Organotypic Culture of Acinar Cells for the Study of Pancreatic Cancer Initiation


Simple Summary
Abstract
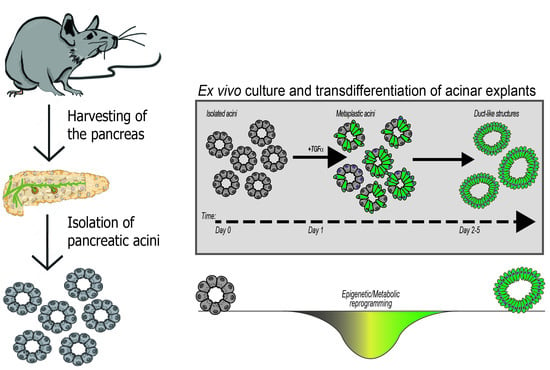
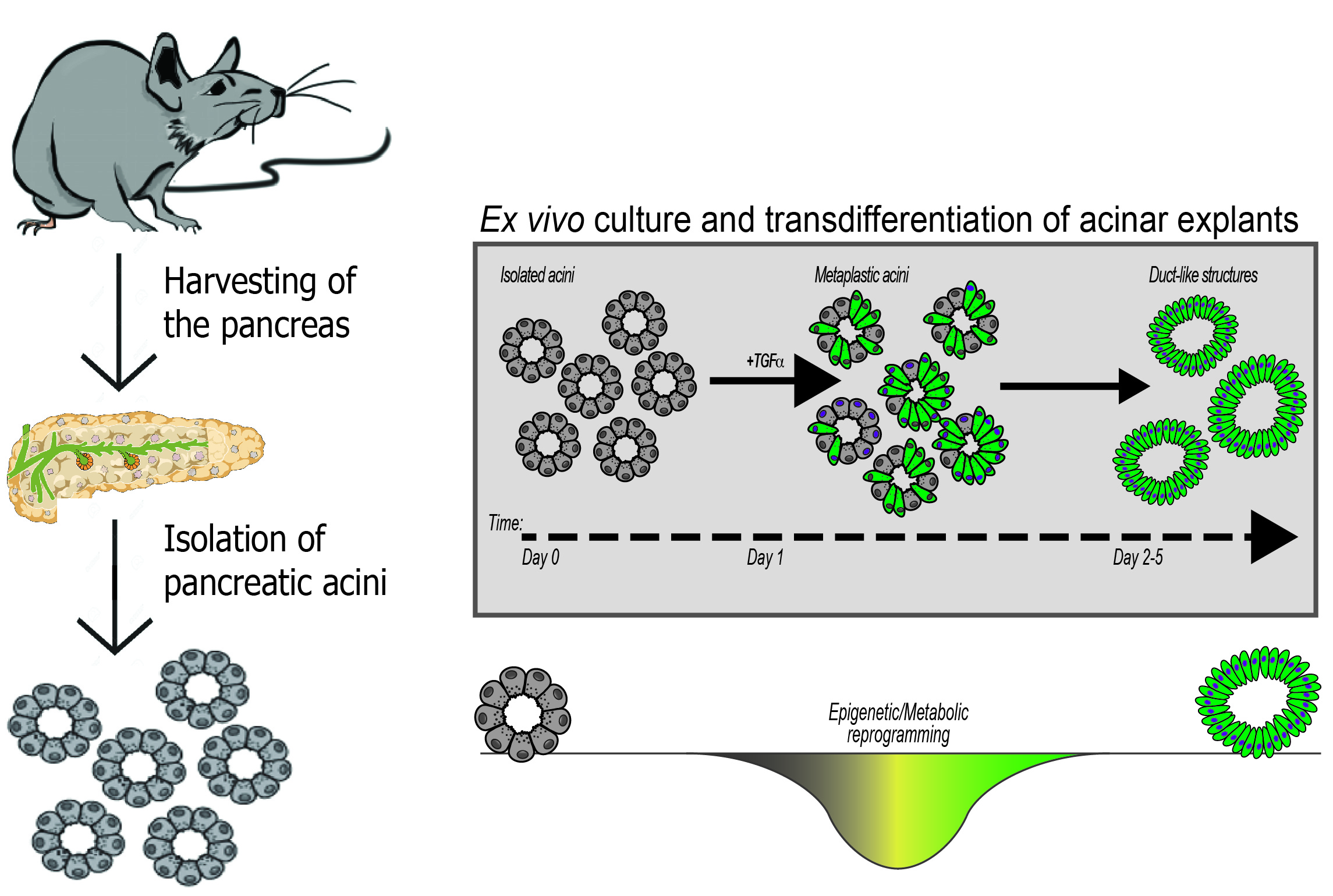{kind=link}
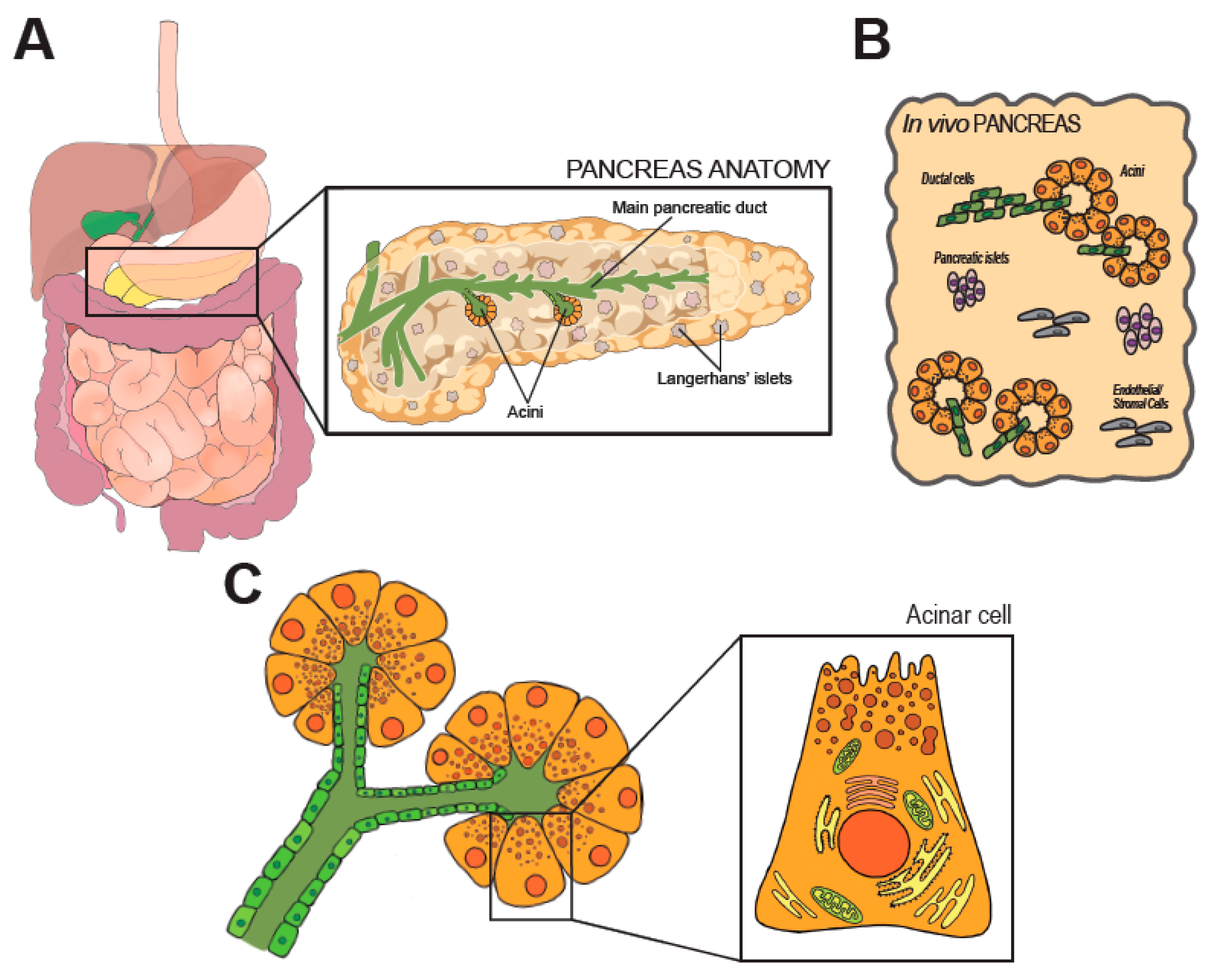{kind=link}
{kind=link}
{kind=link}
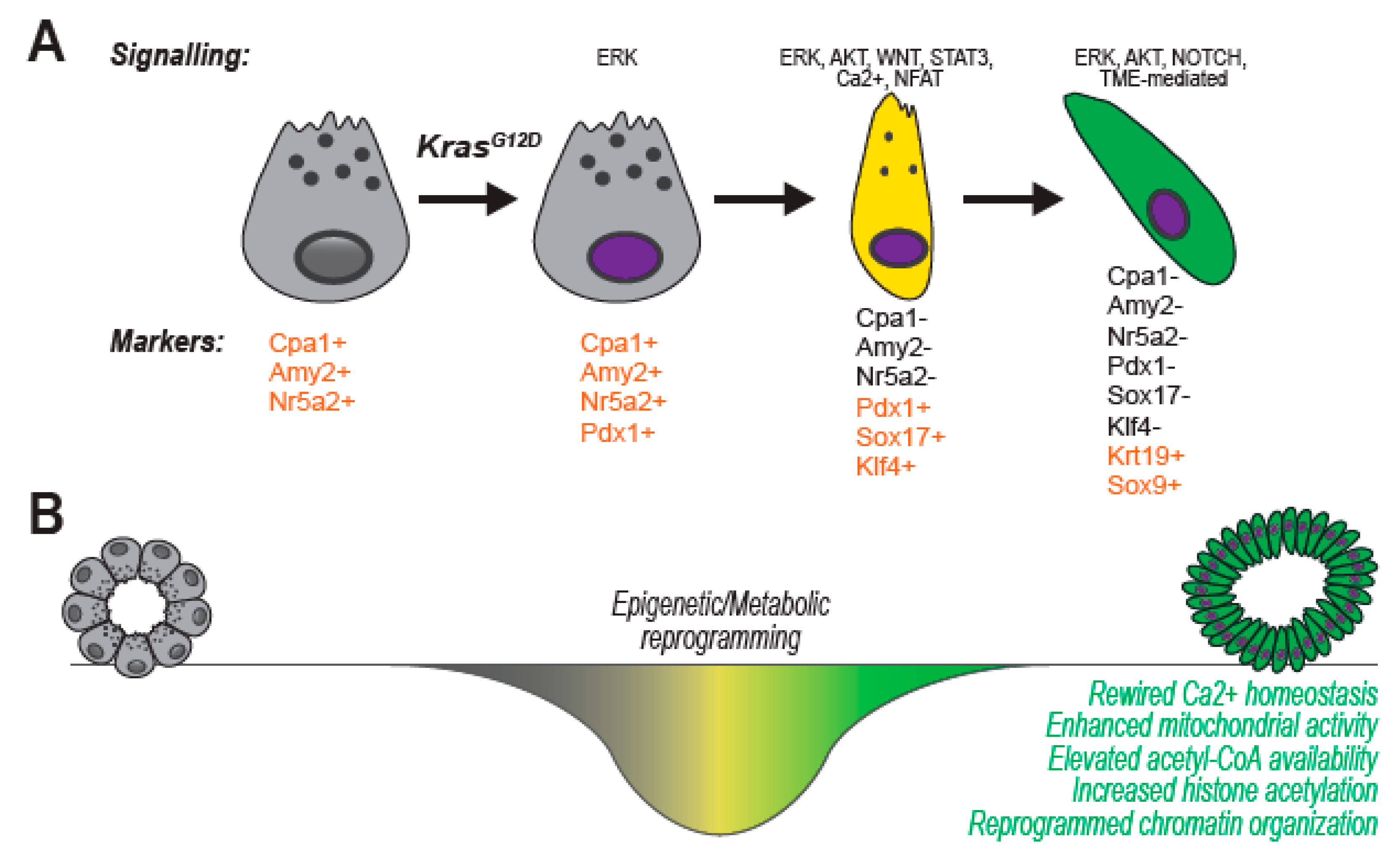{kind=link}
1. Introduction
2. Anatomy and Physiology of the Pancreas
3. Pancreatic Carcinogenesis
4. Organotypic (3D) Acinar Cell Culture
5. 3D Culture of ACs to Study Pancreatic Cancer Initiation
6. Genetic, Epigenetic and Metabolic Alterations Guide Ex-Vivo ADM and Support Tumor Initiation In Vivo
6.1. The Usual Suspects: Tumor-Associated Signaling Pathways Induce ADM in Organotypic Culture
6.2. Master Transcription Factors Shape Cell Identity: Discerning Critical Hubs Using Primary Acinar Cultures
6.3. Epigenetic Regulators Impose Cell Identity and Guide Phenotypic Switch: Epigenetics of Acini-to-Ducts Transition in Primary Cultures
6.4. Rewiring of Cellular Metabolism Supports Ex Vivo ADM through the Production of Anabolic Intermediates
6.5. Non-Cell Autonomous Stimulation of Cultured ACs
6.6. Ex Vivo Modeling of Cell Extrinsic Constrains of Acinar Cell Plasticity
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 13C | Carbon-13 |
| AC | Acinar Cell |
| ADM | Acinar-to-Ductal Metaplasia |
| AMY2 | Amylase-2 |
| ARID1A | AT-Rich Interaction Domain 1A |
| ATAC-seq | Assay for Transposase-Accessible Chromatin-sequencing |
| BCAA | Branched-Chain Amino Acid |
| CCK | Cholecystokinin |
| CCKR | Cholecystokinin Receptor |
| CoA | Coenzyme A |
| CPA1 | Carboxypeptidase-A1 |
| DLCK1 | Doublecortin-like Kinase 1 |
| EGF | Epidermal Growth Factor |
| EGFR | Epidermal Growth Factor Receptor |
| ERK | Extracellular-signal Regulated Kinase |
| FACS | Fluorescence-Activated Cell Sorting |
| GATA6 | GATA-binding factor 6 |
| H2Ak119ub | Histone 2A lysine 119 ubiquitinated |
| H3K27me3 | Histone 3 lysine 27 tri-methylated |
| H3k4me3 | Histone 3 lysine 4 tri-methylated |
| HAT | Histone Acetyl Transferase |
| HSP | Heat-Shock Protein |
| iPSC | inducible Pluripotent Stem Cell |
| KC | Pdx1-Cre; LSL-KRASG12D mice |
| KRAS | Kristen RAt Sarcoma viral oncogene homologue |
| KRT19 | Cytokeratin-19 |
| MAPK | Mitogen-Activated Protein Kinase |
| MEK | MAPK/ERK Kinase |
| Nr5a2 | Nuclear Receptor Subfamily 5 Group A |
| PanIN | Pancreatic Intraepithelial Neoplasia |
| PDA | Pancreatic Ductal Adenocarcinoma |
| PDK1 | 3-Phosphoinositide-dependent kinase 1 |
| PDX1 | Pancreatic-Duodenal Homeobox 1 |
| PI3K | Phosphoinositide 3 Kinase |
| PKB/AKT | Protein Kinase B |
| Ptf1α | Pancreatic Transcription Factor 1a |
| ROS | Reactive oxygen species |
| Rpbjl | Recombination signal binding protein for immunoglobulin kappa J region like |
| sc/snRNA-seq | Single Cell Single Nucleus-RNA sequencing |
| STAT3 | Signal Transducer and activator of Transcription 3 |
| SWI/SNF | SWItch/Sucrose Non-Fermentable |
| TAZ | Transcriptional Co-activator with PDZ-binding Motif |
| TCA | Tricarboxylic Acid |
| TGFα | Transforming Growth Factor α |
| TGFβ | Transforming Growth Factor β |
| Wnt | Wingless-related integration site |
| YAP | Yes-Associated Protein |
References
- Pourshams, A.; Sepanlou, S.G.; Ikuta, K.S.; Bisignano, C.; Safiri, S.; Roshandel, G.; Sharif, M.; Khatibian, M.; Fitzmaurice, C.; Nixon, M.R.; et al. The global, regional, and national burden of pancreatic cancer and its attributable risk factors in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2019, 4, 934–947. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; Koay, E.J.; Chari, S.T.; Maitra, A. Early Detection of Pancreatic Cancer: Opportunities and Challenges. Gastroenterology 2019, 156, 2024–2040. [Google Scholar] [CrossRef]
- Storz, P.; Crawford, H.C. Carcinogenesis of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2020, 158, 2072–2081. [Google Scholar] [CrossRef]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef]
- Maitra, A.; Leach, S.D. Disputed Paternity: The Uncertain Ancestry of Pancreatic Ductal Neoplasia. Cancer Cell 2012, 22, 701–703. [Google Scholar] [CrossRef]
- Cleveland, M.H.; Sawyer, J.M.; Afelik, S.; Jensen, J.; Leach, S.D. Exocrine ontogenies: On the development of pancreatic acinar, ductal and centroacinar cells. Semin. Cell Dev. Biol. 2012, 23, 711–719. [Google Scholar] [CrossRef]
- Murtaugh, L.C.; Keefe, M.D. Regeneration and Repair of the Exocrine Pancreas. Annu. Rev. Physiol. 2015, 77, 229–249. [Google Scholar] [CrossRef]
- Gerasimenko, J.V.; Gerasimenko, O.V.; Petersen, O.H. The role of Ca2+ in the pathophysiology of pancreatitis. J. Physiol. 2014, 592, 269–280. [Google Scholar] [CrossRef]
- Wollny, D.; Zhao, S.; Everlien, I.; Lun, X.; Brunken, J.; Brüne, D.; Ziebell, F.; Tabansky, I.; Weichert, W.; Marciniak-Czochra, A.; et al. Single-Cell Analysis Uncovers Clonal Acinar Cell Heterogeneity in the Adult Pancreas. Dev. Cell 2016, 39, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Tosti, L.; Hang, Y.; Trefzer, T.; Steiger, K.; Ten, F.W.; Lukassen, S.; Ballke, S.; Kuehl, A.A.; Spieckermann, S.; Bottino, R.; et al. Single nucleus RNA sequencing maps acinar cell states in a human pancreas cell atlas. bioRxiv 2019, 733964. [Google Scholar] [CrossRef]
- Jiang, Z.; White, R.A.; Wang, T.C. Adult Pancreatic Acinar Progenitor-like Populations in Regeneration and Cancer. Trends Mol. Med. 2020, 26, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Makohon-Moore, A.P.; Matsukuma, K.; Zhang, M.; Reiter, J.G.; Gerold, J.M.; Jiao, Y.; Sikkema, L.; Attiyeh, M.A.; Yachida, S.; Sandone, C.; et al. Precancerous neoplastic cells can move through the pancreatic ductal system. Nature 2018, 561, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Iacobuzio-Donahue, C.A.; Michael, C.; Baez, P.; Kappagantula, R.; Hooper, J.E.; Hollman, T.J. Cancer biology as revealed by the research autopsy. Nat. Rev. Cancer 2019, 19, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.; Bruns, P.; Behler, N.A.; Chang, L.; Schlitter, A.M.; Cao, J.; Gewies, A.; Ruland, J.; Fritzsche, S.; Valkovskaya, N.; et al. Dynamic landscape of pancreatic carcinogenesis reveals early molecular networks of malignancy. Gut 2018, 67, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Furukawa, T.; Yachida, S.; Nishimura, M.; Seki, A.; Nonaka, K.; Aida, J.; Takubo, K.; Ishiwata, T.; Kimura, W.; et al. The prevalence and clinicopathological characteristics of high-grade pancreatic intraepithelial neoplasia autopsy study evaluating the entire pancreatic parenchyma. Pancreas 2017, 46, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- DeCant, B.T.; Principe, D.R.; Guerra, C.; Pasca di Magliano, M.; Grippo, P.J. Utilizing past and present mouse systems to engineer more relevant pancreatic cancer models. Front. Physiol. 2014, 5, 464. [Google Scholar] [CrossRef]
- Kopp, J.L.; Von Figura, G.; Mayes, E.; Liu, F.; Dubois, C.L.; Iv, J.P.M.; Pan, F.C.; Akiyama, H.; Wright, C.V.E.; Jensen, K.; et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 737–750. [Google Scholar] [CrossRef]
- Guerra, C.; Collado, M.; Navas, C.; Schuhmacher, A.J.; Hernández-Porras, I.; Cañamero, M.; Rodriguez-Justo, M.; Serrano, M.; Barbacid, M. Pancreatitis-Induced Inflammation Contributes to Pancreatic Cancer by Inhibiting Oncogene-Induced Senescence. Cancer Cell 2011, 19, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Storz, P. Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 296–304. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.M.; Brat, D.J.; Wilentz, R.E.; Yeo, C.J.; Cameron, J.L.; Kern, S.E.; Hruban, R.H. Pancreatic intraepithelial neoplasia and infiltrating adenocarcinoma: Analysis of progression and recurrence by DPC4 immunohistochemical labeling. Hum. Pathol. 2001, 32, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Klein, A.P.; Goggins, M.; Maitra, A.; Canto, M.; Ali, S.; Schulick, R.; Palmisano, E.; Hruban, R.H. Increased Prevalence of Precursor Lesions in Familial Pancreatic Cancer Patients. Clin. Cancer Res. 2009, 15, 7737–7743. [Google Scholar] [CrossRef]
- Deramaudt, T.; Rustgi, A.K. Mutant KRAS in the initiation of pancreatic cancer. Biochim. Biophys. Acta Rev. Cancer 2005, 1756, 97–101. [Google Scholar] [CrossRef]
- Di Magliano, M.P.; Logsdon, C.D. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 2013, 144, 1220–1229. [Google Scholar] [CrossRef]
- Shi, C.; Pan, F.C.; Kim, J.N.; Washington, M.K.; Padmanabhan, C.; Meyer, C.T.; Kopp, J.L.; Sander, M.; Gannon, M.; Beauchamp, R.D.; et al. Differential Cell Susceptibilities to KrasG12D in the Setting of Obstructive Chronic Pancreatitis. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 579–594. [Google Scholar] [CrossRef]
- Lee, A.Y.L.; Dubois, C.L.; Sarai, K.; Zarei, S.; Schaeffer, D.F.; Sander, M.; Kopp, J.L. Cell of origin affects tumour development and phenotype in pancreatic ductal adenocarcinoma. Gut 2019, 68, 487–498. [Google Scholar] [CrossRef]
- Biffi, G. Tracing the Origin of Fibroblasts in Pancreatic Cancer. Cell. Mol. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Helms, E.; Onate, M.K.; Sherman, M.H. Fibroblast Heterogeneity in the Pancreatic Tumor Microenvironment. Cancer Discov. 2020, 10, 648–656. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Gore, J.; Korc, M. Pancreatic Cancer Stroma: Friend or Foe? Cancer Cell 2014, 25, 711–712. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.E.; Adoumie, M.; Kim, E.C.; Zhang, Y.; Scales, M.K.; El-Tawil, Y.S.; Shaikh, A.Z.; Wen, H.-J.; Bednar, F.; Allen, B.L.; et al. Differential Contribution of Pancreatic Fibroblast Subsets to the Pancreatic Cancer Stroma. Cell. Mol. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sphyris, N.; Logsdon, C.D.; Harrison, D.J. Improved retention of zymogen granules in cultured murine pancreatic acinar cells and induction of acinar-ductal transdifferentiation in vitro. Pancreas 2005, 30, 148–157. [Google Scholar] [CrossRef]
- Amsterdam, A.; Jamieson, J.D. Structural and functional characterization of isolated pancreatic exocrine cells. Proc. Natl. Acad. Sci. USA 1972, 69, 3028–3032. [Google Scholar] [CrossRef]
- Rooman, I.; Heremans, Y.; Heimberg, H.; Bouwens, L. Modulation of rat pancreatic acinoductal transdifferentiation and expression of PDX-1 in vitro. Diabetologia 2000, 43, 907–914. [Google Scholar] [CrossRef]
- Means, A.L.; Meszoely, I.M.; Suzuki, K.; Miyamoto, Y.; Rustgi, A.K.; Coffey, R.J.; Wright, C.V.E.; Stoffers, D.A.; Leach, S.D. Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development 2005, 132, 3767–3776. [Google Scholar] [CrossRef]
- Gout, J.; Pommier, R.M.; Vincent, D.F.; Kaniewski, B.; Martel, S.; Valcourt, U.; Bartholin, L. Isolation and culture of mouse primary pancreatic acinar cells. J. Vis. Exp. 2013, 1–8. [Google Scholar] [CrossRef]
- Fleming Martinez, A.K.; Storz, P. Mimicking and Manipulating Pancreatic Acinar-to-Ductal Metaplasia in 3-dimensional Cell Culture. J. Vis. Exp. 2019. [Google Scholar] [CrossRef]
- Pinho, A.V.; Rooman, I.; Reichert, M.; De Medts, N.; Bouwens, L.; Rustgi, A.K.; Real, F.X. Adult pancreatic acinar cells dedifferentiate to an embryonic progenitor phenotype with concomitant activation of a senescence programme that is present in chronic pancreatitis. Gut 2011, 60, 958–966. [Google Scholar] [CrossRef]
- Houbracken, I.; de Waele, E.; Lardon, J.; Ling, Z.; Heimberg, H.; Rooman, I.; Bouwens, L. Lineage tracing evidence for transdifferentiation of acinar to duct cells and plasticity of human pancreas. Gastroenterology 2011, 141, 731–741.e4. [Google Scholar] [CrossRef] [PubMed]
- Bläuer, M.; Nordback, I.; Sand, J.; Laukkarinen, J. A novel explant outgrowth culture model for mouse pancreatic acinar cells with long-term maintenance of secretory phenotype. Eur. J. Cell Biol. 2011, 90, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Livshits, G.; Alonso-Curbelo, D.; Morris IV, J.P.; Koche, R.; Saborowski, M.; Wilkinson, J.E.; Lowe, S.W. Arid1a restrains Kras-dependent changes in acinar cell identity. Elife 2018, 7, e35216. [Google Scholar] [CrossRef] [PubMed]
- Westphalen, C.B.; Olive, K.P. Genetically engineered mouse models of pancreatic cancer. Cancer J. 2012, 18, 502–510. [Google Scholar] [CrossRef]
- Brandt, R.; Sell, T.; Lüthen, M.; Uhlitz, F.; Klinger, B.; Riemer, P.; Giesecke-Thiel, C.; Schulze, S.; El-Shimy, I.A.; Kunkel, D.; et al. Cell type-dependent differential activation of ERK by oncogenic KRAS in colon cancer and intestinal epithelium. Nat. Commun. 2019, 10, 2919. [Google Scholar] [CrossRef]
- Poulin, E.J.; Bera, A.K.; Lu, J.; Lin, Y.-J.; Strasser, S.D.; Paulo, J.A.; Huang, T.Q.; Morales, C.; Yan, W.; Cook, J.; et al. Tissue-Specific Oncogenic Activity of KRASA146T. Cancer Discov. 2019. [Google Scholar] [CrossRef]
- Mayers, J.R.; Torrence, M.E.; Danai, L.V.; Papagiannakopoulos, T.; Davidson, S.M.; Bauer, M.R.; Lau, A.N.; Ji, B.W.; Dixit, P.D.; Hosios, A.M.; et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 2016, 353, 1161–1165. [Google Scholar] [CrossRef]
- Shi, G.; DiRenzo, D.; Qu, C.; Barney, D.; Miley, D.; Konieczny, S.F. Maintenance of acinar cell organization is critical to preventing Kras-induced acinar-ductal metaplasia. Oncogene 2013, 32, 1950–1958. [Google Scholar] [CrossRef]
- Martinelli, P.; Madriles, F.; Cañamero, M.; Pau, E.C.S.; del Pozo, N.; Guerra, C.; Real, F.X. The acinar regulator Gata6 suppresses KrasG12V-driven pancreatic tumorigenesis in mice. Gut 2016, 65, 476–486. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Döppler, H.; DelGiorno, K.E.; Zhang, L.; Leitges, M.; Crawford, H.C.; Murphy, M.P.; Storz, P. Mutant KRas-Induced Mitochondrial Oxidative Stress in Acinar Cells Upregulates EGFR Signaling to Drive Formation of Pancreatic Precancerous Lesions. Cell Rep. 2016, 14, 2325–2336. [Google Scholar] [CrossRef]
- Chiou, S.-H.; Winters, I.P.; Wang, J.; Naranjo, S.; Dudgeon, C.; Tamburini, F.B.; Brady, J.J.; Yang, D.; Grüner, B.M.; Chuang, C.-H.; et al. Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Genes Dev. 2015, 29, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, V.; Ayuso, E.; Mallol, C.; Agudo, J.; Casellas, A.; Obach, M.; Muñoz, S.; Salavert, A.; Bosch, F. In vivo genetic engineering of murine pancreatic beta cells mediated by single-stranded adeno-associated viral vectors of serotypes 6, 8 and 9. Diabetologia 2011, 54, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Houbracken, I.; Baeyens, L.; Ravassard, P.; Heimberg, H.; Bouwens, L. Gene delivery to pancreatic exocrine cells in vivo and in vitro. BMC Biotechnol. 2012, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Perera, R.M.; Bardeesy, N. Ready, Set, Go: The EGF Receptor at the Pancreatic Cancer Starting Line. Cancer Cell 2012, 22, 281–282. [Google Scholar] [CrossRef]
- Garcia-Carracedo, D.; Yu, C.-C.; Akhavan, N.; Fine, S.A.; Schönleben, F.; Maehara, N.; Karg, D.C.; Xie, C.; Qiu, W.; Fine, R.L.; et al. Smad4 Loss Synergizes with TGFα Overexpression in Promoting Pancreatic Metaplasia, PanIN Development, and Fibrosis. PLoS ONE 2015, 10, e0120851. [Google Scholar] [CrossRef]
- Means, A.L.; Ray, K.C.; Singh, A.B.; Washington, M.K.; Whitehead, R.H.; Harris, R.C., Jr.; Wright, C.V.E.; Coffey, R.J., Jr.; Leach, S.D. Overexpression of heparin-binding EGF-like growth factor in mouse pancreas results in fibrosis and epithelial metaplasia. Gastroenterology 2003, 124, 1020–1036. [Google Scholar] [CrossRef]
- Song, S.Y.; Gannon, M.; Washington, M.K.; Scoggins, C.R.; Meszoely, I.M.; Goldenring, J.R.; Marino, C.R.; Sandgren, E.P.; Coffey, R.J., Jr.; Wright, C.V.; et al. Expansion of Pdx1-expressing pancreatic epithelium and islet neogenesis in transgenic mice overexpressing transforming growth factor α. Gastroenterology 1999, 117, 1416–1426. [Google Scholar] [CrossRef]
- Ardito, C.M.; Grüner, B.M.; Takeuchi, K.K.; Lubeseder-Martellato, C.; Teichmann, N.; Mazur, P.K.; DelGiorno, K.E.; Carpenter, E.S.; Halbrook, C.J.; Hall, J.C.; et al. EGF Receptor Is Required for KRAS-Induced Pancreatic Tumorigenesis. Cancer Cell 2012, 22, 304–317. [Google Scholar] [CrossRef]
- Navas, C.; Hernández-Porras, I.; Schuhmacher, A.J.; Sibilia, M.; Guerra, C.; Barbacid, M. EGF Receptor Signaling Is Essential for K-Ras Oncogene-Driven Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 22, 318–330. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Wen, H.J.; Ruggeri, J.M.; Takeuchi, K.K.; Zhang, Y.; Pasca di Magliano, M.; Crawford, H.C. Mitogen-activated Protein Kinase Kinase Activity Maintains Acinar-to-Ductal Metaplasia and Is Required for Organ Regeneration in Pancreatitis. Cmgh 2017, 3, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Akanuma, N.; Liu, C.; Naji, A.; Halff, G.A.; Washburn, W.K.; Sun, L.; Wang, P. TGF-β1 promotes acinar to ductal metaplasia of human pancreatic acinar cells. Sci. Rep. 2016, 6, 30904. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Carrer, A.; Trefely, S.; Zhao, S.; Campbell, S.; Norgard, R.J.; Schultz, K.C.; Sidoli, S.; Parris, J.L.D.; Affronti, H.C.; Sivanand, S.; et al. Acetyl-CoA metabolism supports multi-step pancreatic tumorigenesis. Cancer Discov. 2019, 9, 416–435. [Google Scholar] [CrossRef]
- Eser, S.; Reiff, N.; Messer, M.; Seidler, B.; Gottschalk, K.; Dobler, M.; Hieber, M.; Arbeiter, A.; Klein, S.; Kong, B.; et al. Selective requirement of PI3K/PDK1 signaling for kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell 2013, 23, 406–420. [Google Scholar] [CrossRef]
- Baer, R.; Cintas, C.; Dufresne, M.; Cassant-Sourdy, S.; Schönhuber, N.; Planque, L.; Lulka, H.; Couderc, B.; Bousquet, C.; Garmy-Susini, B.; et al. Pancreatic cell plasticity and cancer initiation induced by oncogenic Kras is completely dependent on wild-type PI 3-kinase p110α. Genes Dev. 2014, 28, 2621–2635. [Google Scholar] [CrossRef]
- Shen, J.; Ha, D.P.; Zhu, G.; Rangel, D.F.; Kobielak, A.; Gill, P.S.; Groshen, S.; Dubeau, L.; Lee, A.S. GRP78 haploinsufficiency suppresses acinar-to-ductal metaplasia, signaling, and mutant Kras-driven pancreatic tumorigenesis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, E4020–E4029. [Google Scholar] [CrossRef]
- Lubeseder-Martellato, C.; Alexandrow, K.; Hidalgo-Sastre, A.; Heid, I.; Boos, S.L.; Briel, T.; Schmid, R.M.; Siveke, J.T. Oncogenic KRas-induced Increase in Fluid-phase Endocytosis is Dependent on N-WASP and is Required for the Formation of Pancreatic Preneoplastic Lesions. EBioMedicine 2017, 15, 90–99. [Google Scholar] [CrossRef]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165, 910–920. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef]
- Fukuda, A.; Wang, S.C.; Morris, J.P.; Folias, A.E.; Liou, A.; Kim, G.E.; Akira, S.; Boucher, K.M.; Firpo, M.A.; Mulvihill, S.J.; et al. Stat3 and MMP7 Contribute to Pancreatic Ductal Adenocarcinoma Initiation and Progression. Cancer Cell 2011, 19, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.-M.; Singh, G.; Koenig, A.; Liou, G.-Y.; Storz, P.; Zhang, J.-S.; Regul, L.; Nagarajan, S.; Kühnemuth, B.; Johnsen, S.A.; et al. NFATc1 Links EGFR Signaling to Induction of Sox9 Transcription and Acinar–Ductal Transdifferentiation in the Pancreas. Gastroenterology 2015, 148, 1024–1034.e9. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Döppler, H.; Necela, B.; Edenfield, B.; Zhang, L.; Dawson, D.W.; Storz, P. Mutant KRAS—Induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. 2015, 5, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Sharon, N.; Vanderhooft, J.; Straubhaar, J.; Mueller, J.; Chawla, R.; Zhou, Q.; Engquist, E.N.; Trapnell, C.; Gifford, D.K.; Melton, D.A. Wnt Signaling Separates the Progenitor and Endocrine Compartments during Pancreas Development. Cell Rep. 2019, 27, 2281–2291.e5. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.P.; Wang, S.C.; Hebrok, M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat. Rev. Cancer 2010, 10, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Morris IV, J.P.; Yan, W.; Schofield, H.K.; Gurney, A.; Simeone, D.M.; Millar, S.E.; Hoey, T.; Hebrok, M.; Di Magliano, M.P. Canonical Wnt signaling is required for pancreatic carcinogenesis. Cancer Res. 2013, 73, 4909–4922. [Google Scholar] [CrossRef]
- Di Magliano, M.P.; Sekine, S.; Ermilov, A.; Ferris, J.; Dlugosz, A.A.; Hebrok, M. Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev. 2006, 20, 3161–3173. [Google Scholar] [CrossRef]
- Stanger, B.Z.; Hebrok, M. Control of cell identity in pancreas development and regeneration. Gastroenterology 2013, 144, 1170–1179. [Google Scholar] [CrossRef]
- Reichert, M.; Rustgi, A.K. Science in medicine Pancreatic ductal cells in development, regeneration, and neoplasia. J. Clin. Investig. 2011, 121, 4572–4578. [Google Scholar] [CrossRef]
- Sarkar, A.; Hochedlinger, K. The Sox family of transcription factors: Versatile regulators of stem and progenitor cell fate. Cell Stem Cell 2013, 12, 15–30. [Google Scholar] [CrossRef]
- Delgiorno, K.E.; Hall, J.C.; Takeuchi, K.K.; Pan, F.C.; Halbrook, C.J.; Washington, M.K.; Olive, K.P.; Spence, J.R.; Sipos, B.; Wright, C.V.E.; et al. Identification and manipulation of biliary metaplasia in pancreatic tumors. Gastroenterology 2014, 146, 233–244.e5. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.M.; Alsina, J.; Rasheed, Z.A.; McAllister, F.M.; Fu, Y.Y.; Plentz, R.; Zhang, H.; Pasricha, P.J.; Bardeesy, N.; Matsui, W.; et al. DCLK1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology 2014, 146, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.; Takeuchi, K.K.; Ruggeri, J.M.; Bailey, P.; Chang, D.; Li, J.; Leonhardt, L.; Puri, S.; Hoffman, M.T.; Gao, S.; et al. PDX1 dynamically regulates pancreatic ductal adenocarcinoma initiation and maintenance. Genes Dev. 2016, 30, 2669–2683. [Google Scholar] [CrossRef] [PubMed]
- Miyatsuka, T.; Kaneto, H.; Shiraiwa, T.; Matsuoka, T.; Yamamoto, K.; Kato, K.; Nakamura, Y.; Akira, S.; Takeda, K.; Kajimoto, Y.; et al. Persistent expression of PDX-1 in the pancreas causes acinar-to-ductal metaplasia through Stat3 activation. Genes Dev. 2006, 20, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Von Figura, G.; Morris IV, J.P.; Wright, C.V.E.; Hebrok, M. Nr5a2 maintains acinar cell differentiation and constrains oncogenic Kras-mediated pancreatic neoplastic initiation. Gut 2014, 63, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Cobo, I.; Martinelli, P.; Flández, M.; Bakiri, L.; Zhang, M.; Carrillo-De-Santa-Pau, E.; Jia, J.; Lobo, V.J.S.A.; Megías, D.; Felipe, I.; et al. Transcriptional regulation by NR5A2 links differentiation and inflammation in the pancreas. Nature 2018, 554, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Hoang, C.Q.; Hale, M.A.; Azevedo-Pouly, A.C.; Elsässer, H.P.; Deering, T.G.; Willet, S.G.; Pan, F.C.; Magnuson, M.A.; Wright, C.V.E.; Swift, G.H.; et al. Transcriptional Maintenance of Pancreatic Acinar Identity, Differentiation, and Homeostasis by PTF1A. Mol. Cell. Biol. 2016, 36, 3033–3047. [Google Scholar] [CrossRef]
- Barrero, M.J.; Boué, S.; Izpisúa Belmonte, J.C. Epigenetic Mechanisms that Regulate Cell Identity. Cell Stem Cell 2010, 7, 565–570. [Google Scholar] [CrossRef]
- Wei, D.; Wang, L.; Yan, Y.; Jia, Z.; Gagea, M.; Li, Z.; Zuo, X.; Kong, X.; Huang, S.; Xie, K. KLF4 Is Essential for Induction of Cellular Identity Change and Acinar-to-Ductal Reprogramming during Early Pancreatic Carcinogenesis. Cancer Cell 2016, 29, 324–338. [Google Scholar] [CrossRef]
- Suvà, M.L. Epigenetic Reprogramming in Cancer Mario L. Suvà. Nat. Med. 2013, 1567, 179–192. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Maitra, A.; Ghosh, B.; Zechner, U.; Argani, P.; Iacobuzio-Donahue, C.A.; Sriuranpong, V.; Iso, T.; Meszoely, I.M.; Wolfe, M.S.; et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 2003, 3, 565–576. [Google Scholar] [CrossRef]
- Benitz, S.; Regel, I.; Reinhard, T.; Popp, A.; Schäffer, I.; Raulefs, S.; Kong, B.; Esposito, I.; Michalski, C.W.; Kleeff, J. Polycomb repressor complex 1 promotes gene silencing through H2AK119 mono-ubiquitination in acinar-to-ductal metaplasia and pancreatic cancer cells. Oncotarget 2016, 7, 11424–11433. [Google Scholar] [CrossRef] [PubMed]
- Benitz, S.; Straub, T.; Mahajan, U.M.; Mutter, J.; Czemmel, S.; Unruh, T.; Wingerath, B.; Deubler, S.; Fahr, L.; Cheng, T.; et al. Ring1b-dependent epigenetic remodelling is an essential prerequisite for pancreatic carcinogenesis. Gut 2019, 68, 2007–2018. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Iacobuzio-Donahue, C.A. Evolution and dynamics of pancreatic cancer progression. Oncogene 2013, 32, 5253–5260. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Chiba, T.; Seno, H.; Tsuda, M.; Fukuda, A.; Roy, N.; Hiramatsu, Y.; Leonhardt, L.; Kakiuchi, N.; Hoyer, K.; et al. The BRG1/SOX9 axis is critical for acinar cell–derived pancreatic tumorigenesis Graphical abstract Find the latest version: The BRG1/SOX9 axis is critical for acinar cell—Derived pancreatic tumorigenesis. J. Clin. Investig. 2018, 128, 3475–3489. [Google Scholar] [CrossRef]
- Carrer, A.; Wellen, K.E. Metabolism and epigenetics: A link cancer cells exploit. Curr. Opin. Biotechnol. 2015, 34. [Google Scholar] [CrossRef]
- Shapira, S.N.; Christofk, H.R. Metabolic Regulation of Tissue Stem Cells. Trends Cell Biol. 2020, 30, 566–576. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Lyssiotis, C.A. Review Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef]
- Sidoli, S.; Trefely, S.; Garcia, B.A.; Carrer, A. Integrated Analysis of Acetyl-CoA and Histone Modification via Mass Spectrometry to Investigate Metabolically Driven Acetylation. In Cancer Metabolism: Methods and Protocols; Haznadar, M., Ed.; Springer: New York, NY, USA, 2019; pp. 125–147. ISBN 978-1-4939-9027-6. [Google Scholar]
- Campit, S.E.; Meliki, A.; Youngson, N.A.; Chandrasekaran, S. Nutrient Sensing by Histone Marks: Reading the Metabolic Histone Code Using Tracing, Omics, and Modeling. BioEssays 2020, 42, 2000083. [Google Scholar] [CrossRef]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef]
- Mohammed, A.; Qian, L.; Janakiram, N.B.; Lightfoot, S.; Steele, V.E.; Rao, C.V. Atorvastatin delays progression of pancreatic lesions to carcinoma by regulating PI3/AKT signaling in p48 Cre/+ LSL-Kras G12D/+ mice. Int. J. Cancer 2012, 131, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Neinast, M.D.; Jang, C.; Hui, S.; Murashige, D.S.; Chu, Q.; Morscher, R.J.; Li, X.; Zhan, L.; White, E.; Anthony, T.G.; et al. Quantitative Analysis of the Whole-Body Metabolic Fate of Branched-Chain Amino Acids. Cell Metab. 2019, 29, 417–429.e4. [Google Scholar] [CrossRef] [PubMed]
- Li, J.T.; Yin, M.; Wang, D.; Wang, J.; Lei, M.Z.; Zhang, Y.; Liu, Y.; Zhang, L.; Zou, S.W.; Hu, L.P.; et al. BCAT2-mediated BCAA catabolism is critical for development of pancreatic ductal adenocarcinoma. Nat. Cell Biol. 2020, 22, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Mitochondria: Back to the future. Nat. Rev. Mol. Cell Biol. 2017, 19, 76. [Google Scholar] [CrossRef]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef]
- Serasinghe, M.N.; Wieder, S.Y.; Renault, T.T.; Elkholi, R.; Asciolla, J.J.; Yao, J.L.; Jabado, O.; Hoehn, K.; Kageyama, Y.; Sesaki, H.; et al. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol. Cell 2015, 57, 521–536. [Google Scholar] [CrossRef]
- Yu, M.; Huang, Y.; Deorukhkar, A.; Fujimoto, T.N.; Govindaraju, S.; Molkentine, J.M.; Lin, D.; Kang, Y.; Koay, E.J.; Fleming, J.B.; et al. Mitochondrial Fusion Suppresses Pancreatic Cancer Growth via Reduced Oxidative Metabolism. Available online: https://www.biorxiv.org/content/10.1101/279745v1 (accessed on 11 March 2018).
- Gukovskaya, A.S.; Gukovsky, I.; Jung, Y.; Mouria, M.; Pandol, S.J. Cholecystokinin Induces Caspase Activation and Mitochondrial Dysfunction in Pancreatic Acinar Cells: ROLES IN CELL INJURY PROCESSES OF PANCREATITIS. J. Biol. Chem. 2002, 277, 22595–22604. [Google Scholar] [CrossRef]
- Nagdas, S.; Kashatus, J.A.; Nascimento, A.; Hussain, S.S.; Trainor, R.E.; Pollock, S.R.; Adair, S.J.; Michaels, A.D.; Sesaki, H.; Stelow, E.B.; et al. Drp1 Promotes KRas-Driven Metabolic Changes to Drive Pancreatic Tumor Growth. Cell Rep. 2019, 28, 1845–1859.e5. [Google Scholar] [CrossRef]
- Humpton, T.J.; Alagesan, B.; Denicola, G.M.; Lu, D.; Yordanov, G.N.; Leonhardt, C.S.; Yao, M.A.; Alagesan, P.; Zaatari, M.N.; Park, Y.; et al. Oncogenic KRAS induces NIX-mediated mitophagy to promote pancreatic cancer. Cancer Discov. 2019, 9, 1268–1287. [Google Scholar] [CrossRef]
- Liou, G.Y.; Döppler, H.; Necela, B.; Krishna, M.; Crawford, H.C.; Raimondo, M.; Storz, P. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-??B and MMPs. J. Cell Biol. 2013, 202, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Pitarresi, J.R.; Cuitiño, M.C.; Kladney, R.D.; Woelke, S.A.; Sizemore, G.M.; Nayak, S.G.; Egriboz, O.; Schweickert, P.G.; Yu, L.; et al. Genetic ablation of smoothened in pancreatic fibroblasts increases acinar-ductal metaplasia. Genes Dev. 2016, 30, 1943–1955. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Fujimura, A.; Di Biagio, D.; Frasson, C.; Bresolin, S.; Soligo, S.; Basso, G.; Bicciato, S.; Rosato, A.; et al. Induction of Expandable Tissue-Specific Stem/Progenitor Cells through Transient Expression of YAP/TAZ. Cell Stem Cell 2016, 19, 725–737. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Vicente, R.; Pavón, D.M.; Martín-Padura, I.; Català-Montoro, M.; Díez-Sánchez, A.; Quílez-Álvarez, A.; López, J.A.; Sánchez-Álvarez, M.; Vázquez, J.; Strippoli, R.; et al. Caveolin-1 Modulates Mechanotransduction Responses to Substrate Stiffness through Actin-Dependent Control of YAP. Cell Rep. 2018, 25, 1622–1635.e6. [Google Scholar] [CrossRef] [PubMed]
- Gruber, R.; Panayiotou, R.; Nye, E.; Spencer-Dene, B.; Stamp, G.; Behrens, A. YAP1 and TAZ Control Pancreatic Cancer Initiation in Mice by Direct Up-regulation of JAK–STAT3 Signaling. Gastroenterology 2016, 151, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Labarge, M.A. Context, tissue plasticity, and cancer: Are tumor stem cells also regulated by the microenvironment? Cancer Cell 2005, 7, 17–23. [Google Scholar] [CrossRef]
- Giri, B.; Sethi, V.; Modi, S.; Garg, B.; Banerjee, S.; Saluja, A.; Dudeja, V. Heat shock protein 70 in pancreatic diseases: Friend or foe. J. Surg. Oncol. 2017, 116, 114–122. [Google Scholar] [CrossRef]
- Wagner, A.C.; Weber, H.; Jonas, L.; Nizze, H.; Strowski, M.; Fiedler, F.; Printz, H.; Steffen, H.; Goke, B. Hyperthermia induces heat shock protein expression and protection against cerulein-induced pancreatitis in rats. Gastroenterology 1996, 111, 1333–1342. [Google Scholar] [CrossRef]
- Bhagat, L.; Singh, V.P.; Dawra, R.K.; Saluja, A.K. Sodium arsenite induces heat shock protein 70 expression and protects against secretagogue-induced trypsinogen and NF-κB activation. J. Cell. Physiol. 2008, 215, 37–46. [Google Scholar] [CrossRef]
- Renz, B.W.; Tanaka, T.; Sunagawa, M.; Takahashi, R.; Jiang, Z.; Macchini, M.; Dantes, Z.; Valenti, G.; White, R.A.; Middelhoff, M.A.; et al. Cholinergic Signaling via Muscarinic Receptors Directly and Indirectly Suppresses Pancreatic Tumorigenesis and Cancer Stemness. Cancer Discov. 2018, 8, 1458–1473. [Google Scholar] [CrossRef] [PubMed]
- Westphalen, C.B.; Takemoto, Y.; Tanaka, T.; Macchini, M.; Jiang, Z.; Renz, B.W.; Chen, X.; Ormanns, S.; Nagar, K.; Tailor, Y.; et al. Dclk1 Defines Quiescent Pancreatic Progenitors that Promote Injury-Induced Regeneration and Tumorigenesis. Cell Stem Cell 2016, 18, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Maddipati, R.; Stanger, B.Z. Pancreatic Cancer Metastases Harbor Evidence of Polyclonality. Cancer Discov. 2015, 5, 1086–1097. [Google Scholar] [CrossRef]
- Lugea, A.; Waldron, R.T.; Mareninova, O.A.; Shalbueva, N.; Deng, N.; Su, H.Y.; Thomas, D.D.; Jones, E.K.; Messenger, S.W.; Yang, J.; et al. Human Pancreatic Acinar Cells: Proteomic Characterization, Physiologic Responses, and Organellar Disorders in ex Vivo Pancreatitis. Am. J. Pathol. 2017, 187, 2726–2743. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paoli, C.; Carrer, A. Organotypic Culture of Acinar Cells for the Study of Pancreatic Cancer Initiation. Cancers 2020, 12, 2606. https://doi.org/10.3390/cancers12092606
Paoli C, Carrer A. Organotypic Culture of Acinar Cells for the Study of Pancreatic Cancer Initiation. Cancers. 2020; 12(9):2606. https://doi.org/10.3390/cancers12092606
Chicago/Turabian StylePaoli, Carlotta, and Alessandro Carrer. 2020. "Organotypic Culture of Acinar Cells for the Study of Pancreatic Cancer Initiation" Cancers 12, no. 9: 2606. https://doi.org/10.3390/cancers12092606
APA StylePaoli, C., & Carrer, A. (2020). Organotypic Culture of Acinar Cells for the Study of Pancreatic Cancer Initiation. Cancers, 12(9), 2606. https://doi.org/10.3390/cancers12092606