deficient phenotype can serve as a good predictive biomarker of clinical response to ICI therapy.
11. T Cell Epitopes Associated with Impaired Peptide Processing
T cell epitopes associated with impaired peptide processing (TEIPP) constitute a unique, alternative repertoire of CD8+ T cell epitopes. TEIPP peptides are non-mutated self-antigens arising from housekeeping genes and emerge only in immune-edited tumors with low MHCI expression and defects in the APM as functional TAP seems to prevent their presentation. Their processing can also be conducted by alternative routes, such as the signal peptide route or the convertase family. TEIPP peptides are thought to be present within the ER of cells carrying intact TAP but cannot be presented due to their competition with the large flow of TAP-pumped peptides that are normally loaded onto MHCI molecules. A CD8+ T cell subset was discovered that selectively recognizes and targets tumor cells with defects in their APM and not cells with proficient APM. This T cell subset is positively selected in the thymus but remains in a naïve state in the periphery so it is not affected by tolerance [249,250,251]. The ppCT16–25 peptide derived from the signal peptide of pre-procalcitonin was the first human tumor epitope identified whose surface expression is associated with impaired TAP transporter function [252]. Moreover, in a recent study, 16 different HLA-A*02:01 presented TEIPP peptides were identified in mouse tumor models with defects in TAP transporter [253]. In addition, successful targeting of immune-escaped tumour variants by TEIPP-specific T cells was demonstrated [253]. TEIPP could be considered as tumor-specific neoantigens since their surface presentation is favored only under conditions of TAP dysfunction.
Recent work has highlighted that dysfunction of another APM component, ERAP1 (or ERAAP in mouse) can also lead to up-regulation of non-classical MHC class Ib molecules that normally present peptides from the signal sequence of MHCI [254]. Presentation by these non-classical MHC led to robust CD8+ responses [254]. Interestingly, ERAP1 downregulation affected the immunopeptidome of both classical and non-classical MHCI [255]. It was thus proposed that MHC class Ib presentation of signal sequence peptides may constitute a mechanism for immune surveillance for the dysfunction of the aminopeptidase trimming component of the APM [256].
12. Strategies for Enhancing ICI Therapy Effectiveness: The Role of Antigen Presentation
Despite impressive clinical results, resistance to ICI immunotherapy is commonly a bottleneck in the successful treatment of several cancer types. Many approaches are currently under investigation aiming to surmount resistance to ICIs and improve clinical outcomes, often focusing on combining various therapeutic modalities (traditional therapies, other immunotherapy regiments as well as molecularly targeted therapies) on a checkpoint inhibitor backbone [257]. From a mechanism point of view, ongoing approaches aim to promote antigen processing and presentation, improve tumor antigen release and neoantigen supply, enhance T cell priming, expansion, survival, and effector functions, make the TME more friendly for immune cells, attenuate tumor-induced immunosuppressive factors, and promote proinflammatory/immunogenic pathways [258,259,260]. Moreover, there is significant evidence that gut microbiota diversity and composition can affect ICI responses and resistance in many cancers, by educating local and systemic immune responses, enhancing beneficial effects of metabolites, and dampening immune-related side-effects [261]. A summary of ongoing pre-clinical and clinical efforts focusing on overcoming resistance to ICI therapies is shown in Figure 3.
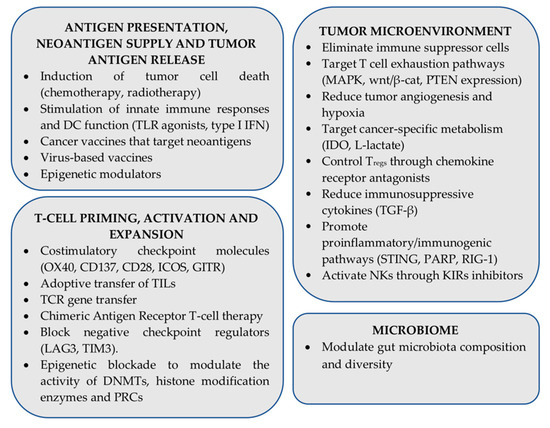
Figure 3.
Combinatorial strategies under investigation that aim to enhance efficacy of immune checkpoint inhibitor (ICI) immunotherapy (PRC: Polycomb repressive complex).
It is becoming increasingly clear that antigen processing and presentation is both central to cancer immune evasion and also a key puzzle piece in cancer immunotherapy. In order, however, to be able to manipulate APP to enhance cancer immunotherapy regiments, it is first necessary to understand the exact mechanisms by which APP is altered in cancer. Tumor cells can manipulate antigen presentation either by altering the cellular proteome or any of the components of the APP machinery. Therapeutically, several of the components of the APP machinery could be targeted in order to enhance the immunogenicity of cancer: the ubiquitin-proteasome degradation pathway, cytosolic peptidases, the TAP transporter, the peptide loading complex, peptide editing chaperones such as Tapasin or TAPBPR, ER aminopeptidases, and the MHCI themselves. In addition, induction of changes in the cellular proteome can regulate antigen presentation. In one study, researchers demonstrated a correlation between protein homeostasis and tumor antigen presentation by showing IFNγ-independent changes the MHCI peptide repertoire by low-level inhibition of the Heat Shock Protein Hsp90 [262]. Recently, Ilca and colleagues used a soluble form of the peptide editor TAPBPR and found an efficient way to bypass the peptides that are naturally presented and load onto tumour cells immunogenic peptides that resulted in robust immune responses [263].
One component of the APP that appears amenable to pharmacological targeting are the ER aminopeptidases ERAP1 and ERAP2 and the cross-presentation related aminopeptidase Insulin-regulated aminopeptidase IRAP. ERAP1 and ERAP2 appear to have a significant amount of specialization for antigen processing, whereas IRAP participates in additional biological processes including T cell receptor signaling [264]. Both ERAP1 and ERAP2 have been shown to be downregulated in some cancers [265], play key roles in the shaping of the cellular immunopeptidome [85], and their activity has been associated with changes in anti-cancer immune responses [101]. Furthermore, ERAP1 inhibitors have been shown to regulate the immunopeptidome [86] and elicit antitumor cytotoxic responses [90,91,94]. IRAP has been shown to be important in cross-presentation [266] and an IRAP inhibitor to be able to enhance cytotoxic responses ex vivo [267]. Furthermore, the development of inhibitors for these enzymes has reached significant maturity [268,269,270]. However, the synergism between inhibition of intracellular antigen processing by aminopeptidases and enhancement of antitumor immunity by ICI has not been explored yet.
A potential synergism between aminopeptidase inhibition and ICI is depicted in Figure 4. As shown in panel A, an immune-evading tumor can be using ERAP1/ERAP2 to destroy tumor-associated antigenic peptides and over-expresses immune checkpoints such as PD-L1 to avoid T cell responses. Therapeutic interventions using ICIs, such as a-PD-1, can help promote T cell re-engagement but lack of presentation of appropriate tumor-associated antigenic peptides represents a bottleneck on antitumor cytotoxic responses (Panel B). Inhibition of ERAP1 or ERAP2, can help reactivate such responses by protecting tumor-associated antigenic peptides from degradation (Panel C). While this combinatorial approach is promising, it has not been experimentally evaluated and can suffer from a number of serious caveats since it cannot address exclusion of T cells from the TME or other means of T cell inactivation and antigen presentation silencing. Thus, it may be limited to specific cases or require combination with additional immunotherapy approaches. Given however the important role of APP in antitumor responses and the multitude of combinatorial cancer immunotherapy approaches currently under investigation, the modulation of intracellular antigen processing by aminopeptidase inhibitors is highly likely to find an application in enhancing tumor antigenicity.
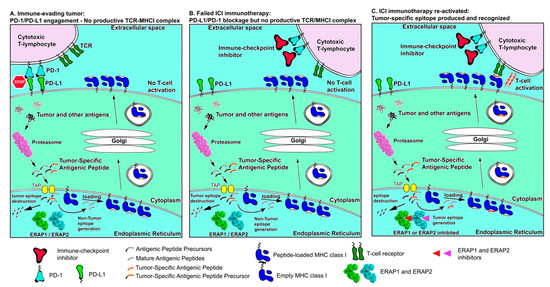
Figure 4.
Schematic representation of antigen processing and presentation in cancer immune evasion and immune re-activation by ICIs and manipulation of intracellular antigen processing. (Panel A) tumor antigens are processed by the proteasome but then destroyed by ER aminopeptidases ERAP1 or ERAP2 resulting in lack of presentation on the cell surface. Overexpression of PD-L1 on the cancer cell surface downregulates cytotoxic T-lymphocyte responses. Synergism between these two mechanisms results in efficient immune evasion by the tumor. (Panel B) Immune-checkpoint inhibitors can help activate T cells but lack of tumor antigen presentation limits cytotoxic responses. (Panel C) inhibition of ERAP1 and ERAP2 can help rescue tumor-associated antigenic peptides from destruction and promote their presentation, which, in combination with ICI treatment, can help re-activate T cell cytotoxic responses against the tumor.
13. Concluding Remarks
Expanding the benefits of cancer immunotherapy with ICIs to more patients and cancer types is probably one of the most urgent challenges in modern cancer therapy. The initial enthusiasm with ICI clinical successes gradually gave way to the realization that the interplay between the immune system and cancer is extremely complex and poorly understood. Many facets of this interplay have to synergize to circumvent the established evolutionary immune evasion of cancer. Not surprisingly, many immunotherapy approaches under investigation aim to combine multiple modulations of the immune system, including T cells and the TME, to achieve synergistic therapeutic effects. Antigen processing and presentation is undoubtably a key component in the immune evasion by cancer and thus its modulation constitutes a highly promising avenue for therapy. Still, being only one part of a larger puzzle, time will tell if manipulation of antigen presentation will be an effective monotherapy or it will find its place as a component of combination immunotherapy.
Funding
A.M acknowledges financial support from the Hellenic Foundation for Research and Innovation (H.F.R.I) post-doctoral grant no 303.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank Angeliki Chroni for critical advice and editing of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| APC | antigen presenting cell |
| APP(M) | antigen processing and presentation (machinery) |
| CTA | cancer testis antigen |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| DC | dendritic cell |
| HLA | human leukocyte antigen |
| IC(I) | immune checkpoint (inhibitor) |
| LN | lymph node |
| MMR | mismatch repair |
| NK | natural killer cell |
| NSCLC | non-small cell lung cancer |
| PAMPs/DAMPs | pathogen/damage associated molecular patterns |
| PD-1 | programmed cell death protein 1 |
| MHCI | Major Histocompatibility Complex class I |
| TCR | T cell receptor |
| TIL | Tumor infiltrating Lymphocyte |
| TMB | tumor mutational burden |
| TME | tumor microenvironment |
References
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Mechanisms of Tumor Cell-Intrinsic Immune Evasion. Annu. Rev. Cancer Biol. 2018, 2, 213–228. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to T cell–mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.; Baylot, V.; Gutgemann, I.; Eilers, M.; et al. MYC Regulates the Anti-Tumor Immune Response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef]
- Ji, H.; Ramsey, M.R.; Hayes, D.N.; Fan, C.; McNamara, K.; Kozlowski, P.; Torrice, C.; Wu, M.C.; Shimamura, T.; Perera, S.A.; et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007, 448, 807–810. [Google Scholar] [CrossRef]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Al Bakir, M.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanić, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen directed immune escape in lung cancer evolution Rachel. Nature 2020, 567, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Dupage, M.; Mazumbar, C.; Schmidt, L.M.; Cheung, A.F.; Jacks, T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature 2012, 482, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Shea, L.K.; Hundal, J.; et al. Cancer Exome Analysis Reveals a T Cell Dependent Mechanism of Cancer Immunoediting Hirokazu. Nature 2013, 482, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Montesion, M.; Murugesan, K.; Jin, D.X.; Sharaf, R.; Sanchez, N.; Guria, A.; Minker, M.; Li, G.; Fisher, V.; Sokol, E.S.; et al. Somatic HLA class I loss is a widespread mechanism of immune evasion which refines the use of tumor mutational burden as a biomarker of checkpoint inhibitor response. Cancer Discov. 2020. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e11. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e9. [Google Scholar] [CrossRef]
- Vigan, S.; Perreau, M.; Pantaleo, G.; Harari, A. Positive and negative regulation of cellular immune responses in physiologic conditions and diseases. Clin. Dev. Immunol. 2012, 2012. [Google Scholar] [CrossRef]
- Chen, L. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef]
- Linsley, P.S.; Greene, J.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors [published erratum appears in Immunity 1995 Feb;2(2):following 203]. Immunity 1994, 1, 793–801. [Google Scholar] [CrossRef]
- Van Der Merwe, A.P.; Bodian, D.L.; Daenke, S.; Linsley, P.; Davis, S.J. Affinity and Very Fast Kinetics. J. Exp. Med. 1997, 185, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Egen, J.G.; Allison, J.P. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity 2002, 16, 23–35. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 1–17. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.; Gajewski, T.F.; Mackensen, A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: Implications for tumor immunotherapy. Cancer Immunol. Immunother. 2005, 54, 307–314. [Google Scholar] [CrossRef]
- Weiner, L.; Murray, J.; Shuptrine, C. Antibody-based immunotherapy of cancer: New insights, new targets. Cell 2012, 148, 1081–1084. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2016, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Kvistborg, P.; Philips, D.; Kelderman, S.; Hageman, L.; Ottensmeier, C.; Joseph-Pietras, D.; Welters, M.J.P.; Van Der Burg, S.; Kapiteijn, E.; Michielin, O.; et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci. Transl. Med. 2014, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yost, K.; Satpathy, A.; Wells, D.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.; Granja, J.; Sarin, K.; Brown, R.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Godec, J.; Odorizzi, P.M.; Brown, K.E.; Yates, K.B.; Ngiow, S.F.; Burke, K.P.; Maleri, S.; Grande, S.M.; Francisco, L.M.; et al. The PD-1 Pathway Regulates Development and Function of Memory CD8+ T Cells following Respiratory Viral Infection. Cell Rep. 2020, 31, 107827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ave, H. Advances of FDA A pproved D rugs that T arget PD-1 and PD-L1 for Cancer Immunotherapy. Am. J. Cancer Sci. 2019, 7, 18–31. [Google Scholar]
- Ramagopal, U.A.; Liu, W.; Garrett-Thomson, S.C.; Bonanno, J.B.; Yan, Q.; Srinivasan, M.; Wong, S.C.; Bell, A.; Mankikar, S.; Rangan, V.S.; et al. Structural basis for cancer immunotherapy by the first-in-class checkpoint inhibitor ipilimumab. Proc. Natl. Acad. Sci. USA 2017, 114, 4223–4232. [Google Scholar] [CrossRef]
- Selby, M.J.; Engelhardt, J.J.; Quigley, M.; Henning, K.A.; Chen, T.; Srinivasan, M.; Korman, A.J. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res. 2013, 1, 32–42. [Google Scholar] [CrossRef]
- Bulliard, Y.; Jolicoeur, R.; Windman, M.; Rue, S.M.; Ettenberg, S.; Knee, D.A.; Wilson, N.S.; Dranoff, G.; Brogdon, J.L. Activating fc γ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J. Exp. Med. 2013, 210, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Robert, L.; Tsoi, J.; Wang, X.; Emerson, R.; Homet, B.; Chodon, T.; Mok, S.; Huang, R.R.; Cochran, A.J.; Comin-Anduix, B.; et al. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin. Cancer Res. 2014, 20, 2424–2432. [Google Scholar] [CrossRef]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Emma, J.; Taylor, M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef]
- Wang, S.; He, Z.; Wang, X.; Li, H.; Liu, X.S. Antigen presentation and tumor immunogenicity in cancer immunotherapy response prediction. Elife 2019, 8, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Blum, S.J. Pathways of Antigen Processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, F.A.; Oettgen, H.C. Adaptive immunity. J. Allergy Clin. Immunol. 2010, 125, S33–S40. [Google Scholar] [CrossRef] [PubMed]
- Yewdell, J.W.; Hollý, J. DRiPs get molecular. Curr. Opin. Immunol. 2020, 64, 130–136. [Google Scholar] [CrossRef]
- Rock, K.L.; York, I.A.; Goldberg, A.L. Post-proteasomal antigen processing for major histocompatibility complex class I presentation. Nat. Immunol. 2004, 5, 670–677. [Google Scholar] [CrossRef]
- Jensen, P.E. Recent advances in antigen processing and presentation. Nat. Immunol. 2007, 8, 1041–1048. [Google Scholar] [CrossRef]
- Blees, A.; Januliene, D.; Hofmann, T.; Koller, N.; Schmidt, C.; Trowitzsch, S.; Moeller, A.; Tampé, R. Structure of the human MHC-I peptide-loading complex. Nature 2017, 551, 525–528. [Google Scholar] [CrossRef]
- Lee, M.Y.; Jeon, J.W.; Sievers, C.; Allen, C.T. Antigen processing and presentation in cancer immunotherapy. J. Immunother. Cancer 2020, 8, 1–15. [Google Scholar] [CrossRef]
- Saveanu, L.; Carroll, O.; Lindo, V.; Del Val, M.; Lopez, D.; Lepelletier, Y.; Greer, F.; Schomburg, L.; Fruci, D.; Niedermann, G.; et al. Concerted peptide trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the endoplasmic reticulum. Nat. Immunol. 2005, 6, 689–697. [Google Scholar] [CrossRef]
- Morozov, G.I.; Zhao, H.; Mage, M.G.; Boyd, L.F.; Jiang, J.; Dolan, M.A.; Venna, R.; Norcross, M.A.; McMurtrey, C.P.; Hildebrand, W.; et al. Interaction of TAPBPR, a tapasin homolog, with MHC-I molecules promotes peptide editing. Proc. Natl. Acad. Sci. USA 2016, 113, E1006–E1015. [Google Scholar] [CrossRef]
- Seliger, B.; Maeurer, M.J.; Ferrone, S. Antigen-processing machinery breakdown and tumor growth. Immunol. Today 2000, 21, 455–464. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wangmo, D.; Robertson, M.; Subramanian, S. Acquired resistance to immune checkpoint blockade therapies. Cancers 2020, 12, 1161. [Google Scholar] [CrossRef]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Cho, H.; Perry, C.; Song, J.; Ylaya, K.; Lee, H.; Kim, J.H. Downregulation of ERp57 expression is associated with poor prognosis in early-stage cervical cancer. Biomarkers 2013, 18, 573–579. [Google Scholar] [CrossRef]
- Hasim, A.; Abudula, M.; Aimiduo, R.; Ma, J.Q.; Jiao, Z.; Akula, G.; Wang, T.; Abudula, A. Post-Transcriptional and Epigenetic Regulation of Antigen Processing Machinery (APM) Components and HLA-I in Cervical Cancers from Uighur Women. PLoS ONE 2012, 7, e44952. [Google Scholar] [CrossRef]
- Leys, C.M.; Nomura, S.; LaFleur, B.J.; Ferrone, S.; Kaminishi, M.; Montgomery, E.; Goldenring, J.R. Expression and prognostic significance of prothymosin-α and ERp57 in human gastric cancer. Surgery 2007, 141, 41–50. [Google Scholar] [CrossRef]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef]
- Cathro, H.P.; Smolkin, M.E.; Theodorescu, D.; Jo, V.Y.; Ferrone, S.; Frierson, H.F., Jr. Relationship between HLA class I antigen processing machinery component expression and the clinicopathologic characteristics of bladder carcinomas. Cancer Immunol. Immunother. 2012, 59, 465–472. [Google Scholar] [CrossRef]
- Arshad, N.; Cresswell, P. Tumor-associated calreticulin variants functionally compromise the peptide loading complex and impair its recruitment of MHC-I. J. Biol. Chem. 2018, 293, 9555–9569. [Google Scholar] [CrossRef]
- Mehta, A.M.; Jordanova, E.S.; Kenter, G.G.; Ferrone, S.; Fleuren, G.J. Association of antigen processing machinery and HLA class I defects with clinicopathological outcome in cervical carcinoma. Cancer Immunol. Immunother. 2008, 57, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B.; Höhne, A.; Knuth, A.; Bernhard, H.; Ehring, B.; Tampé, R.; Huber, C. Reduced membrane major histocompatibility complex class I density and stability in a subset of human renal cell carcinomas with low TAP and LMP expression. Clin. Cancer Res. 1996, 2, 1427–1433. [Google Scholar] [PubMed]
- Ling, A.; Löfgren-Burström, A.; Larsson, P.; Li, X.; Wikberg, M.L.; Öberg, Å.; Stenling, R.; Edin, S.; Palmqvist, R. TAP1 down-regulation elicits immune escape and poor prognosis in colorectal cancer. Oncoimmunology 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Meissner, M.; Reichert, T.E.; Kunkel, M.; Gooding, W.; Whiteside, T.L.; Ferrone, S.; Seliger, B. Defects in the human leukocyte antigen class I antigen-processing machinery in head and neck squamous cell carcinoma: Association with clinical outcome. Clin. Cancer Res. 2005, 11, 2552–2560. [Google Scholar] [CrossRef] [PubMed]
- Lazaridou, M.-F.; Massa, C.; Handke, D.; Mueller, A.; Friedrich, M.; Subbarayan, K.; Tretbar, S.; Dummer, R.; Koelblinger, P.; Seliger, B. Identification of microRNAs Targeting the Transporter Associated with Antigen Processing TAP1 in Melanoma. J. Clin. Med. 2020, 9, 2690. [Google Scholar] [CrossRef]
- Dissemond, J.; Kothen, T.; Mörs, J.; Weimann, T.K.; Lindeke, A.; Goos, M.; Wagner, S.N. Downregulation of tapasin expression in progressive human malignant melanoma. Arch. Derm. Res. 2003, 295, 43–49. [Google Scholar] [CrossRef]
- Shionoya, Y.; Kanaseki, T.; Miyamoto, S.; Tokita, S.; Hongo, A.; Kikuchi, Y.; Kochin, V.; Watanabe, K.; Horibe, R.; Saijo, H.; et al. Loss of tapasin in human lung and colon cancer cells and escape from tumor-associated antigen-specific CTL recognition. Oncoimmunology 2017, 6, 1–11. [Google Scholar] [CrossRef]
- Sokol, L.; Koelzer, V.H.; Rau, T.T.; Karamitopoulou, E.; Zlobec, I.; Lugli, A. Loss of tapasin correlates with diminished CD8+ T-cell immunity and prognosis in colorectal cancer. J. Transl. Med. 2015, 13, 1–6. [Google Scholar] [CrossRef]
- Turnquist, H.R.; Kohlgraf, K.G.; McIlhaney, M.M.; Lee Mosley, R.; Hollingsworth, M.A.; Solheim, J.C. Tapasin decreases immune responsiveness to a model tumor antigen. J. Clin. Immunol. 2004, 24, 462–470. [Google Scholar] [CrossRef]
- Chang, C.C.; Pirozzi, G.; Wen, S.H.; Chung, I.H.; Chiu, B.L.; Errico, S.; Luongo, M.; Lombardi, M.L.; Ferrone, S. Multiple structural and epigenetic defects in the human leukocyte antigen class I antigen presentation pathway in a recurrent metastatic melanoma following immunotherapy. J. Biol. Chem. 2015, 290, 26562–26575. [Google Scholar] [CrossRef]
- Fruci, D.; Ferracuti, S.; Limongi, M.Z.; Cunsolo, V.; Giorda, E.; Fraioli, R.; Sibilio, L.; Carroll, O.; Hattori, A.; van Endert, P.M.; et al. Expression of Endoplasmic Reticulum Aminopeptidases in EBV-B Cell Lines from Healthy Donors and in Leukemia/Lymphoma, Carcinoma, and Melanoma Cell Lines. J. Immunol. 2006, 176, 4869–4879. [Google Scholar] [CrossRef] [PubMed]
- Compagnone, M.; Cifaldi, L.; Fruci, D. Regulation of ERAP1 and ERAP2 genes and their disfunction in human cancer. Hum. Immunol. 2019, 80, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, Y.; Yan, Z.; Dai, S.; Liu, S.; Wang, X.; Wang, J.; Zhang, X.; Shi, L.; Yao, Y. Polymorphisms in endoplasmic reticulum aminopeptidase genes are associated with cervical cancer risk in a Chinese Han population. BMC Cancer 2020, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Stratikos, E.; Stamogiannos, A.; Zervoudi, E.; Fruci, D. A role for naturally occurring alleles of Endoplasmic Reticulum Aminopeptidases in tumor immunity and cancer predisposition. Front. Oncol. 2014, 4, 1–10. [Google Scholar] [CrossRef]
- López de Castro, J.A. How ERAP1 and ERAP2 Shape the Peptidomes of Disease-Associated MHC-I Proteins. Front. Immunol. 2018, 9, 2463. [Google Scholar] [CrossRef]
- Koumantou, D.; Barnea, E.; Martin-Esteban, A.; Maben, Z.; Papakyriakou, A.; Mpakali, A.; Kokkala, P.; Pratsinis, H.; Georgiadis, D.; Stern, L.J.; et al. Editing the immunopeptidome of melanoma cells using a potent inhibitor of endoplasmic reticulum aminopeptidase 1 (ERAP1). Cancer Immunol. Immunother. 2019, 68, 1245–1261. [Google Scholar] [CrossRef]
- Mehta, A.M.; Jordanova, E.S.; Corver, W.E.; van Wezel, T.; Uh, H.-W.; Kenter, G.G.; Fleuren, G.J. Single Nucleotide Polymorphisms in Antigen Processing Machinery Component ERAP1 Significantly Associate with Clinical Outcome in Cervical Carcinoma. Genes. Chromosomes Cancer 2009, 48, 410–418. [Google Scholar] [CrossRef]
- Kamphausen, E.; Kellert, C.; Abbas, T.; Akkad, N.; Tenzer, S.; Pawelec, G.; Schild, H.; Van Endert, P.; Seliger, B. Distinct molecular mechanisms leading to deficient expression of ER-resident aminopeptidases in melanoma. Cancer Immunol. Immunother. 2010, 59, 1273–1284. [Google Scholar] [CrossRef]
- Cifaldi, L.; Romania, P.; Lorenzi, S.; Locatelli, F.; Fruci, D. Role of endoplasmic reticulum aminopeptidases in health and disease: From infection to cancer. Int. J. Mol. Sci. 2012, 13, 8338–8352. [Google Scholar] [CrossRef]
- James, E.; Bailey, I.; Sugiyarto, G.; Elliott, T. Induction of Protective Antitumor Immunity through Attenuation of ERAAP Function. J. Immunol. 2013, 190, 5839–5846. [Google Scholar] [CrossRef]
- Zervoudi, E.; Saridakis, E.; Birtley, J.R.; Seregin, S.S.; Reeves, E.; Kokkala, P.; Aldhamen, Y.A.; Amalfitano, A.; Mavridis, I.M.; James, E.; et al. Rationally designed inhibitor targeting antigentrimming aminopeptidases enhances antigen presentation and cytotoxic T-cell responses. Proc. Natl. Acad. Sci. USA 2013, 110, 19890–19895. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.; Ebstein, F.; Bürger, E.; Textoris-Taube, K.; Gorny, X.; Urban, S.; Zhao, F.; Dannenberg, T.; Sucker, A.; Keller, C.; et al. The proteasome immunosubunits, PA28 and ER-aminopeptidase 1 protect melanoma cells from efficient MART-126-35-specific T-cell recognition. Eur. J. Immunol. 2015, 45, 3257–3268. [Google Scholar] [CrossRef] [PubMed]
- Cifaldi, L.; Romania, P.; Falco, M.; Lorenzi, S.; Meazza, R.; Petrini, S.; Andreani, M.; Pende, D.; Locatelli, F.; Fruci, D. ERAP1 regulates natural killer cell function by controlling the engagement of inhibitory receptors. Cancer Res. 2015, 75, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Cifaldi, L.; Lo Monaco, E.; Forloni, M.; Giorda, E.; Lorenzi, S.; Petrini, S.; Tremante, E.; Pende, D.; Locatelli, F.; Giacomini, P.; et al. Natural killer cells efficiently reject lymphoma silenced for the endoplasmic reticulum aminopeptidase associated with antigen processing. Cancer Res. 2011, 71, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Kuo, I.C.; Kao, H.K.; Huang, Y.; Wang, C.I.; Yi, J.S.; Liang, Y.; Liao, C.T.; Yen, T.C.; Wu, C.C.; Chang, K.P. Endoplasmic reticulum aminopeptidase 2 involvement in metastasis of oral cavity squamous cell carcinoma discovered by proteome profiling of primary cancer cells. Oncotarget 2017, 8, 61698–61708. [Google Scholar] [CrossRef]
- Warthan, M.D.; Washington, S.L.; Franzese, S.E.; Ramus, R.M.; Kim, K.R.; York, T.P.; Stratikos, E.; Strauss, J.F.; Lee, E.D. The role of endoplasmic reticulum aminopeptidase 2 in modulating immune detection of choriocarcinoma. Biol. Reprod. 2018, 98, 309–322. [Google Scholar] [CrossRef]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In Vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef]
- Lim, Y.W.; Chen-Harris, H.; Mayba, O.; Lianoglou, S.; Wuster, A.; Bhangale, T.; Khan, Z.; Mariathasan, S.; Daemen, A.; Reeder, J.; et al. Germline genetic polymorphisms influence tumor gene expression and immune cell infiltration. Proc. Natl. Acad. Sci. USA 2018, 115, E11701–E11710. [Google Scholar] [CrossRef]
- Reeves, E.; Wood, O.; Ottensmeier, C.H.; King, E.V.; Thomas, G.J.; Elliott, T.; James, E. HPV Epitope Processing Differences Correlate with ERAP1 Allotype and Extent of CD8þ T-cell Tumor Infiltration in OPSCC. Cancer Immunol. Res. 2019, 7, 1202–1213. [Google Scholar] [CrossRef]
- Reeves, E.; Islam, Y.; James, E. ERAP1: A Potential Therapeutic Target for a Myriad of Diseases. Expert Opin. Ther. Targets 2020, 24, 535–544. [Google Scholar] [CrossRef]
- Stratikos, E. Modulating antigen processing for cancer immunotherapy. Oncoimmunology 2014, 3, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2016, 7, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; Testi, M.G.; Pasetto, M.; Picchio, M.C.; Innamorati, G.; Mazzocco, M.; Ugel, S.; Cingarlini, S.; Bronte, V.; Zanovello, P.; et al. IFN-γ-mediated upmodulation of MHC class I expression activates tumor-specific immune response in a mouse model of prostate cancer. Vaccine 2010, 28, 3548–3557. [Google Scholar] [CrossRef] [PubMed]
- Sveen, A.; Johannessen, B.; Tengs, T.; Danielsen, S.A.; Eilertsen, I.A.; Lind, G.E.; Berg, K.C.G.; Leithe, E.; Meza-Zepeda, L.A.; Domingo, E.; et al. Multilevel genomics of colorectal cancers with microsatellite instability-clinical impact of JAK1 mutations and consensus molecular subtype 1. Genome Med. 2017, 9, 1–16. [Google Scholar] [CrossRef]
- Karpf, A.R.; Jones, D.A. Reactivating the expression of methylation silenced genes in human cancer. Oncogene 2002, 21, 5496–5503. [Google Scholar] [CrossRef]
- Burr, M.L.; Sparbier, C.E.; Chan, K.L.; Chan, Y.C.; Kersbergen, A.; Lam, E.Y.N.; Azidis-Yates, E.; Vassiliadis, D.; Bell, C.C.; Gilan, O.; et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell 2019, 36, 385–401.e8. [Google Scholar] [CrossRef]
- Campoli, M.; Ferrone, S. HLA antigen changes in malignant cells: Epigenetic mechanisms and biologic significance. Oncogene 2008, 27, 5869–5885. [Google Scholar] [CrossRef]
- Setiadi, A.F.; Omilusik, K.; David, M.D.; Seipp, R.P.; Hartikainen, J.; Gopaul, R.; Choi, K.B.; Jefferies, W.A. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Cancer Res. 2008, 68, 9601–9607. [Google Scholar] [CrossRef]
- Zingg, D.; Arenas-Ramirez, N.; Sahin, D.; Rosalia, R.A.; Antunes, A.T.; Haeusel, J.; Sommer, L.; Boyman, O. The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Immunotherapy. Cell Rep. 2017, 20, 854–867. [Google Scholar] [CrossRef]
- Dunn, G.P.; Sheehan, K.C.F.; Old, L.J.; Schreiber, R.D. IFN unresponsiveness in LNCaP cells due to the lack of JAK1 gene expression. Cancer Res. 2005, 65, 3447–3453. [Google Scholar] [CrossRef]
- Yoshihama, S.; Roszik, J.; Downs, I.; Meissner, T.B.; Vijayan, S.; Chapuy, B.; Sidiq, T.; Shipp, M.A.; Lizee, G.A.; Kobayashi, K.S. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 5999–6004. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.; Fan, K.; Paschen, A.; Reker Hardrup, S.; Ferrone, S.; Nghiem, P.; Ugurel, S.; Schrama, D.; Becker, J.C. Epigenetic priming restores the HLA class-I antigen processing machinery expression in Merkel cell carcinoma. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ugurel, S.; Spassova, I.; Wohlfarth, J.; Drusio, C.; Cherouny, A.; Melior, A.; Sucker, A.; Zimmer, L.; Ritter, C.; Schadendorf, D.; et al. MHC class-I downregulation in PD-1/PD-L1 inhibitor refractory Merkel cell carcinoma and its potential reversal by histone deacetylase inhibition: A case series. Cancer Immunol. Immunother. 2019, 68, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.; Modur, V.; Senovilla, L.; Kroemer, G.; Komurov, K. Suppression of tumor antigen presentation during aneuploid tumor evolution contributes to immune evasion. Oncoimmunology 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.S. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef]
- Angell, T.E.; Lechner, M.G.; Jang, J.K.; LoPresti, J.S.; Epstein, A.L. MHC class I loss is a frequent mechanism of immune escape in papillary thyroid cancer that is reversed by interferon and selumetinib treatment in Vitro. Clin. Cancer Res. 2014, 20, 6034–6044. [Google Scholar] [CrossRef]
- Vermeulen, C.F.W.; Jordanova, E.S.; Zomerdijk-Nooijen, Y.A.; Ter Haar, N.T.; Peters, A.A.W.; Fleuren, G.J. Frequent HLA class I loss is an early event in cervical carcinogenesis. Hum. Immunol. 2005, 66, 1167–1173. [Google Scholar] [CrossRef]
- Ferrone, S.; Marincola, F.M. Loss of HLA class I antigens by melanoma cells: Molecular mechanisms, functional significance and clinical relevance. Trends Neurosci. 1993, 16, 487–494. [Google Scholar] [CrossRef]
- Aptsiauri, N.; Ruiz-Cabello, F.; Garrido, F. The transition from HLA-I positive to HLA-I negative primary tumors: The road to escape from T-cell responses. Curr. Opin. Immunol. 2018, 51, 123–132. [Google Scholar] [CrossRef]
- Garrido, F.; Aptsiauri, N. Cancer immune escape: MHC expression in primary tumours versus metastases. Immunology 2019, 158, 255–266. [Google Scholar] [CrossRef]
- Garrido, F.; Perea, F.; Bernal, M.; Sánchez-Palencia, A.; Aptsiauri, N.; Ruiz-Cabello, F. The escape of cancer from T cell-mediated immune surveillance: HLA class I loss and tumor tissue architecture. Vaccines 2017, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Kochan, G.; Escors, D.; Breckpot, K.; Guerrero-Setas, D. Role of non-classical MHC class I molecules in cancer immunosuppression. Oncoimmunology 2013, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.A. HLA-mediated tumor escape mechanisms that may impair immunotherapy clinical outcomes via T-cell activation (Review). Oncol. Lett. 2017, 14, 4415–4427. [Google Scholar] [CrossRef] [PubMed]
- Bradley, S.D.; Chen, Z.; Melendez, B.; Talukder, A.; Khalili, J.S.; Rodriguez-Cruz, T.; Liu, S.; Whittington, M.; Deng, W.; Li, F.; et al. BRAFV600E co-opts a conserved MHC class I internalization pathway to diminish antigen presentation and CD8+ T-cell recognition of melanoma. Cancer Immunol. Res. 2015, 3, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy Promotes Immune Evasion of Pancreatic Cancer by Degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Cui, X.; Zhuo, Y.; Li, H.; Ha, C.; Xin, L.; Ren, Y.; Zhang, W.; Sun, X.; et al. Oncoprotein SND1 hijacks nascent MHC-I heavy chain to ER-associated degradation, leading to impaired CD8+ T cell response in tumor. Sci. Adv. 2020, 6, 1–16. [Google Scholar] [CrossRef]
- Romero, I.; Martinez, M.; Garrido, C.; Collado, A.; Algarra, I.; Garrido, F.; Garcia-Lora, A.M. The tumour suppressor Fhit positively regulates MHC class I expression on cancer cells. J. Pathol. 2012, 227, 367–379. [Google Scholar] [CrossRef]
- Garancher, A.; Suzuki, H.; Haricharan, S.; Chau, L.Q.; Masihi, M.B.; Rusert, J.M.; Norris, P.S.; Carrette, F.; Romero, M.M.; Morrissy, S.A.; et al. Tumor necrosis factor overcomes immune evasion in p53-mutant medulloblastoma. Nat. Neurosci. 2020, 1–12. [Google Scholar] [CrossRef]
- Kriegsman, B.A.; Vangala, P.; Chen, B.J.; Meraner, P.; Brass, A.L.; Garber, M.; Rock, K.L. Frequent Loss of IRF2 in Cancers Leads to Immune Evasion through Decreased MHC Class I Antigen Presentation and Increased PD-L1 Expression. J. Immunol. 2019, 203, 1999–2010. [Google Scholar] [CrossRef]
- Huang, L.; Malu, S.; McKenzie, J.A.; Andrews, M.C.; Talukder, A.H.; Tieu, T.; Karpinets, T.; Haymaker, C.; Forget, M.A.; Williams, L.J.; et al. The RNA-binding protein MEX£B mediates resistance to cancer immunotherapy by downregulating HLA-a expression. Clin. Cancer Res. 2018, 24, 3366–3376. [Google Scholar] [CrossRef]
- Li, H.; Xiong, H.G.; Xiao, Y.; Yang, Q.C.; Yang, S.C.; Tang, H.C.; Zhang, W.F.; Sun, Z.J. Long Non-coding RNA LINC02195 as a Regulator of MHC I Molecules and Favorable Prognostic Marker for Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kulski, J.K. Long Noncoding RNA HCP5, a Hybrid HLA Class I Endogenous Retroviral Gene: Structure, Expression, and Disease Associations. Cells 2019, 8, 480. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lin, T.Y.; Chen, L.; Liu, Y.; Dian, M.J.; Hao, W.C.; Lin, X.L.; Li, X.Y.; Li, Y.L.; Lian, M.; et al. Mir-19 regulates the expression of interferon-induced genes and mhc class i genes in human cancer cells. Int. J. Med. Sci. 2020, 17, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Eichmüller, S.B.; Osen, W.; Mandelboim, O.; Seliger, B. Immune Modulatory microRNAs Involved in Tumor Attack and Tumor Immune Escape. J. Natl. Cancer Inst. 2017, 109, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Samstein, R.M.; Valero, C.; Chan, T.A.; Morris, L.G.T. Tumor mutational burden as a predictive biomarker for checkpoint inhibitor immunotherapy. Hum. Vaccines Immunother. 2020, 16, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Stokidis, S.; Fortis, S.P.; Kogionou, P.; Anagnostou, T.; Perez, S.A.; Baxevanis, C.N. HLA class I allele expression and clinical outcome in De Novo metastatic prostate cancer. Cancers 2020, 12, 1623. [Google Scholar] [CrossRef] [PubMed]
- Chowell, D. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2017, 47, 549–562. [Google Scholar] [CrossRef]
- Marty, R.; Kaabinejadian, S.; Rossell, D.; Slifker, M.J.; van de Haar, J.; Engin, H.B.; de Prisco, N.; Ideker, T.; Hildebrand, W.H.; Font-Burgada, J.; et al. MHC-I Genotype Restricts the Oncogenic Mutational Landscape. Cell 2017, 171, 1272–1283.e15. [Google Scholar] [CrossRef]
- Hurwitz, A.A.; Kwon, E.D.; Van Elsas, A. Costimulatory wars: The tumor menace. Curr. Opin. Immunol. 2000, 12, 589–596. [Google Scholar] [CrossRef]
- Kurts, C.; Cannarile, M.; Klebba, I.; Brocker, T. Cutting Edge: Dendritic Cells Are Sufficient to Cross-Present Self-Antigens to CD8 T Cells In Vivo. J. Immunol. 2001, 166, 1439–1442. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.B.; Schuler, G. Immature, semi-mature and fully mature dendritic cells: Which signals induce tolerance or immunity? Trends Immunol. 2002, 23, 445–449. [Google Scholar] [CrossRef]
- Reis, E.; Sousa, C. Dendritic cells in a mature age. Nat. Rev. Immunol. 2006, 6, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Worbs, T.; Hammerschmidt, S.I.; Förster, R. Dendritic cell migration in health and disease. Nat. Rev. Immunol. 2017, 17, 30–48. [Google Scholar] [CrossRef]
- Gardner Dendritic Cells and Cancer Immunity. Trends Immunol. 2016, 37, 855–865. [CrossRef]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Ma, Y.; Shurin, G.V.; Peiyuan, Z.; Shurin, M.R. Dendritic cells in the cancer microenvironment. J. Cancer 2013, 4, 36–44. [Google Scholar] [CrossRef]
- Mueller, S.N.; Gebhardt, T.; Carbone, F.R.; Heath, W.R. Memory T cell subsets, migration patterns, and tissue residence. Annu. Rev. Immunol. 2013, 31, 137–161. [Google Scholar] [CrossRef]
- Preynat-Seauve, O.; Schuler, P.; Contassot, E.; Beermann, F.; Huard, B.; French, L.E. Tumor-Infiltrating Dendritic Cells Are Potent Antigen-Presenting Cells Able to Activate T Cells and Mediate Tumor Rejection. J. Immunol. 2006, 176, 61–67. [Google Scholar] [CrossRef]
- van Mierlo, G.J.D.; Boonman, Z.F.H.M.; Dumortier, H.M.H.; den Boer, A.T.; Fransen, M.F.; Nouta, J.; van der Voort, E.I.H.; Offringa, R.; Toes, R.E.M.; Melief, C.J.M. Activation of Dendritic Cells That Cross-Present Tumor-Derived Antigen Licenses CD8 + CTL to Cause Tumor Eradication. J. Immunol. 2004, 173, 6753–6759. [Google Scholar] [CrossRef]
- Henrickson, S.E.; Perro, M.; Loughhead, S.M.; Senman, B.; Quigley, M.; Alexe, G.; Iannacone, M.; Flynn, M.P.; Jesneck, J.L.; Imam, S.; et al. Antigen availability determines CD8+ T cell-dendritic cell interaction kinetics and memory fate decisions. Immunity 2014, 39, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Bonaccorsi, I.; Campana, S.; Morandi, B.; Ferlazzo, G. Acquisition and Presentation of Tumor Antigens by Dendritic Cells. Crit. Rev. Immunol. 2015, 35, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Sofopoulos, M.; Fortis, S.P.; Vaxevanis, C.K.; Sotiriadou, N.N.; Arnogiannaki, N.; Ardavanis, A.; Vlachodimitropoulos, D.; Perez, S.A.; Baxevanis, C.N. The prognostic significance of peritumoral tertiary lymphoid structures in breast cancer. Cancer Immunol. Immunother. 2019, 68, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T. Getting to the source: Dendritic cells as therapeutic reagents for the treatment of patients with cancer. Ann. Surg. 1997, 226, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Stoitzner, P.; Green, L.K.; Jung, J.Y.; Price, K.M.; Atarea, H.; Kivell, B.; Ronchese, F. Inefficient presentation of tumor-derived antigen by tumor-infiltrating dendritic cells. Cancer Immunol. Immunother. 2008, 57, 1665–1673. [Google Scholar] [CrossRef]
- Mcdonnell, A.M.; Lesterhuis, W.J.; Khong, A.; Nowak, A.K.; Lake, R.A.; Currie, A.J.; Robinson, B.W.S. Tumor-infiltrating dendritic cells exhibit defective cross-presentation of tumor antigens, but is reversed by chemotherapy. Eur. J. Immunol. 2015, 45, 49–59. [Google Scholar] [CrossRef]
- Veglia, F.; Tyurin, V.A.; Mohammadyani, D.; Blasi, M.; Duperret, E.K.; Donthireddy, L.; Hashimoto, A.; Kapralov, A.; Amoscato, A.; Angelini, R.; et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Zhuang, Y.; Liu, C.; Liu, J.; Li, G. Resistance mechanism of PD-1/PD-L1 blockade in the cancer-immunity cycle. Onco. Targets 2020, 13, 83–94. [Google Scholar] [CrossRef]
- Scarlett, U.K.; Rutkowski, M.R.; Rauwerdink, A.M.; Fields, J.; Escovar-Fadul, X.; Baird, J.; Cubillos-Ruiz, J.R.; Jacobs, A.C.; Gonzalez, J.L.; Weaver, J.; et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J. Exp. Med. 2012, 209, 495–506. [Google Scholar] [CrossRef]
- Sánchez-Paulete, A.R.; Teijeira, A.; Cueto, F.J.; Garasa, S.; Pérez-Gracia, J.L.; Sánchez-Arráez, A.; Sancho, D.; Melero, I. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann. Oncol. 2017, 28, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Bandola-Simon, J.; Roche, P.A. Dysfunction of antigen processing and presentation by dendritic cells in cancer. Mol. Immunol. 2019, 113, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Baxevanis, C.N.; Fortis, S.P.; Perez, S.A. The balance between breast cancer and the immune system: Challenges for prognosis and clinical benefit from immunotherapies. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Cao, W.; Ramakrishnan, R.; Tuyrin, V.A.; Veglia, F.; Condamine, T.; Amoscato, A.; Mohammadyani, D.; Johnson, J.J.; Zhang, L.M.; Klein-Seetharaman, J.; et al. Correction: Oxidized Lipids Block Antigen Cross-Presentation by Dendritic Cells in Cancer. J. Immunol. 2014, 192, 4935. [Google Scholar] [CrossRef]
- Hargadon, K.M. Tumor-altered dendritic cell function: Implications for anti-tumor immunity. Front. Immunol. 2013, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.Y.; Bolton, H.; Kim, J.W.; Silveira, P.A.; Fromm, P.D.; Clark, G.J. On the other side: Manipulating the immune checkpoint landscape of dendritic cells to enhance cancer immunotherapy. Front. Oncol. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Alloatti, A.; Rookhuizen, D.C.; Joannas, L.; Carpier, J.M.; Iborra, S.; Magalhaes, J.G.; Yatim, N.; Kozik, P.; Sancho, D.; Albert, M.L.; et al. Critical role for Sec22b-dependent antigen crosspresentation in antitumor immunity. J. Exp. Med. 2017, 214, 2231–2241. [Google Scholar] [CrossRef]
- Sánchez-Paulete, A.R.; Cueto, F.J.; Martínez-lópez, M.; Labiano, S.; Rodridriguez-Ruiz, M.E.; Jure-Kunkel, M.; Azpilikueta, A.; Aznar, A.M.; Quetglas, J.I.; Sancho, D.; et al. Cancer immunotherapy with immunomodulatory anti-CD137 and anti-PD-1 monoclonal antibodies requires Batf3-dependent dendritic cells. Cancer Discov. 2016, 6, 71–79. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Dammeijer, F.; Lau, S.P.; van Eijck, C.H.J.; van der Burg, S.H.; Aerts, J.G.J.V. Rationally combining immunotherapies to improve efficacy of immune checkpoint blockade in solid tumors. Cytokine Growth Factor Rev. 2017, 36, 5–15. [Google Scholar] [CrossRef]
- Kadam, P.; Sharma, S. Pd-1 immune checkpoint blockade promotes therapeutic cancer vaccine to eradicate lung cancer. Vaccines 2020, 8, 317. [Google Scholar] [CrossRef] [PubMed]
- Antonios, J.P.; Soto, H.; Everson, R.G.; Orpilla, J.; Moughon, D.; Shin, N.; Sedighim, S.; Yong, W.H.; Li, G.; Cloughesy, T.F.; et al. PD-1 blockade enhances the vaccination-induced immune response in glioma. JCI Insight 2019, 1, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Roenblatt, J.; Glotzbecker, B.; Millis, H.; Baldev, V.; Tzachanis, D.; Levine, J.D.; Joyce, R.M.; Wellenstein, K.; Keefe, W.; Schickler, M.; et al. PD-1 blockade by CT-011, anti PD-1 antibody, enhances ex-vivo T cell responses to autologous dendritic/myeloma fusion vaccine. J. Immunother. 2011, 34, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Xi, H.; Ju, S.; Zhang, X. Blockade of PD-1/PD-L1 immune checkpoint during DC vaccination induces potent protective immunity against breast cancer in hu-SCID mice. Cancer Lett. 2013, 336, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Shimizu, K.; Iyoda, T.; Ueda, S.; Shinga, J.; Mochizuki, Y.; Watanabe, T.; Ohara, O.; Fujii, S. ichiro PD-L1 Expression Affects Neoantigen Presentation. iScience 2020, 23, 101238. [Google Scholar] [CrossRef]
- Linette, G.P.; Becker-Hapak, M.; Skidmore, Z.L.; Baroja, M.L.; Xu, C.; Hundal, J.; Spencer, D.H.; Fu, W.; Cummins, C.; Robnett, M.; et al. Immunological ignorance is an enabling feature of the oligo-clonal T cell response to melanoma neoantigens. Proc. Natl. Acad. Sci. USA 2019, 116, 23662–23670. [Google Scholar] [CrossRef]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Jordan, S.; Casanova-acebes, M.; Agudo, J.; Tung, N.; Chakarov, S.; Hogstad, B.; Bosenberg, M.; et al. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef]
- Hammerich, L.; Marron, T.U.; Upadhyay, R.; Svensson-Arvelund, J.; Dhainaut, M.; Hussein, S.; Zhan, Y.; Ostrowski, D.; Yellin, M.; Marsh, H.; et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat. Med. 2019, 25, 814–824. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Kubiniok, P.; Kovalchik, K.; Ma, Q.; Duquette, J.D.; Mongrain, I.; Deutsch, E.W.; Peters, B.; Sette, A.; Sirois, I.; et al. The Human Immunopeptidome Project: A roadmap to predict and treat immune diseases Running title: Next-generation immunopeptidomics. Mol. Cell Proteom. 2020, 19, 31–49. [Google Scholar] [CrossRef]
- Backert, L.; Kowalewski, D.J.; Walz, S.; Schuster, H.; Berlin, C.; Neidert, M.C.; Schemionek, M.; Brümmendorf, T.H.; Vucinic, V.; Niederwieser, D.; et al. A meta-analysis of HLA peptidome composition in different hematological entities: Entity-specific dividing lines and “panleukemia” antigens. Oncotarget 2017, 8, 43915–43924. [Google Scholar] [CrossRef] [PubMed]
- Bassani-Sternberg, M.; Coukos, G. Mass spectrometry-based antigen discovery for cancer immunotherapy. Curr. Opin. Immunol. 2016, 41, 9–17. [Google Scholar] [CrossRef]
- Schuster, H.; Peper, J.K.; Bösmüller, H.C.; Röhle, K.; Backert, L.; Bilich, T.; Ney, B.; Löffler, M.W.; Kowalewski, D.J.; Trautwein, N.; et al. The immunopeptidomic landscape of ovarian carcinomas. Proc. Natl. Acad. Sci. USA 2017, 114, E9942–E9951. [Google Scholar] [CrossRef] [PubMed]
- Loffler, M.W.; Kowalewski, D.J.; Backert, L.; Bernhardt, J.; Adam, P.; Schuster, H.; Dengler, F.; Backes, D.; Kopp, H.G.; Beckert, S.; et al. Mapping the HLA ligandome of colorectal cancer reveals an imprint of malignant cell transformation. Cancer Res. 2018, 78, 4627–4641. [Google Scholar] [CrossRef] [PubMed]
- Feola, S.; Chiaro, J.; Martins, B.; Cerullo, V. Uncovering the tumor antigen landscape: What to know about the discovery process. Cancers 2020, 12, 1660. [Google Scholar] [CrossRef]
- Kanaseki, T.; Torigoe, T. Proteogenomics: Advances in cancer antigen research. Immunol. Med. 2019, 42, 65–70. [Google Scholar] [CrossRef]
- Admon, A.; Bassani-Sternberg, M. The human immunopeptidome project, a suggestion for yet another postgenome next big thing. Mol. Cell. Proteom. 2011, 10, 1–4. [Google Scholar] [CrossRef]
- Pfammatter, S.; Bonneil, E.; Lanoix, J.; Vincent, K.; Hardy, M.P.; Courcelles, M.; Perreault, C.; Thibault, P. Extending the Comprehensiveness of Immunopeptidome Analyses Using Isobaric Peptide Labeling. Anal. Chem. 2020, 92, 9194–9204. [Google Scholar] [CrossRef]
- Coulie, P.G.; Van Den Eynde, B.J.; Van Der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Ilyas, S.; Yang, J.C. Landscape of Tumor Antigens in T Cell Immunotherapy. J. Immunol. 2015, 195, 5117–5122. [Google Scholar] [CrossRef]
- Zhang, M.; Fritsche, J.; Roszik, J.; Williams, L.J.; Peng, X.; Chiu, Y.; Tsou, C.C.; Hoffgaard, F.; Goldfinger, V.; Schoor, O.; et al. RNA editing derived epitopes function as cancer antigens to elicit immune responses. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Lill, J.R. MHC class I presented antigens from malignancies: A perspective on analytical characterization & immunogenicity. J. Proteom. 2018, 191, 48–57. [Google Scholar] [CrossRef]
- Engelhard, V.H.; Obeng, R.C.; Cummings, K.L.; Petroni, G.R.; Ambakhutwala, A.L.; Chianese-Bullock, K.A.; Smith, K.T.; Lulu, A.; Varhegyi, N.; Smolkin, M.E.; et al. MHC-restricted phosphopeptide antigens: Preclinical validation and first-in-humans clinical trial in participants with high-risk melanoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.I.; Yewdell, J.W.; Yang, J.C. Immune recognition of a human renal cancer antigen through post-translational protein splicing. Nature 2004, 427, 252–256. [Google Scholar] [CrossRef]
- Liepe, J.; Marino, F.; Sidney, J.; Jeko, A.; Bunting, D.E.; Sette, A.; Kloetzel, P.M.; Stumpf, M.P.H.; Heck, A.J.R.; Mishto, M. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science 2016, 354, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Mylonas, R.; Beer, I.; Iseli, C.; Chong, C.; Pak, H.S.; Gfeller, D.; Coukos, G.; Xenarios, I.; Müller, M.; Bassani-Sternberg, M. Estimating the contribution of proteasomal spliced peptides to the HLA-I ligandome. Mol. Cell. Proteom. 2018, 17, 2347–2357. [Google Scholar] [CrossRef]
- Chong, C.; Müller, M.; Pak, H.S.; Harnett, D.; Huber, F.; Grun, D.; Leleu, M.; Auger, A.; Arnaud, M.; Stevenson, B.J.; et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Laumont, C.M.; Daouda, T.; Laverdure, J.P.; Bonneil, É.; Caron-Lizotte, O.; Hardy, M.P.; Granados, D.P.; Durette, C.; Lemieux, S.; Thibault, P.; et al. Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Fratta, E.; Coral, S.; Covre, A.; Parisi, G.; Colizzi, F.; Danielli, R.; Marie Nicolay, H.J.; Sigalotti, L.; Maio, M. The biology of cancer testis antigens: Putative function, regulation and therapeutic potential. Mol. Oncol. 2011, 5, 164–182. [Google Scholar] [CrossRef]
- Gibbs, Z.A.; Whitehurst, A.W. Emerging Contributions of Cancer/Testis Antigens to Neoplastic Behaviors. Trends Cancer 2018, 4, 701–712. [Google Scholar] [CrossRef]
- Faramarzi, S.; Ghafouri-Fard, S. Melanoma: A prototype of cancer-testis antigen-expressing malignancies. Immunotherapy 2017, 9, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Arenas-Ramirez, N.; Sahin, D.; Boyman, O. Epigenetic mechanisms of tumor resistance to immunotherapy. Cell. Mol. Life Sci. 2018, 75, 4163–4176. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Kudchadkar, R.R.; Yu, B.; Gallenstein, D.; Horak, C.E.; Inzunza, H.D.; Zhao, X.; Martinez, A.J.; Wang, W.; Gibney, G.; et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J. Clin. Oncol. 2013, 31, 4311–4318. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Gnjatic, S.; Li, H.; Powel, S.; Gallardo, H.F.; Ritter, E.; Ku, G.Y.; Jungbluth, A.A.; Segal, N.H.; Rasalan, T.S.; et al. CTLA-4 blockade enhances polyfunctional NY-ESO-1 specific T cell responses in metastatic melanoma patients with clinical benefit. Proc. Natl. Acad. Sci. USA 2008, 105, 20410–20415. [Google Scholar] [CrossRef] [PubMed]
- Roszik, J.; Wang, W.-L.; Livingston, J.A.; Roland, C.L.; Ravi, V.; Yee, C.; Hwu, P.; Futreal, A.; Lazar, A.J.; Patel, S.R.; et al. Overexpressed PRAME is a potential immunotherapy target in sarcoma subtypes. Clin. Sarcoma Res. 2017, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ishak, C.A.; Classon, M.; De Carvalho, D.D. Deregulation of Retroelements as an Emerging Therapeutic Opportunity in Cancer. Trends Cancer 2018, 4, 583–597. [Google Scholar] [CrossRef]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef]
- Smith, C.C.; Beckermann, K.E.; Bortone, D.S.; Cubas, A.A.; Bixby, L.M.; Lee, S.J.; Panda, A.; Ganesan, S.; Bhanot, G.; Wallen, E.M.; et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J. Clin. Investig. 2018, 128, 4804–4820. [Google Scholar] [CrossRef]
- Luo, W.; Wang, X.; Kageshita, T.; Wakasugi, S.; Karpf, A.R.; Ferrone, S. Regulation of high molecular weight-melanoma associated antigen (HMW-MAA) gene expression by promoter DNA methylation in human melanoma cells. Oncogene 2006, 25, 2873–2884. [Google Scholar] [CrossRef][Green Version]
- Qamra, A.; Xing, M.; Padmanabhan, N.; Kwok, J.J.T.; Zhang, S.; Xu, C.; Leong, Y.S.; Lim, A.P.L.; Tang, Q.; Ooi, W.F.; et al. Epigenomic promoter alterations amplify gene isoform and immunogenic diversity in gastric adenocarcinoma. Cancer Discov. 2017, 7, 630–651. [Google Scholar] [CrossRef]
- Srivastava, P.K.; Duan, F. Harnessing the antigenic fingerprint of each individual cancer for immunotherapy of human cancer: Genomics shows a new way and its challenges. Cancer Immunol. Immunother. 2013, 62, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Vormehr, M.; Diken, M.; Boegel, S.; Kreiter, S.; Türeci, Ö.; Sahin, U. Mutanome directed cancer immunotherapy. Curr. Opin. Immunol. 2016, 39, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yu, L.; Wei, X.; Wei, Y. Role of tumor gene mutations in treatment response to immune checkpoint blockades. Precis. Clin. Med. 2019, 2, 100–109. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef]
- Riaz, N.; Morris, L.; Havel, J.J.; Makarov, V.; Desrichard, A.; Chan, T.A. The role of neoantigens in response to immune checkpoint blockade. Int. Immunol. 2016, 28, 411–419. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Geukes Foppen, M.H.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2016, 352, 207–212. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, M.; Miller, V.; Stephens, P.J.; Daniels, G.A. Response to Immunotherapy in Diverse Cancers. Mol. Cancer 2018, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Subudhi, S.K.; Vence, L.; Zhao, H.; Blando, J.; Yadav, S.S.; Xiong, Q.; Reuben, A.; Aparicio, A.; Corn, P.G.; Chapin, B.F.; et al. Neoantigen responses, immune correlates, and favorable outcomes after ipilimumab treatment of patients with prostate cancer. Sci. Transl. Med. 2020, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Xu, H.; Xu, X.; Guo, T.; Ge, W. High tumor mutation burden predicts better efficacy of immunotherapy: A pooled analysis of 103078 cancer patients. Oncoimmunology 2019, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhang, T.; Li, J.; Lin, J.; Liang, W.; Huang, W.; Wan, N.; Jiang, J. Association between tumor mutation burden (TMB) and outcomes of cancer patients treated with PD-1/ PD-L1 inhibitions: A meta-analysis. Front. Pharm. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Kowanetz, M.; Zou, W.; Shames, D.S.; Cummings, C.; Rizvi, N.; Spira, A.I.; Frampton, G.M.; Leveque, V.; Flynn, S.; Mocci, S.; et al. Tumor mutation load assessed by FoundationOne (FM1) is associated with improved efficacy of atezolizumab (atezo) in patients with advanced NSCLC. Ann. Oncol. 2016, 27, vi23. [Google Scholar] [CrossRef]
- Van Rooij, N.; Van Buuren, M.M.; Philips, D.; Velds, A.; Toebes, M.; Heemskerk, B.; Van Dijk, L.J.A.; Behjati, S.; Hilkmann, H.; El Atmioui, D.; et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J. Clin. Oncol. 2013, 31. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.B.; Abu-Akeel, M.; et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 2018, 33, 843–852.e4. [Google Scholar] [CrossRef]
- Samstein, R.M. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Yarchoan, M.; Johnson III, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Alborelli, I.; Leonards, K.; Rothschild, S.I.; Leuenberger, L.P.; Savic Prince, S.; Mertz, K.D.; Poechtrager, S.; Buess, M.; Zippelius, A.; Läubli, H.; et al. Tumor mutational burden assessed by targeted NGS predicts clinical benefit from immune checkpoint inhibitors in non-small cell lung cancer. J. Pathol. 2020, 250, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xu, J.; Du, C.; Wu, Y.; Xia, D.; Lv, W.; Hu, J. The Predictive Value of Tumor Mutation Burden on Efficacy of Immune Checkpoint Inhibitors in Cancers: A Systematic Review and Meta-Analysis. Front. Oncol. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Hoff, J.; Powles, T.; Van Der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; Fleming, M.T.; Petrylak, D.P.; Perez-gracia, J.L.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362. [Google Scholar] [CrossRef]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; Van Den Heuvel, M.M.; Ciuleanu, T.E.; Badin, F.; et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef]
- Johnson, D.B.; Frampton, G.M.; Rioth, M.J.; Yusko, E.; Xu, Y.; Guo, X.; Ennis, R.C.; Fabrizio, D.; Chalmers, Z.R.; Greenbowe, J.; et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol. Res. 2017, 4, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The therapeutic significance of mutational signatures from DNA repair deficiency in cancer. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schrock, A.B.; Ouyang, C.; Sandhu, J.; Sokol, E.; Jin, D.; Ross, J.S.; Miller, V.A.; Lim, D.; Amanam, I.; Chao, J.; et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann. Oncol. 2019, 30, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Gregory, P. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas Theo. Cell Rep. 2018, 23, 239–254. [Google Scholar] [CrossRef]
- Chae, Y.K.; Anker, J.F.; Oh, M.S.; Bais, P.; Namburi, S.; Agte, S.; Giles, F.J.; Chuang, J.H. Mutations in DNA repair genes are associated with increased neoantigen burden and a distinct immunophenotype in lung squamous cell carcinoma. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rospo, G.; Lorenzato, A.; Amirouchene-Angelozzi, N.; Magrì, A.; Cancelliere, C.; Corti, G.; Negrino, C.; Amodio, V.; Montone, M.; Bartolini, A.; et al. Evolving neoantigen profiles in colorectal cancers with DNA repair defects. Genome Med. 2019, 11, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magri, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ciombor, K.K.; Goldberg, R.M. Hypermutated Tumors and Immune Checkpoint Inhibition. Drugs 2018, 78, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Azad, N.S.; Laheru, D.; Biedrzycki, B.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef]
- Marijt, K.A.; Van Der Burg, S.H.; van Hall, T. TEIPP peptides: Exploration of unTAPped cancer antigens. Oncoimmunology 2019, 8, 1–3. [Google Scholar] [CrossRef]
- Marijt, K.A.; Doorduijn, E.M.; van Hall, T. TEIPP antigens for T-cell based immunotherapy of immune-edited HLA class Ilow cancers. Mol. Immunol. 2019, 113, 43–49. [Google Scholar] [CrossRef]
- Seidel, U.J.E.; Oliveira, C.C.; Lampen, M.H.; Van Hall, T. A novel category of antigens enabling CTL immunity to tumor escape variants: Cinderella antigens. Cancer Immunol. Immunother. 2012, 61, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Durgeau, A.; El Hage, F.; Vergnon, I.; Validire, P.; de Montpréville, V.; Besse, B.; Soria, J.-C.; van Hall, T.; Mami-Chouaib, F. Different Expression Levels of the TAP Peptide Transporter Lead to Recognition of Different Antigenic Peptides by Tumor-Specific CTL. J. Immunol. 2011, 187, 5532–5539. [Google Scholar] [CrossRef]
- Marijt, K.A.; Blijleven, L.; Verdegaal, E.M.E.; Kester, M.G.; Kowalewski, D.J.; Rammensee, H.G.; Stevanović, S.; Heemskerk, M.H.M.; Van Der Burg, S.H.; Van Hall, T. Identification of non-mutated neoantigens presented by TAP-deficient tumors. J. Exp. Med. 2018, 215, 2325–2337. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, N.A.; Gonzalez, F.; Shastri, N. Non-classical MHC class Ib-restricted cytotoxic T cells monitor antigen processing in the endoplasmic reticulum Niranjana. Nat. Immunol. 2012, 13, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, N.A.; de Verteuil, D.A.; Sriranganadane, D.; Yahyaoui, W.; Thibault, P.; Perreault, C. Shastr ERAAP shapes the peptidome associated with classical and non-classical MHC class I molecules. J. Immunol. 2016, 197, 1035–1043. [Google Scholar] [CrossRef]
- Nagarajan, N.A.; Shastri, N. Immune surveillance for ERAAP dysfunction. Mol. Immunol. 2013, 55, 120–122. [Google Scholar] [CrossRef]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef]
- Puccini, A.; Battaglin, F.; Iaia, M.L.; Lenz, H.J.; Salem, M.E. Overcoming resistance to anti-PD1 and anti-PD-L1 treatment in gastrointestinal malignancies. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Li, Y.; Liu, J.; Gao, L.; Liu, Y.; Meng, F.; Li, X.; Qin, F.X.F. Targeting the tumor microenvironment to overcome immune checkpoint blockade therapy resistance. Immunol. Lett. 2020, 220, 88–96. [Google Scholar] [CrossRef]
- Trojaniello, C.; Vitale, M.G.; Scarpato, L.; Esposito, A.; Ascierto, P.A. Melanoma immunotherapy: Strategies to overcome pharmacological resistance. Expert. Rev. Anticancer. 2020, 20, 289–304. [Google Scholar] [CrossRef]
- Huang, J.; Jiang, Z.; Wang, Y.; Fan, X.; Cai, J.; Yao, X.; Liu, L.; Huang, J.; He, J.; Xie, C.; et al. Modulation of gut microbiota to overcome resistance to immune checkpoint blockade in cancer immunotherapy. Curr. Opin. Pharm. 2020, 54, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, A.M.; Stopfer, L.; Lee, S.; Gaglia, G.; Sandel, D.; Santagata, S.; Lin, N.U.; Trepel, J.B.; White, F.; Jacks, T.; et al. Rebalancing protein homeostasis enhances tumor antigen presentation. Clin. Cancer Res. 2019, 25, 6392–6405. [Google Scholar] [CrossRef] [PubMed]
- Tudor Ilca, F.; Neerincx, A.; Wills, M.R.; De La Roche, M.; Boyle, L.H. Utilizing TAPBPR to promote exogenous peptide loading onto cell surface MHC I molecules. Proc. Natl. Acad. Sci. USA 2018, 115, 9353–9361. [Google Scholar] [CrossRef] [PubMed]
- Evnouchidou, I.; Weimershaus, M.; Saveanu, L.; van Endert, P. ERAP1–ERAP2 Dimerization Increases Peptide-Trimming Efficiency. J. Immunol. 2014, 193, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Fruci, D.; Giacomini, P.; Nicotra, M.R.; Forloni, M.; Fraioli, R.; Saveanu, L.; Endert, P.V.A.N. Altered Expression of Endoplasmic Reticulum Aminopeptidases ERAP1 and ERAP2 in Transformed Non-Lymphoid Human Tissues. J. Cell. Physiol. 2008, 216, 742–749. [Google Scholar] [CrossRef]
- Saveanu, L. IRAP Identifies an Endosomal Compartment Required for MHC Class I Cross-Presentation. Science 2009, 325, 213–217. [Google Scholar] [CrossRef]
- Papakyriakou, A.; Zervoudi, E.; Tsoukalidou, S.; Mauvais, F.X.; Sfyroera, G.; Mastellos, D.C.; Van Endert, P.; Theodorakis, E.A.; Vourloumis, D.; Stratikos, E. 3,4-diaminobenzoic acid derivatives as inhibitors of the oxytocinase subfamily of m1 aminopeptidases with immune-regulating properties. J. Med. Chem. 2015, 58, 1524–1543. [Google Scholar] [CrossRef]
- Georgiadis, D.; Ziotopoulou, A.; Kaloumenou, E.; Lelis, A.; Papasava, A. The Discovery of Insulin-Regulated Aminopeptidase (IRAP) Inhibitors: A Literature Review. Front. Pharm. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Liddle, J.; Hutchinson, J.P.; Kitchen, S.; Rowland, P.; Neu, M.; Cecconie, T.; Holmes, D.S.; Jones, E.; Korczynska, J.; Koumantou, D.; et al. Targeting the Regulatory Site of ER Aminopeptidase 1 Leads to the Discovery of a Natural Product Modulator of Antigen Presentation. ACS Appl. Mater. Interfaces 2020. [Google Scholar] [CrossRef]
- Georgiadis, D.; Mpakali, A.; Koumantou, D.; Stratikos, E. Inhibitors of ER Aminopeptidase 1 and 2: From Design to Clinical Application. Curr. Med. Chem. 2019, 26, 1–14. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).