Paradoxical Role of AT-rich Interactive Domain 1A in Restraining Pancreatic Carcinogenesis

 , , , and
, , , and
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
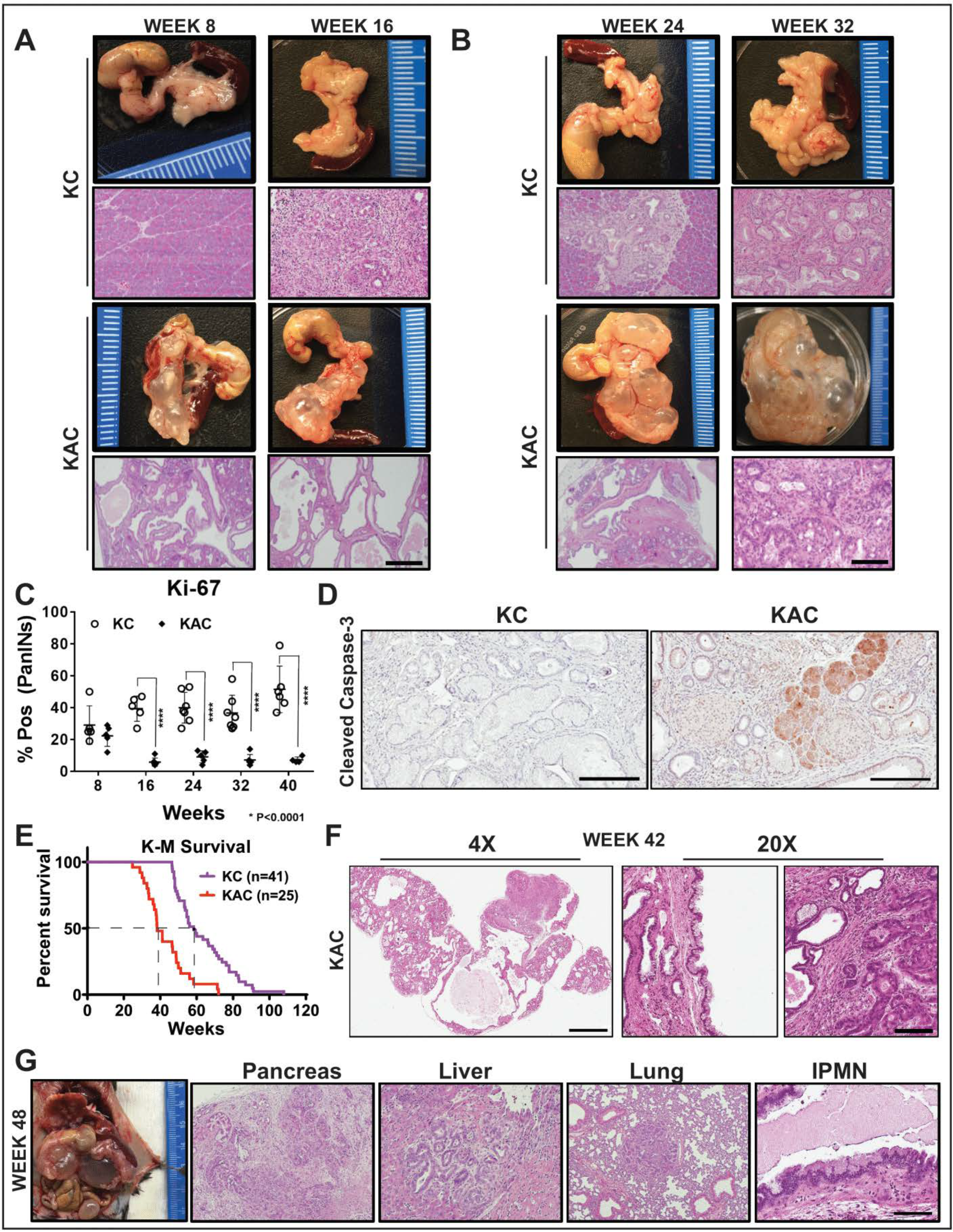
2.1. Loss of Arid1a Restrains Expansion and Progression of Ras-induced Pancreatic Precursor Lesions
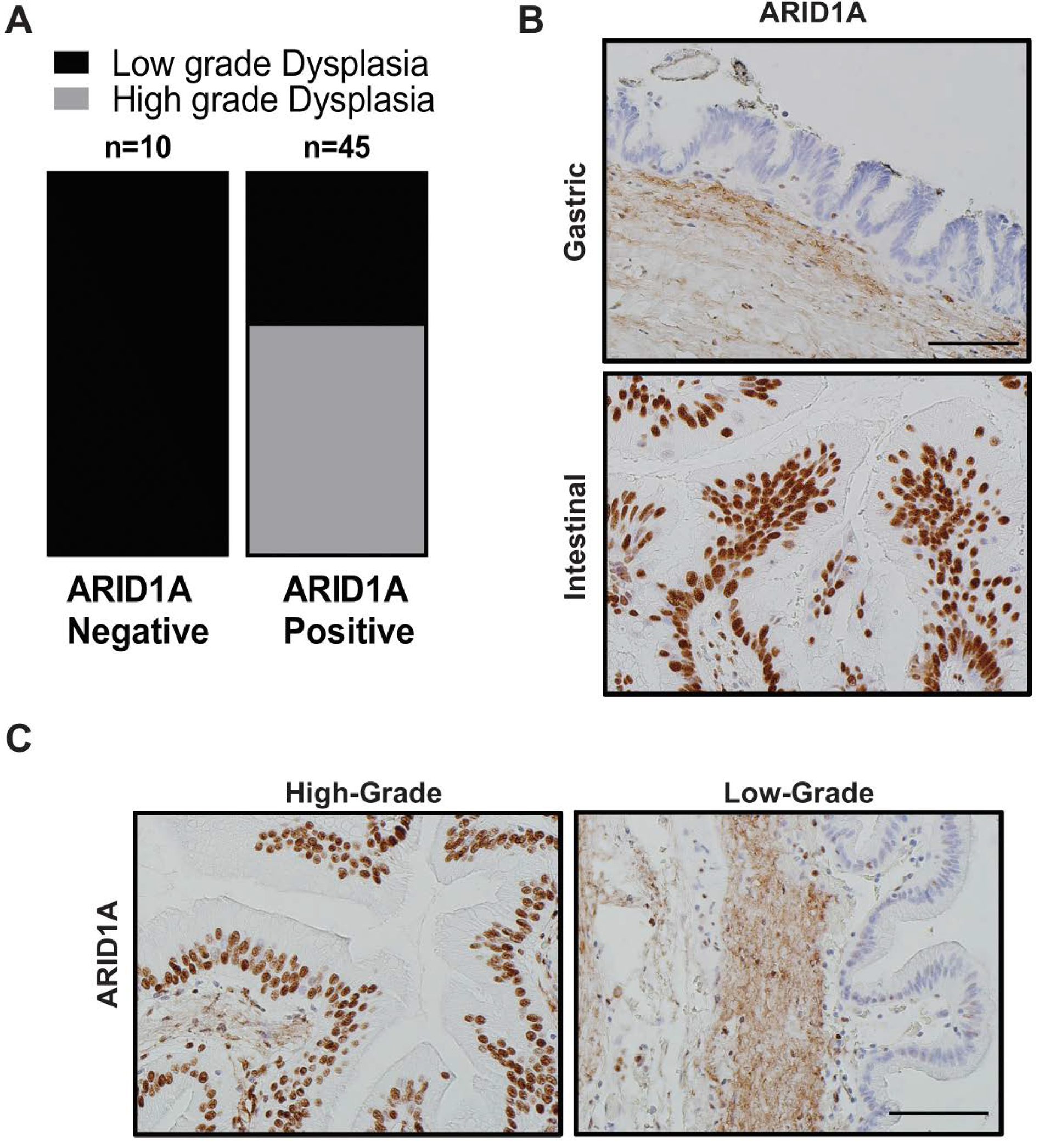
2.2. Loss of ARID1A Expression in Human IPMNs with Low-Grade Dysplasia
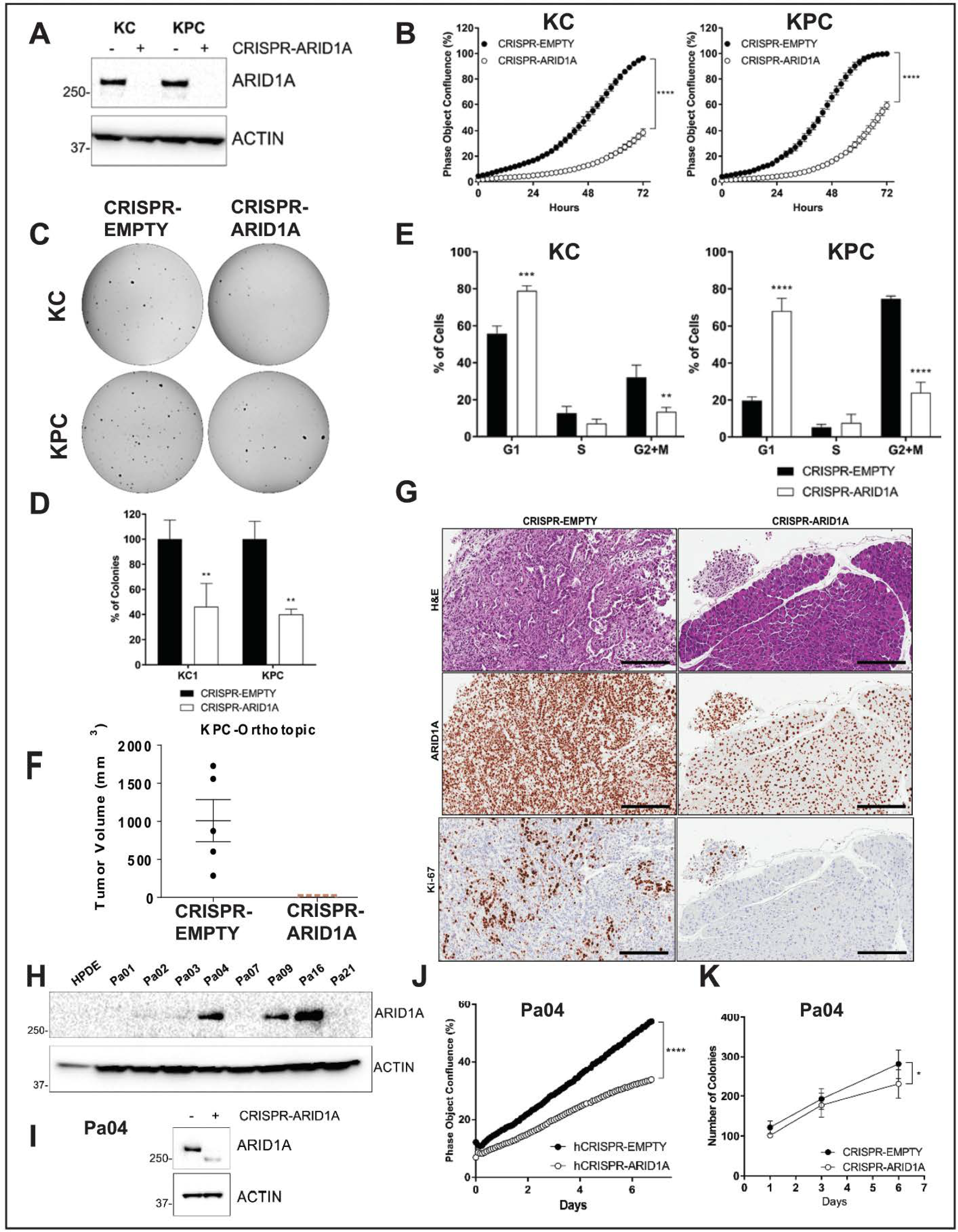
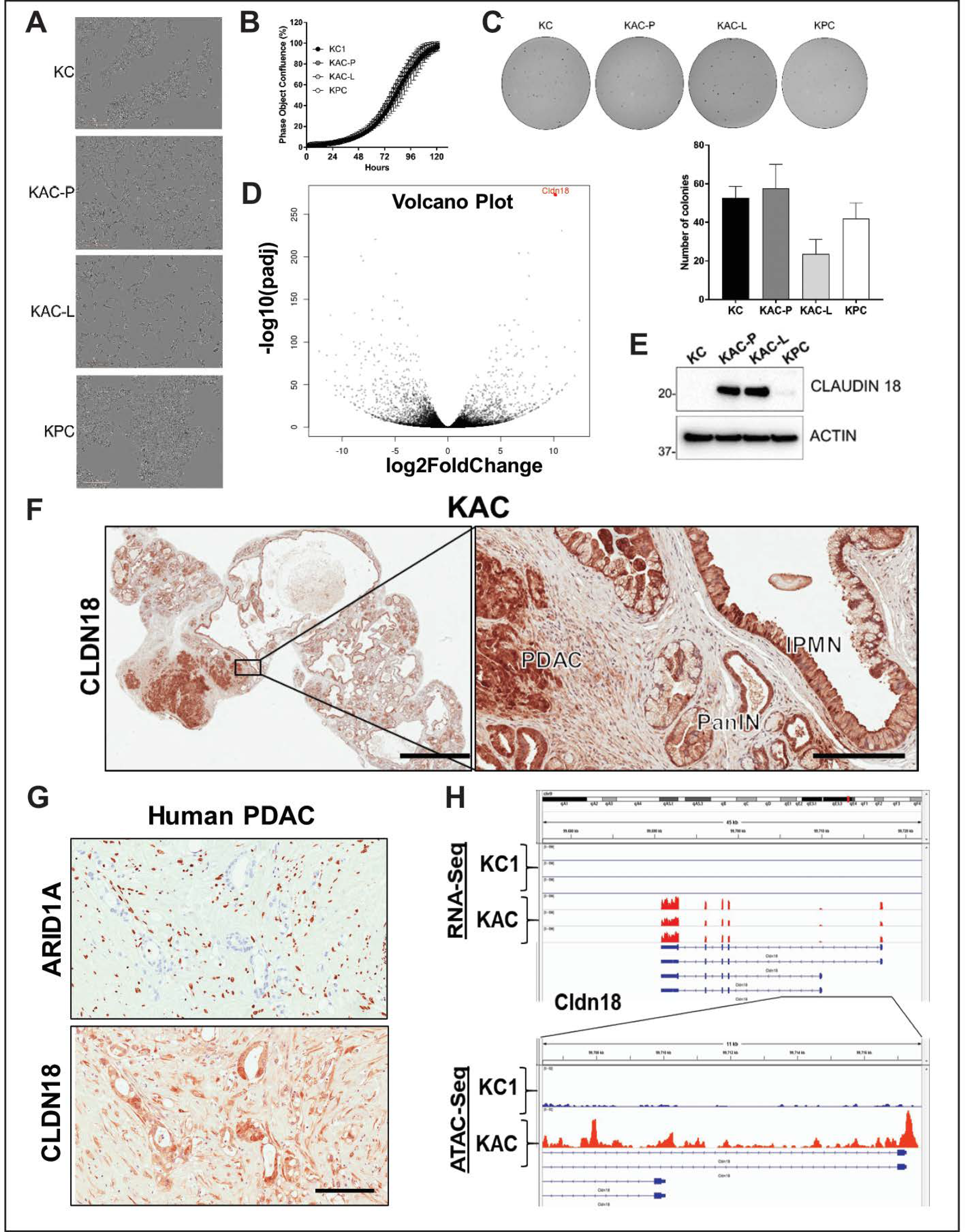
2.3. Arid1a Loss is Detrimental to the Growth of Established PDAC Lines
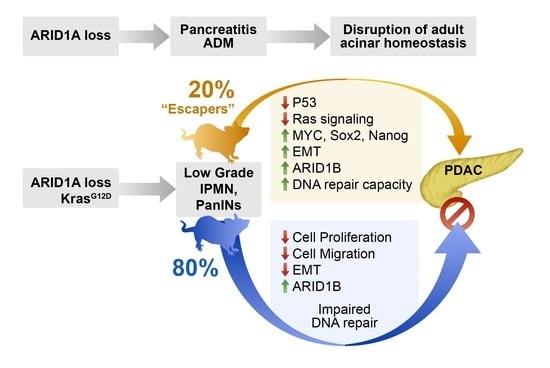
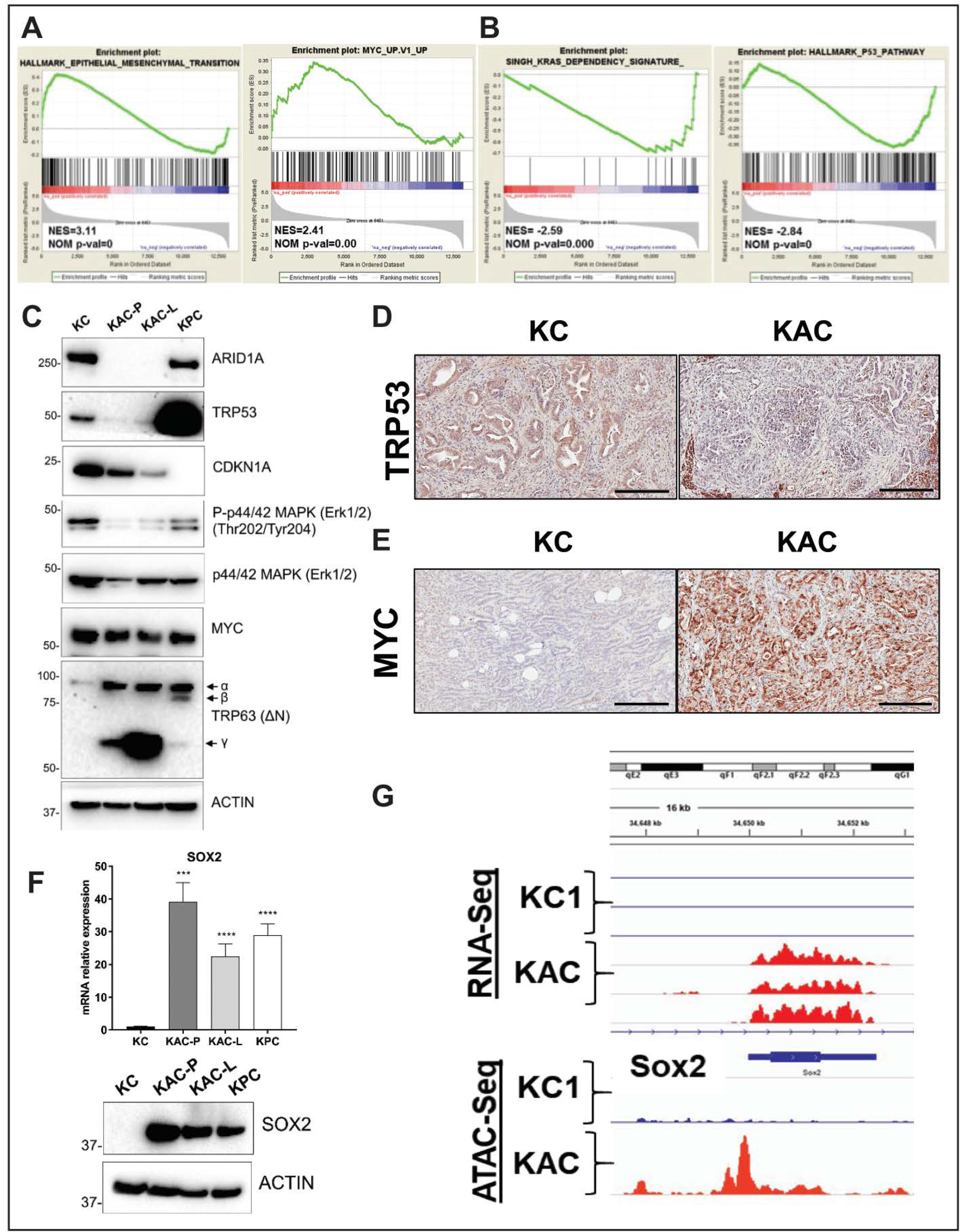
2.4. Identification of “Escaper” Mechanisms in Autochthonous Arid1a-null PDAC Cells
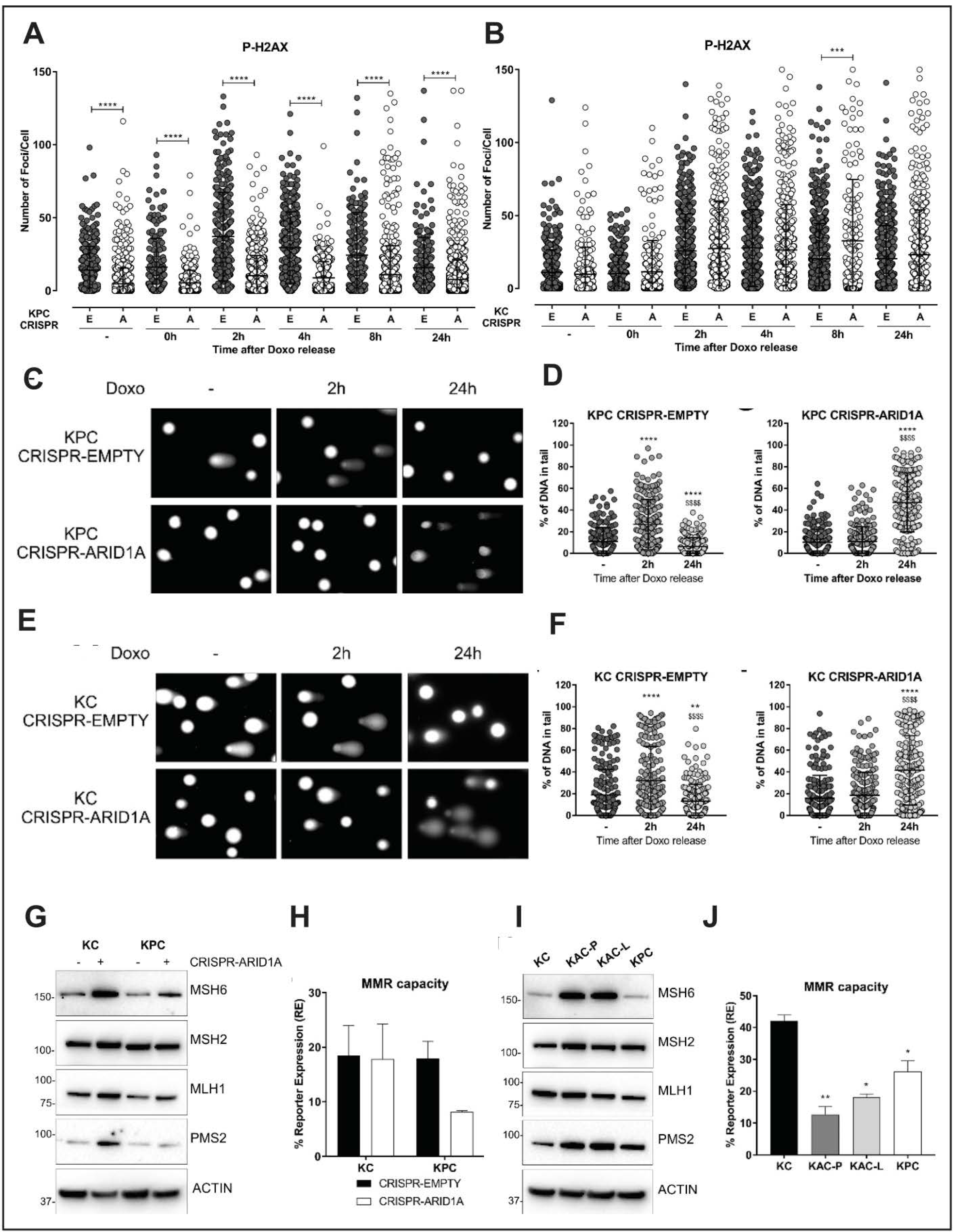
2.5. Impaired DNA Damage Repair as a Potential Mechanism Restraining Neoplastic Progression in Arid1a Null Cells
2.6. Loss of Arid1a Causes Impaired Mismatch Repair (MMR) in PDAC Cells
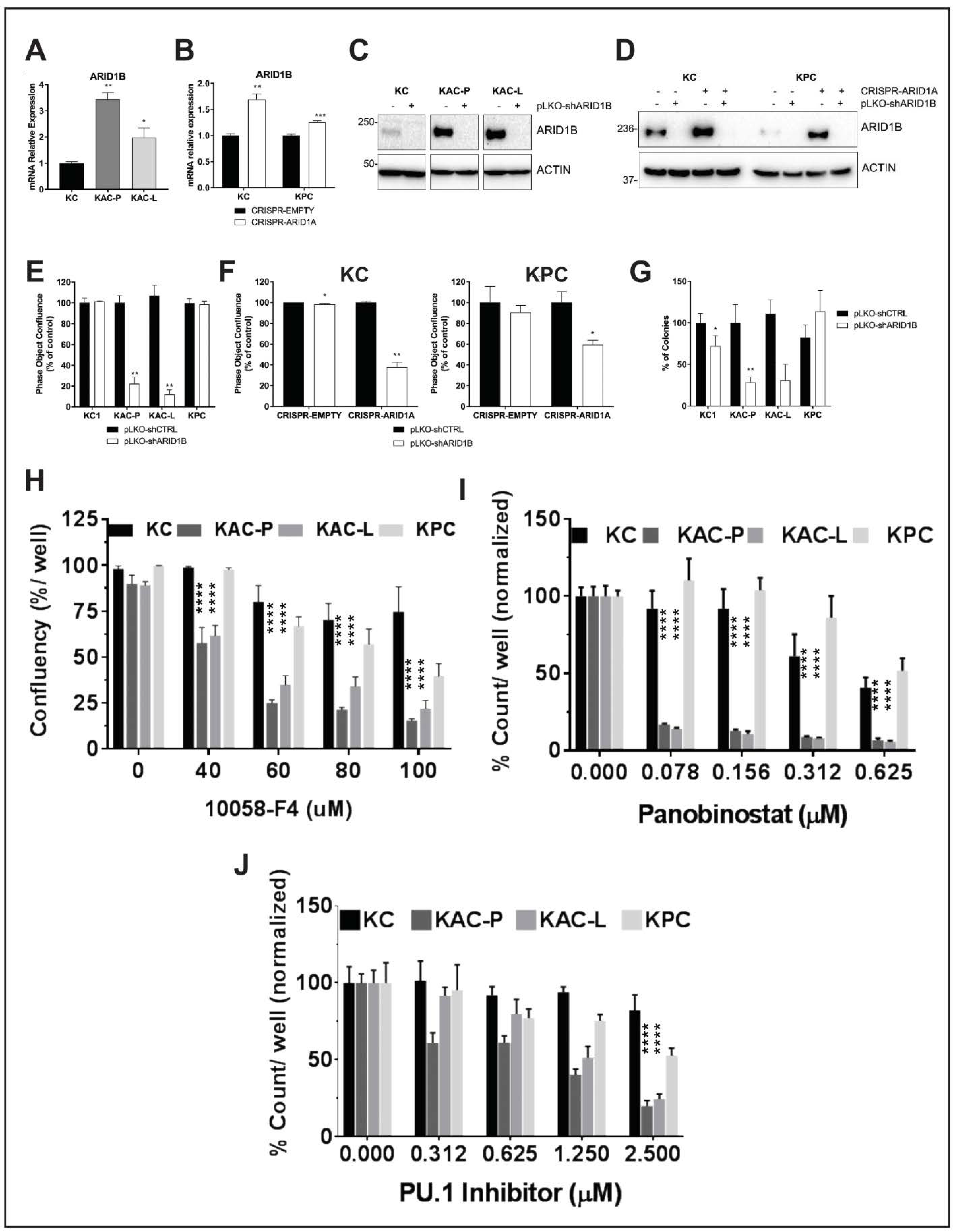
2.7. Synthetic Lethal Targeting of Arid1a Loss in PDAC
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. ARID1A Deletion by CRISPR/Cas9
4.3. Determination of Cell Growth and Morphology
4.4. 3D Cultures for Colony Formation
4.5. Migration Assay
4.6. Immunofluorescent (IF) Staining for Foci Formation
4.7. Alkaline Comet Assay
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; et al. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Fukuda, A.; Ogawa, S.; Maruno, T.; Takada, Y.; Tsuda, M.; Hiramatsu, Y.; Araki, O.; Nagao, M.; Yoshikawa, T.; et al. ARID1A Maintains differentiation of pancreatic ductal cells and inhibits development of pancreatic ductal adenocarcinoma in mice. Gastroenterology 2018, 155, 194–209.e2. [Google Scholar] [CrossRef] [PubMed]
- Livshits, G.; Alonso-Curbelo, D.; Morris, J.P.; Koche, R.; Saborowski, M.; Wilkinson, J.E.; Lowe, S.W. Arid1a restrains Kras-dependent changes in acinar cell identity. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Nassour, I.; Xiao, S.; Zhang, S.; Luo, X.; Lee, J.; Li, L.; Sun, X.; Nguyen, L.H.; Chuang, J.-C.; et al. SWI/SNF component ARID1A restrains pancreatic neoplasia formation. Gut 2018, 68, 1259–1270. [Google Scholar] [CrossRef]
- Wang, W.; Friedland, S.C.; Guo, B.; O’Dell, M.R.; Alexander, W.B.; Whitney-Miller, C.L.; Agostini-Vulaj, D.; Huber, A.R.; Myers, J.R.; Ashton, J.; et al. ARID1A, a SWI/SNF subunit, is critical to acinar cell homeostasis and regeneration and is a barrier to transformation and epithelial-mesenchymal transition in the pancreas. Gut 2018, 68, 1245–1258. [Google Scholar] [CrossRef]
- Kuboki, Y.; Fischer, C.G.; Guthrie, V.B.; Huang, W.; Yu, J.; Chianchiano, P.; Hosoda, W.; Zhang, H.; Zheng, L.; Shao, X.; et al. Single-cell sequencing defines genetic heterogeneity in pancreatic cancer precursor lesions. J. Pathol. 2019, 247, 347–356. [Google Scholar] [CrossRef]
- Tan, M.C.B.; Basturk, O.; Brannon, A.R.; Bhanot, U.; Scott, S.N.; Bouvier, N.; LaFemina, J.; Jarnagin, W.R.; Berger, M.F.; Klimstra, D.; et al. GNAS and KRAS mutations define separate progression pathways in intraductal papillary mucinous neoplasm-associated carcinoma. J. Am. Coll. Surg. 2015, 220, 845–854.e1. [Google Scholar] [CrossRef]
- Jackson, E.L.; Willis, N.; Mercer, K.; Bronson, R.T.; Crowley, D.; Montoya, R.; Jacks, T.; Tuveson, D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001, 15, 3243–3248. [Google Scholar] [CrossRef]
- Tanaka, M.; Shibahara, J.; Fukushima, N.; Shinozaki, A.; Umeda, M.; Ishikawa, S.; Kokudo, N.; Fukayama, M. Claudin-18 Is an Early-Stage Marker of Pancreatic Carcinogenesis. J. Histochem. Cytochem. 2011, 59, 942–952. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Y.; Peng, Y.; Zheng, X.; Fan, S.; Yi, Y.; Zeng, P.; Chen, H.; Kang, H.; Zhang, Y.; et al. ΔNp63α down-regulates c-Myc modulator MM1 via E3 ligase HERC3 in the regulation of cell senescence. Cell Death Differ. 2018, 25, 2118–2129. [Google Scholar] [CrossRef] [PubMed]
- Herreros-Villanueva, M.; Zhang, J.-S.; Koenig, A.; Abel, E.V.; Smyrk, T.C.; Bamlet, W.; De Narvajas, A.A.-M.; Gomez, T.S.; Simeone, D.M.; Bujanda, L.; et al. SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogenesis 2013, 2, e61. [Google Scholar] [CrossRef] [PubMed]
- Jeter, C.R.; Liu, B.; Lu, Y.; Chao, H.-P.; Zhang, D.; Liu, X.; Chen, X.; Li, Q.; Rycaj, K.; Calhoun-Davis, T.; et al. NANOG reprograms prostate cancer cells to castration resistance via dynamically repressing and engaging the AR/FOXA1 signaling axis. Cell Discov. 2016, 2, 16041. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Park, J.Y.; Park, K.H.; Chung, H.W.; Bang, S.; Park, S.W.; Song, S.Y. Oct4 and Nanog expression Is associated with early stages of pancreatic carcinogenesis. Pancreas 2010, 39, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun, M.; Mannoor, K.; Cao, J.; Qadri, F.; Song, X. SOX2 in cancer stemness: Tumor malignancy and therapeutic potentials. J. Mol. Cell Boil. 2018, 12, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Park, E.-J.; Lee, H.-S.; Kim, S.J.; Hur, S.-K.; Imbalzano, A.N.; Kwon, J. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting γ-H2AX induction. EMBO J. 2006, 25, 3986–3997. [Google Scholar] [CrossRef]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef]
- Shen, J.; Peng, Y.; Wei, L.; Zhang, W.; Yang, L.; Lan, L.; Kapoor, P.; Ju, Z.; Mo, Q.; Shih, I.-M.; et al. ARID1A deficiency impairs the dna damage checkpoint and sensitizes cells to parp inhibitors. Cancer Discov. 2015, 5, 752–767. [Google Scholar] [CrossRef]
- Kurz, E.U.; Douglas, P.; Lees-Miller, S.P. Doxorubicin activates atm-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J. Biol. Chem. 2004, 279, 53272–53281. [Google Scholar] [CrossRef]
- Huelsenbeck, S.C.; Schorr, A.; Roos, W.P.; Huelsenbeck, J.; Henninger, C.; Kaina, B.; Fritz, G. Rac1 Protein signaling is required for dna damage response stimulated by topoisomerase II Poisons. J. Boil. Chem. 2012, 287, 38590–38599. [Google Scholar] [CrossRef]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2013, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Gibson, S.L.; Narayanan, L.; Hegan, D.C.; Buermeyer, A.B.; Liskay, R.M.; Glazer, P.M. Overexpression of the DNA mismatch repair factor, PMS2, confers hypermutability and DNA damage tolerance. Cancer Lett. 2006, 244, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Nagel, Z.D.; Margulies, C.M.; Chaim, I.A.; McRee, S.K.; Mazzucato, P.; Ahmad, A.; Abo, R.P.; Butty, V.L.; Forget, A.L.; Samson, L.D. Multiplexed DNA repair assays for multiple lesions and multiple doses via transcription inhibition and transcriptional mutagenesis. Proc. Natl. Acad. Sci. USA 2014, 111, E1823–E1832. [Google Scholar] [CrossRef]
- Zhao, D.; Depinho, R.A. Synthetic essentiality: Targeting tumor suppressor deficiencies in cancer. BioEssays 2017, 39, 1700076. [Google Scholar] [CrossRef] [PubMed]
- Pulice, J.L.; Kadoch, C. Composition and function of mammalian SWI/SNF chromatin remodeling complexes in human disease. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 53–60. [Google Scholar] [CrossRef]
- Helming, K.C.; Wang, X.; Wilson, B.G.; Vazquez, F.; Haswell, J.R.; Manchester, H.E.; Kim, Y.; Kryukov, G.V.; Ghandi, M.; Aguirre, A.J.; et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med. 2014, 20, 251–254. [Google Scholar] [CrossRef]
- Huang, M.-J.; Cheng, Y.-C.; Liu, C.-R.; Lin, S.; Liu, H.E. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp. Hematol. 2006, 34, 1480–1489. [Google Scholar] [CrossRef]
- Gupta, S.V.; Albertson, D.J.; Parnell, T.J.; Butterfield, A.; Weston, A.; Pappas, L.M.; Dalley, B.; O’Shea, J.M.; Lowrance, W.T.; Cairns, B.R.; et al. Histone deacetylase inhibition has targeted clinical benefit in ARID1A-mutated advanced urothelial carcinoma. Mol. Cancer Ther. 2018, 18, 185–195. [Google Scholar] [CrossRef]
- Antony-Debré, I.; Paul, A.; Leite, J.; Mitchell, K.; Kim, H.M.; Carvajal, L.A.; Todorova, T.I.; Huang, K.; Kumar, A.; Farahat, A.A.; et al. Pharmacological inhibition of the transcription factor PU.1 in leukemia. J. Clin. Investig. 2017, 127, 4297–4313. [Google Scholar] [CrossRef]
- Sun, X.; Wang, S.C.; Wei, Y.; Luo, X.; Jia, Y.; Li, L.; Gopal, P.; Zhu, M.; Nassour, I.; Chuang, J.-C.; et al. Arid1a has context-dependent oncogenic and tumor suppressor functions in liver cancer. Cancer Cell 2018, 33, 151–152. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, J.; Lin, H.; Jin, R.; Liu, J.; Liu, X.; Meng, N.; Cai, X. The Clinicopathologic significance of baf250a (ARID1A) Expression in hepatocellular carcinoma. Pathol. Oncol. Res. 2015, 22, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Mathur, R.; Alver, B.H.; Roman, A.K.S.; Wilson, B.G.; Wang, X.; Agoston, A.T.; Park, P.J.; Shivdasani, R.A.; Roberts, C.W.M. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet. 2016, 49, 296–302. [Google Scholar] [CrossRef]
- Sen, M.; Wang, X.; Hamdan, F.H.; Rapp, J.; Eggert, J.; Kosinsky, R.L.; Wegwitz, F.; Kutschat, A.P.; Younesi, F.S.; Gaedcke, J.; et al. ARID1A facilitates KRAS signaling-regulated enhancer activity in an AP1-dependent manner in colorectal cancer cells. Clin. Epigenet. 2019, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- De Wilde, R.F.; Edil, B.H.; Hruban, R.H.; Maitra, A. Well-differentiated pancreatic neuroendocrine tumors: From genetics to therapy. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 199–208. [Google Scholar] [CrossRef]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef]
- Ho, J.S.L.; Ma, W.; Mao, D.Y.L.; Benchimol, S. P53-dependent transcriptional repression of c-myc is required for g1 cell cycle arrest. Mol. Cell. Boil. 2005, 25, 7423–7431. [Google Scholar] [CrossRef]
- Cho, H.D.; Lee, J.E.; Jung, H.Y.; Oh, M.-H.; Lee, J.-H.; Jang, S.-H.; Kim, K.-J.; Han, S.W.; Kim, S.Y.; Kim, H.J.; et al. Loss of tumor suppressor arid1a protein expression correlates with poor prognosis in patients with primary breast cancer. J. Breast Cancer 2015, 18, 339–346. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Roeth, A.A.; Zhang, Y.; Yang, A.; Mashadova, O.; Asara, J.M.; Wang, X.; Bronson, R.T.; Lyssiotis, C.A.; Ying, H.; et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun. 2018, 9, 4945. [Google Scholar] [CrossRef]
- Hayes, T.K.; Neel, N.F.; Hu, C.; Gautam, P.; Chenard, M.; Long, B.; Aziz, M.; Kassner, M.; Bryant, K.L.; Pierobon, M.; et al. Long-term erk inhibition in KRAS-mutant pancreatic cancer is associated with myc degradation and senescence-like growth suppression. Cancer Cell 2016, 29, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Carugo, A.; Tepper, J.; Robinson, F.S.; Li, L.; Svelto, M.; Nezi, L.; Corti, D.; Minelli, R.; Pettazzoni, P.; et al. Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature 2017, 542, 362–366. [Google Scholar] [CrossRef]
- Farrell, A.S.; Joly, M.M.; Allen-Petersen, B.L.; Worth, P.J.; Lanciault, C.; Sauer, D.; Link, J.; Pelz, C.; Heiser, L.M.; Morton, J.P.; et al. MYC regulates ductal-neuroendocrine lineage plasticity in pancreatic ductal adenocarcinoma associated with poor outcome and chemoresistance. Nat. Commun. 2017, 8, 1728. [Google Scholar] [CrossRef] [PubMed]
- Ideno, N.; Yamaguchi, H.; Okumura, T.; Huang, J.; Brun, M.J.; Ho, M.L.; Suh, J.; Gupta, S.; Maitra, A.; Ghosh, B. A pipeline for rapidly generating genetically engineered mouse models of pancreatic cancer using in vivo CRISPR-Cas9-mediated somatic recombination. Lab. Investig. 2019, 99, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Hodges, C.; Kirkland, J.G.; Crabtree, G.R. The many roles of BAF (mSWI/SNF) and PBAF complexes in cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026930. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Ui, A.; Kanno, S.-I.; Ogiwara, H.; Nagase, T.; Kohno, T.; Yasui, A. SWI/SNF factors required for cellular resistance to DNA damage include ARID1A and ARID1B and show interdependent protein stability. Cancer Res. 2014, 74, 2465–2475. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Chui, M.H.; Rahmanto, Y.S.; Yu, Z.-C.; Shamanna, R.A.; Bellani, M.A.; Gaillard, S.; Ayhan, A.; Viswanathan, A.; Seidman, M.M.; et al. Loss of ARID1A in tumor cells renders selective vulnerability to combined ionizing radiation and PARP inhibitor therapy. Clin. Cancer Res. 2019, 25, 5584–5594. [Google Scholar] [CrossRef]
- Storz, P. KRas, ROS and the initiation of pancreatic cancer. Small GTPases 2016, 8, 38–42. [Google Scholar] [CrossRef]
- DeNicola, G.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Stein, T.I.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017. [Google Scholar] [CrossRef]
- Scuto, A.; Kirschbaum, M.; Kowolik, C.; Kretzner, L.; Juhasz, A.; Atadja, P.; Pullarkat, V.; Bhatia, R.; Forman, S.; Yen, Y.; et al. The novel histone deacetylase inhibitor, LBH589, induces expression of DNA damage response genes and apoptosis in Ph—Acute lymphoblastic leukemia cells. Blood 2008, 111, 5093–5100. [Google Scholar] [CrossRef] [PubMed]
- Bitler, B.G.; Wu, S.; Park, P.H.; Hai, Y.; Aird, K.M.; Wang, Y.; Zhai, Y.; Kossenkov, A.V.; Vara-Ailor, A.; Iii, F.J.R.; et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nature 2017, 19, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Pramanik, D.; Mukherjee, R.; Campbell, N.; Elumalai, S.; De Wilde, R.F.; Hong, S.-M.; Goggins, M.G.; De Jesus-Acosta, A.; Laheru, D.; et al. Molecular determinants of retinoic acid sensitivity in pancreatic cancer. Clin. Cancer Res. 2011, 18, 280–289. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferri-Borgogno, S.; Barui, S.; McGee, A.M.; Griffiths, T.; Singh, P.K.; Piett, C.G.; Ghosh, B.; Bhattacharyya, S.; Singhi, A.; Pradhan, K.; et al. Paradoxical Role of AT-rich Interactive Domain 1A in Restraining Pancreatic Carcinogenesis. Cancers 2020, 12, 2695. https://doi.org/10.3390/cancers12092695
Ferri-Borgogno S, Barui S, McGee AM, Griffiths T, Singh PK, Piett CG, Ghosh B, Bhattacharyya S, Singhi A, Pradhan K, et al. Paradoxical Role of AT-rich Interactive Domain 1A in Restraining Pancreatic Carcinogenesis. Cancers. 2020; 12(9):2695. https://doi.org/10.3390/cancers12092695
Chicago/Turabian StyleFerri-Borgogno, Sammy, Sugata Barui, Amberly M. McGee, Tamara Griffiths, Pankaj K. Singh, Cortt G. Piett, Bidyut Ghosh, Sanchari Bhattacharyya, Aatur Singhi, Kith Pradhan, and et al. 2020. "Paradoxical Role of AT-rich Interactive Domain 1A in Restraining Pancreatic Carcinogenesis" Cancers 12, no. 9: 2695. https://doi.org/10.3390/cancers12092695
APA StyleFerri-Borgogno, S., Barui, S., McGee, A. M., Griffiths, T., Singh, P. K., Piett, C. G., Ghosh, B., Bhattacharyya, S., Singhi, A., Pradhan, K., Verma, A., Nagel, Z., Maitra, A., & Gupta, S. (2020). Paradoxical Role of AT-rich Interactive Domain 1A in Restraining Pancreatic Carcinogenesis. Cancers, 12(9), 2695. https://doi.org/10.3390/cancers12092695