Practical Review on Preclinical Human 3D Glioblastoma Models: Advances and Challenges for Clinical Translation


Abstract
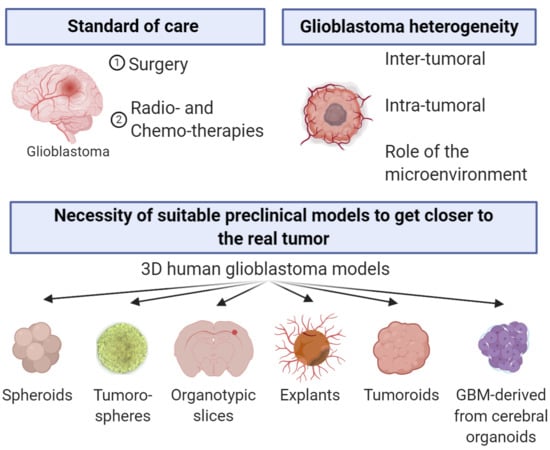
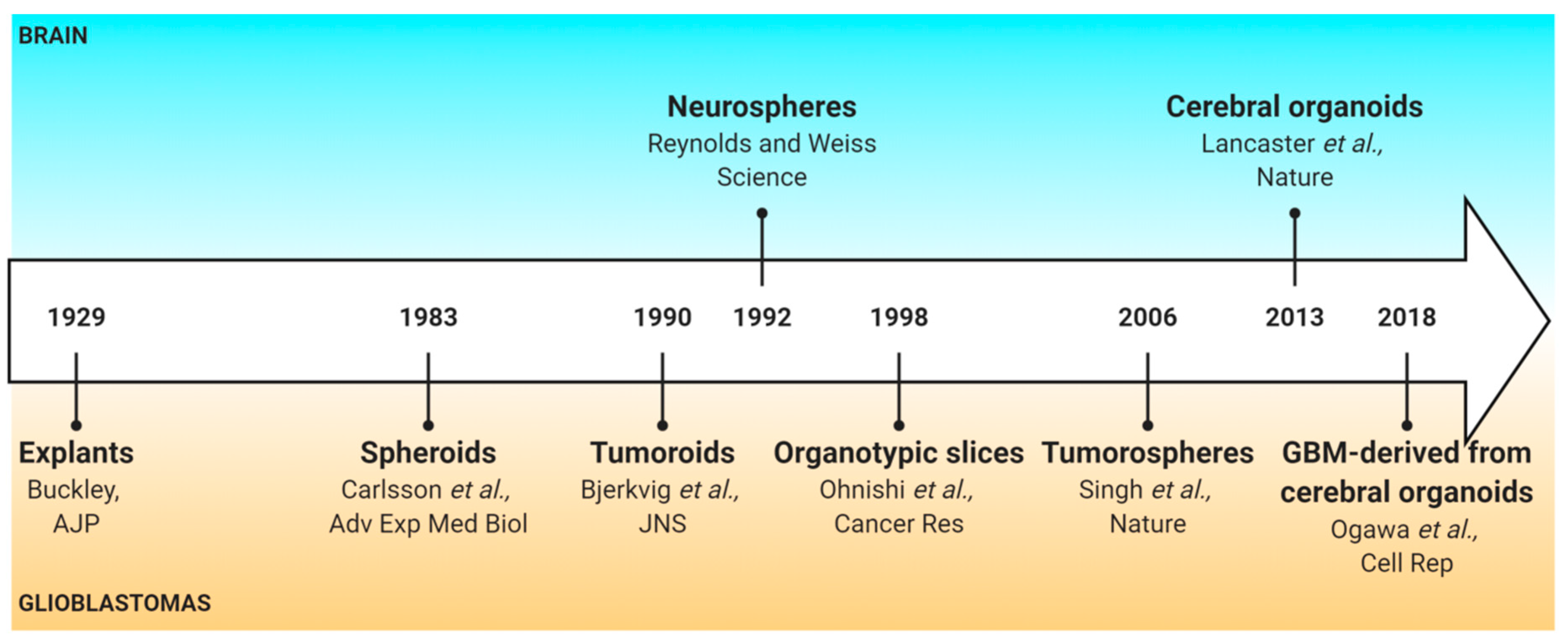
1. Introduction
2. Sphere-Based Models
2.1. Spheroids
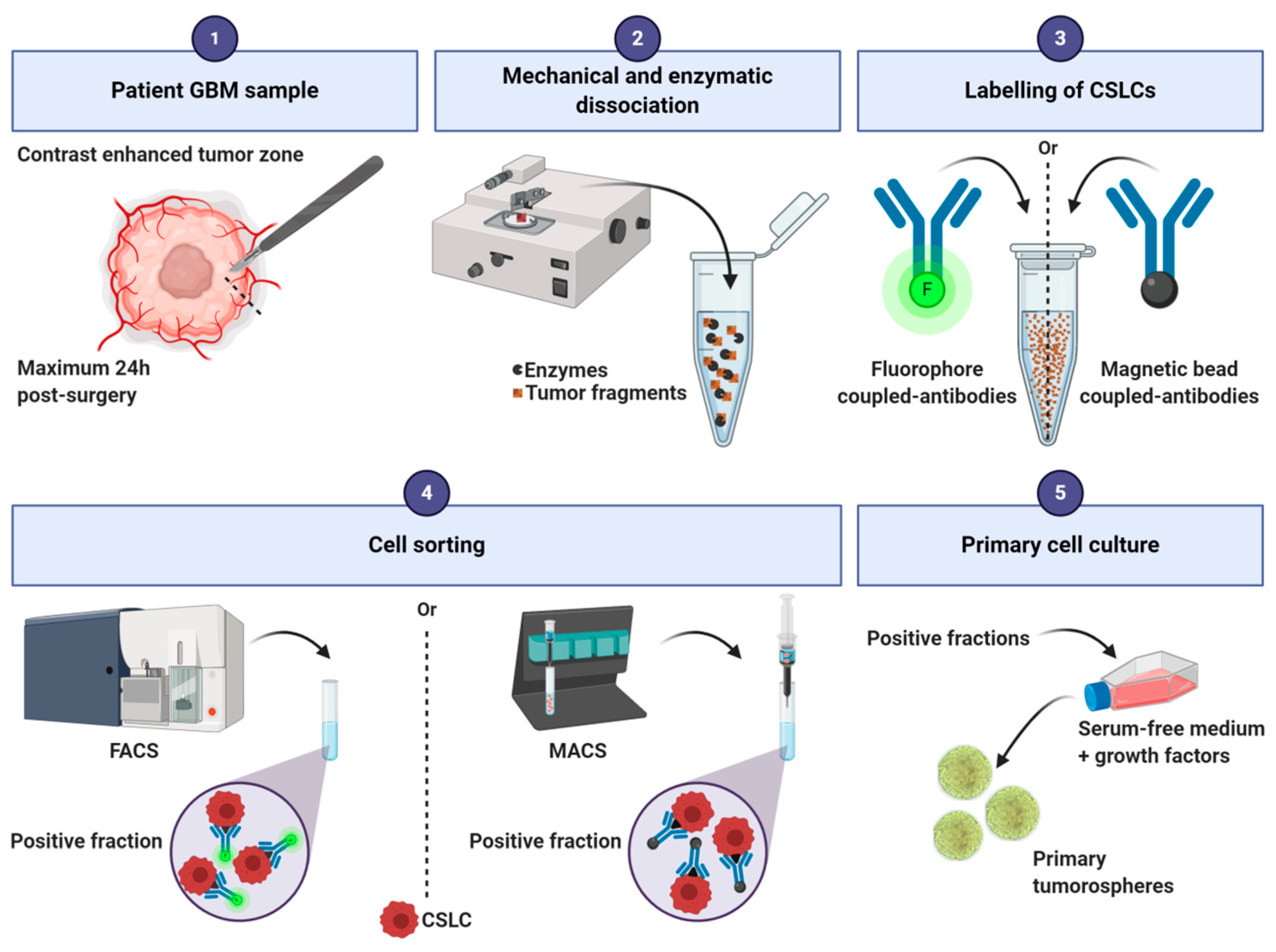
2.2. Tumorospheres
3. Organotypic Cultures
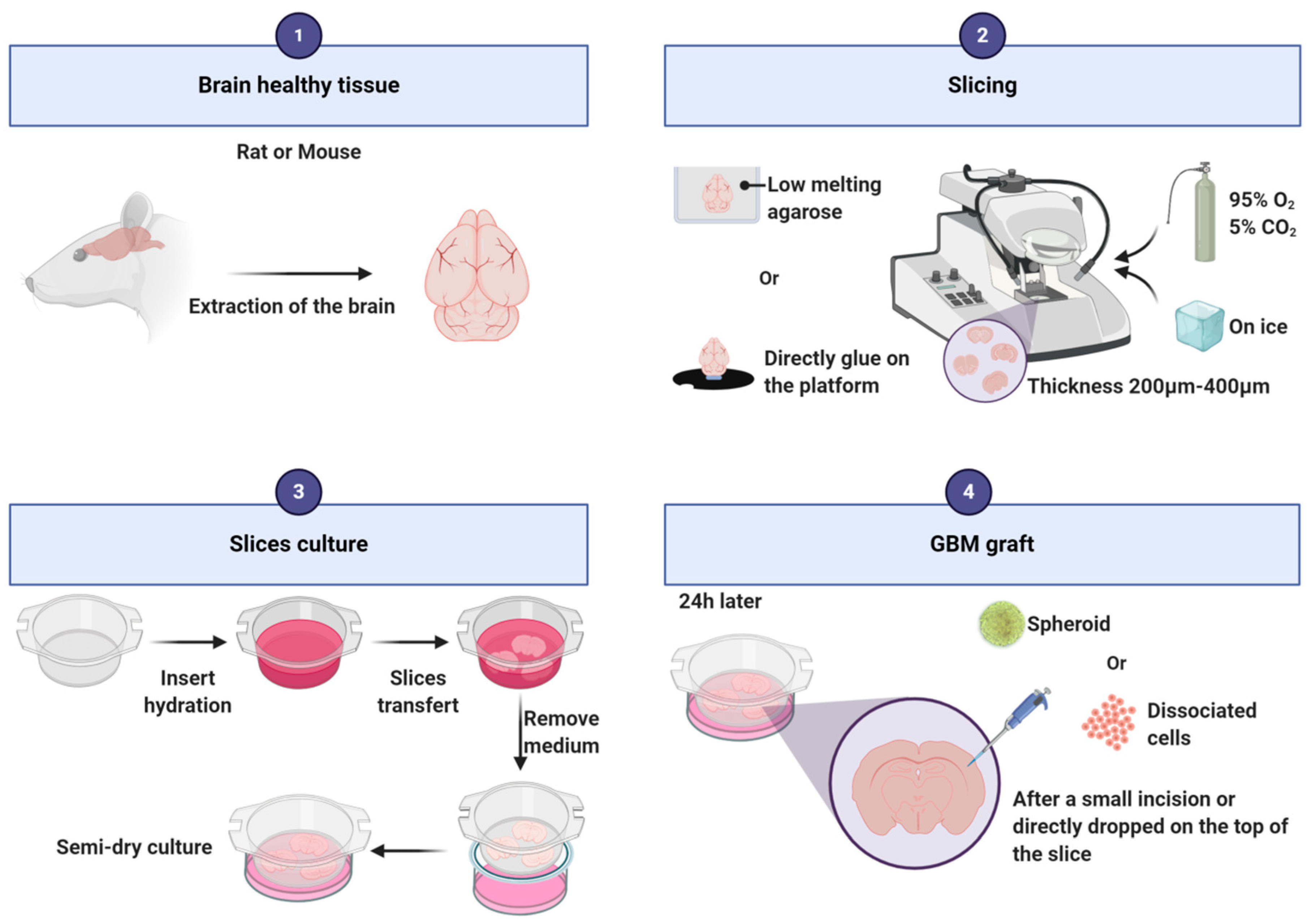
3.1. Organotypic Slice Model
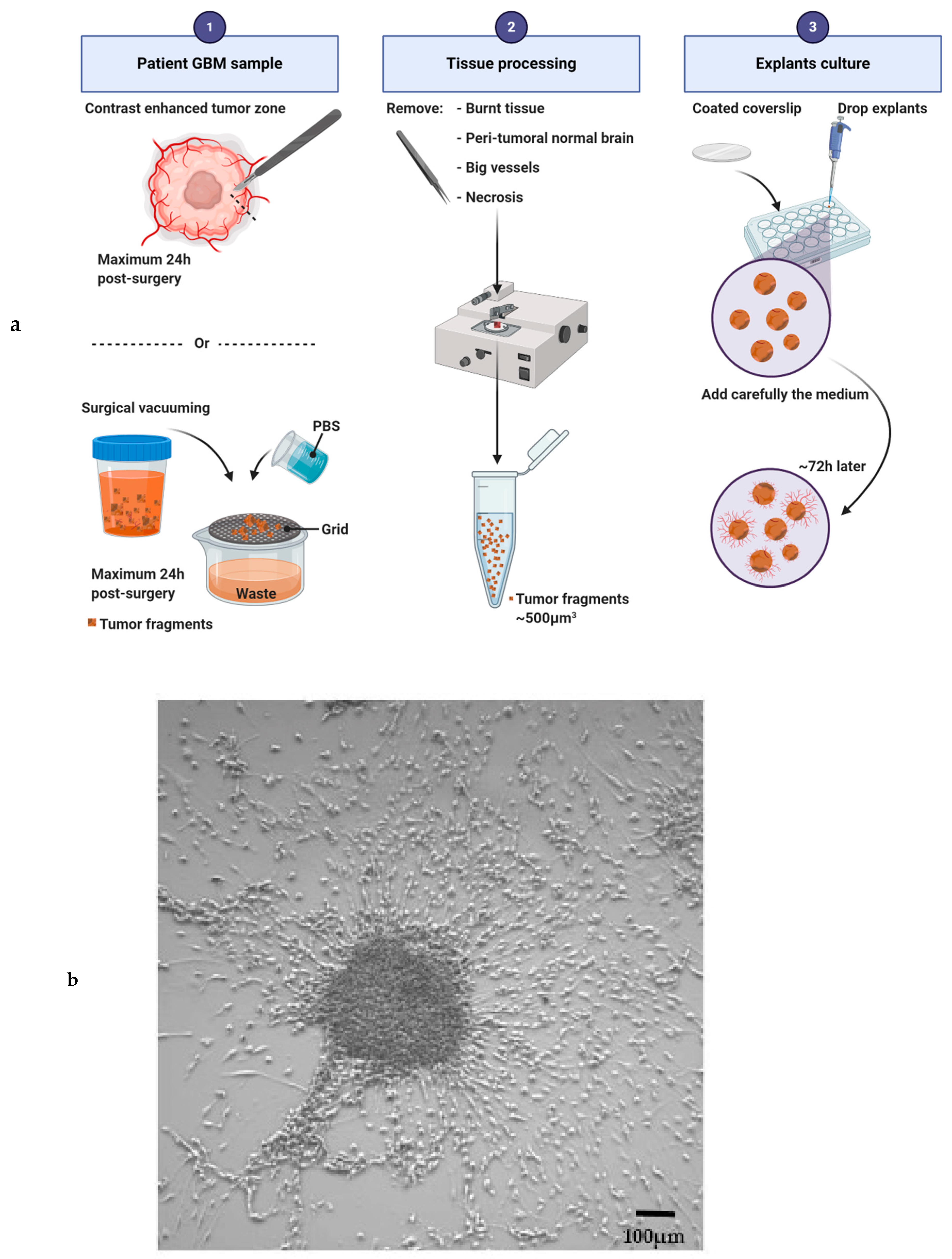
3.2. Explants
4. Organoids
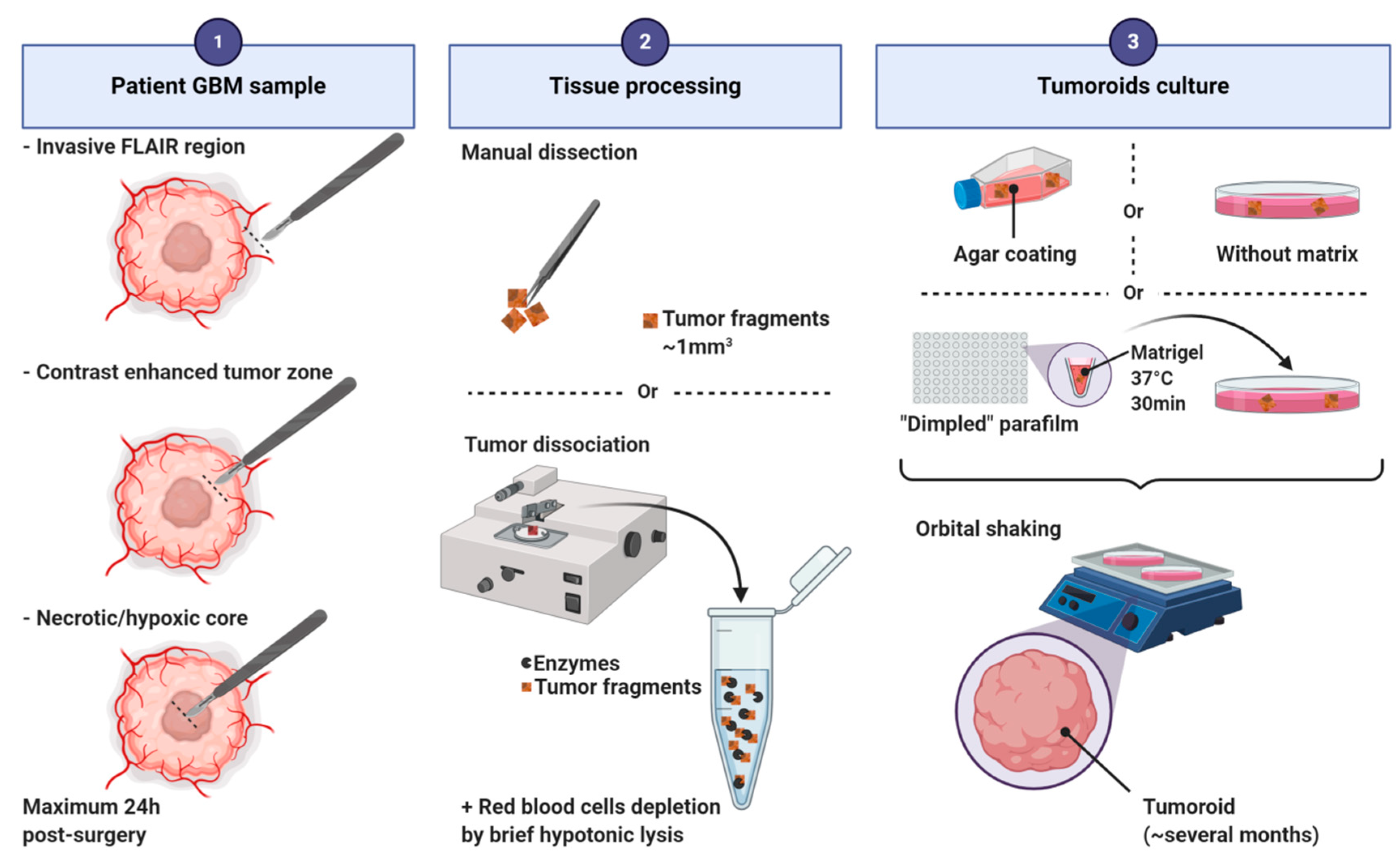
4.1. Tumoroids
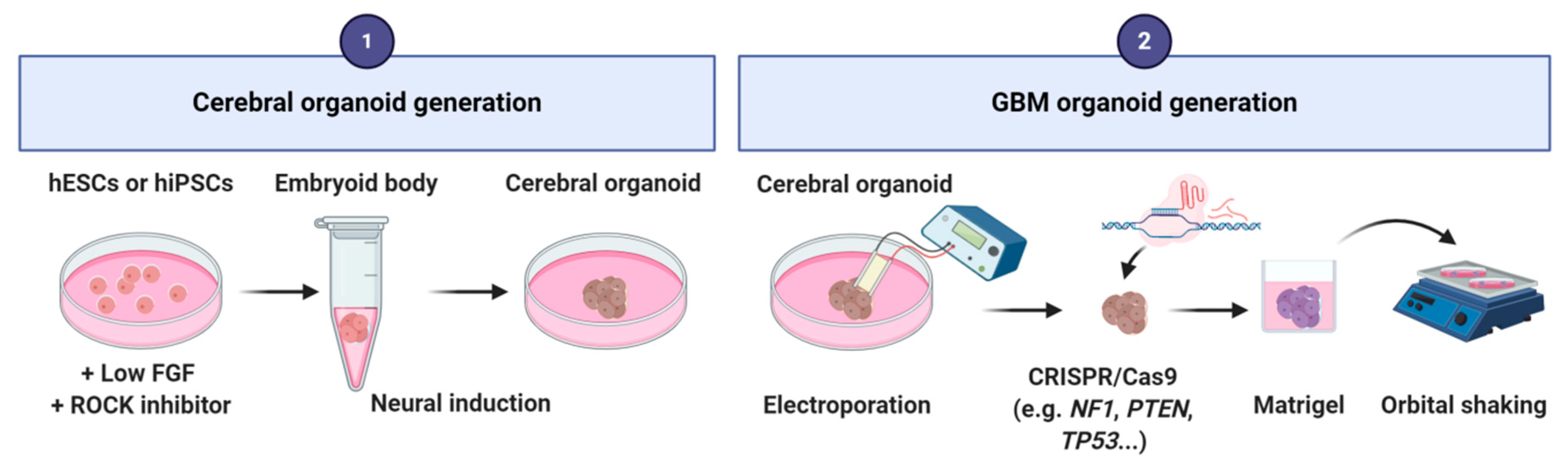
4.2. GBM-Derived from Cerebral Organoids
5. Discussion
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2012, 60, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Bonavia, R.; Inda, M.-M.; Cavenee, W.; Furnari, F. Heterogeneity Maintenance in Glioblastoma: A social network. Cancer Res. 2011, 71, 4055–4060. [Google Scholar] [CrossRef]
- Godlewski, J.; Krichevsky, A.M.; Johnson, M.D.; Chiocca, E.A.; Bronisz, A. Belonging to a network--microRNAs, extracellular vesicles, and the glioblastoma microenvironment. Neuro Oncol. 2015, 17, 652–662. [Google Scholar] [CrossRef]
- Buckley, R.C. Tissue Culture Studies of the Glioblastoma Multiforme. Am. J. Pathol 1929, 5, 467–472. [Google Scholar]
- Carlsson, J.; Nilsson, K.; Westermark, B.; Pontén, J.; Sundström, C.; Larsson, E.; Bergh, J.; Påhlman, S.; Busch, C.; Collins, V.P. Formation and growth of multicellular spheroids of human origin. Int. J. Cancer 1983, 31, 523–533. [Google Scholar] [CrossRef]
- Kolchinsky, A.; Roninson, I.B. Drug resistance conferred by MDR1 expression in spheroids formed by glioblastoma cell lines. Anticancer Res. 1997, 17, 3321–3327. [Google Scholar]
- Liu, M.; Inoue, K.; Leng, T.; Guo, S.; Xiong, Z. TRPM7 channels regulate glioma stem cell through STAT3 and Notch signaling pathways. Cell. Signal. 2014, 26, 2773–2781. [Google Scholar] [CrossRef]
- Jack, G.D.; Cabrera, M.C.; Manning, M.L.; Slaughter, S.M.; Potts, M.; Helm, R.F. Activated stress response pathways within multicellular aggregates utilize an autocrine component. Cell. Signal. 2007, 19, 772–781. [Google Scholar] [CrossRef]
- Tchoghandjian, A.; Baeza, N.; Colin, C.; Cayre, M.; Metellus, P.; Beclin, C.; Ouafik, L.; Figarella-Branger, D. A2B5 cells from human glioblastoma have cancer stem cell properties. Brain Pathol. 2010, 20, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Binder, Z.A.; Wilson, K.M.; Salmasi, V.; Orr, B.A.; Eberhart, C.G.; Siu, I.-M.; Lim, M.; Weingart, J.D.; Quinones-Hinojosa, A.; Bettegowda, C.; et al. Establishment and Biological Characterization of a Panel of Glioblastoma Multiforme (GBM) and GBM Variant Oncosphere Cell Lines. PLoS ONE 2016, 11, e150271. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Hau, P.; Proescholdt, M.; Lohmeier, A.; Wischhusen, J.; Oefner, P.J.; Aigner, L.; Brawanski, A.; Bogdahn, U.; Beier, C.P. CD133(+) and CD133(-) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007, 67, 4010–4015. [Google Scholar] [CrossRef]
- Kersting, G. Tissue culture and the classification of brain tumours. Acta Neurochir. Wien. 1964, 11, 68–74. [Google Scholar] [CrossRef]
- Rubinstein, L.J.; Herman, M.M.; Foley, V.L. In Vitro Characteristics of Human Glioblastomas Maintained in Organ Culture Systems. Am. J. Pathol. 1973, 71, 61–80. [Google Scholar] [PubMed]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found in Vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204. [Google Scholar] [CrossRef]
- De Witt Hamer, P.C.; Van Tilborg, A.A.G.; Eijk, P.P.; Sminia, P.; Troost, D.; Van Noorden, C.J.F.; Ylstra, B.; Leenstra, S. The genomic profile of human malignant glioma is altered early in primary cell culture and preserved in spheroids. Oncogene 2008, 27, 2091–2096. [Google Scholar] [CrossRef]
- Bjerkvig, R.; Tønnesen, A.; Laerum, O.D.; Backlund, E.-O. Multicellular tumor spheroids from human gliomas maintained in organ culture. J. Neurosurg. 1990, 72, 463–475. [Google Scholar] [CrossRef]
- Mahesparan, R.; Tysnes, B.B.; Read, T.-A.; Enger, P.-Ø.; Bjerkvig, R.; Lund-Johansen, M. Extracellular matrix-induced cell migration from glioblastoma biopsy specimens in vitro. Acta Neuropathol. 1999, 97, 231–239. [Google Scholar] [CrossRef]
- Paulus, W.; Huettner, C.; Tonn, J.C. Collagens, integrins and the mesenchymal drift in glioblastomas: A comparison of biopsy specimens, spheroid and early monolayer cultures. Int. J. Cancer 1994, 58, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Aaberg-Jessen, C.; Nørregaard, A.; Christensen, K.; Pedersen, C.B.; Andersen, C.; Kristensen, B.W. Invasion of primary glioma- and cell line-derived spheroids implanted into corticostriatal slice cultures. Int. J. Clin. Exp. Pathol. 2013, 6, 546–560. [Google Scholar] [PubMed]
- Munthe, S.; Halle, B.; Boldt, H.B.; Christiansen, H.; Schmidt, S.; Kaimal, V.; Xu, J.; Zabludoff, S.; Mollenhauer, J.; Poulsen, F.R.; et al. Shift of microRNA profile upon glioma cell migration using patient-derived spheroids and serum-free conditions. J. Neurooncol. 2017, 132, 45–54. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hamer Philip, C.D.W.; Jonker, A.; Leenstra, S.; Ruijter, J.M.; Noorden, C.J.F.V. Quantification of Viability in Organotypic Multicellular Spheroids of Human Malignant Glioma using Lactate Dehydrogenase Activity: A Rapid and Reliable Automated Assay. J. Histochem. Cytochem. 2005, 53, 23–34. [Google Scholar] [CrossRef]
- Kaaijk, P.; Troostt, D.; Dast, P.K.; Leenstra, S.; Bosch, D.A. Long-term culture of organotypic multicellular glioma spheroids: A good culture model for studying gliomas. Neuropathol. Appl. Neurobiol. 1995, 21, 386–391. [Google Scholar] [CrossRef]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019, 26, 3203–3211. [Google Scholar] [CrossRef]
- Bian, S.; Repic, M.; Guo, Z.; Kavirayani, A.; Burkard, T.; Bagley, J.A.; Krauditsch, C.; Knoblich, J.A. Genetically engineered cerebral organoids model brain tumour formation. Nat. Methods 2018, 15, 631–639. [Google Scholar] [CrossRef]
- Sutherland, R.M.; McCredie, J.A.; Inch, W.R. Growth of multicell spheroids in tissue culture as a model of nodular carcinomas. J. Natl. Cancer Inst. 1971, 46, 113–120. [Google Scholar]
- Alessandri, K.; Sarangi, B.R.; Gurchenkov, V.V.; Sinha, B.; Kießling, T.R.; Fetler, L.; Rico, F.; Scheuring, S.; Lamaze, C.; Simon, A.; et al. Cellular capsules as a tool for multicellular spheroid production and for investigating the mechanics of tumor progression in vitro. Proc. Natl. Acad. Sci. USA 2013, 110, 14843–14848. [Google Scholar] [CrossRef]
- Kaufman, L.J.; Brangwynne, C.P.; Kasza, K.E.; Filippidi, E.; Gordon, V.D.; Deisboeck, T.S.; Weitz, D.A. Glioma Expansion in Collagen I Matrices: Analyzing Collagen Concentration-Dependent Growth and Motility Patterns. Biophys. J. 2005, 89, 635–650. [Google Scholar] [CrossRef] [PubMed]
- Civita, P.; Leite, D.M.; Pilkington, G.J. Pre-Clinical Drug Testing in 2D and 3D Human in Vitro Models of Glioblastoma Incorporating Non-Neoplastic Astrocytes: Tunneling Nano Tubules and Mitochondrial Transfer Modulates Cell Behavior and Therapeutic Respons. Int. J. Mol. Sci. 2019, 20, 6017. [Google Scholar] [CrossRef] [PubMed]
- Leite, D.M.; Baskovic, B.Z.; Civita, P.; Neto, C.; Gumbleton, M.; Pilkington, G.J. A human co-culture cell model incorporating microglia supports glioblastoma growth and migration, and confers resistance to cytotoxics. FASEB J. 2020, 34, 1710–1727. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, J.; Acker, H. Relations between pH, oxygen partial pressure and growth in cultured cell spheroids. Int. J. Cancer 1988, 42, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Glimelius, B.; Norling, B.; Nederman, T.; Carlsson, J. Extracellular matrices in multicellular spheroids of human glioma origin: Increased incorporation of proteoglycans and fibronectin as compared to monolayer cultures. APMIS 1988, 96, 433–444. [Google Scholar] [CrossRef]
- Khaitan, D.; Chandna, S.; Arya, M.; Dwarakanath, B. Establishment and characterization of multicellular spheroids from a human glioma cell line; Implications for tumor therapy. J. Transl. Med. 2006, 4, 12. [Google Scholar] [CrossRef]
- Oraiopoulou, M.-E.; Tampakaki, M.; Tzamali, E.; Tamiolakis, T.; Makatounakis, V.; Vakis, A.F.; Zacharakis, G.; Sakkalis, V.; Papamatheakis, J. A 3D tumor spheroid model for the T98G Glioblastoma cell line phenotypic characterization. Tissue Cell 2019, 59, 39–43. [Google Scholar] [CrossRef]
- Ahn, H.-Y.; Hadizadeh, K.R.; Seul, C.; Yun, Y.-P.; Vetter, H.; Sachinidis, A. Epigallocathechin-3 Gallate Selectively Inhibits the PDGF-BB–induced Intracellular Signaling Transduction Pathway in Vascular Smooth Muscle Cells and Inhibits Transformation of sis-transfected NIH 3T3 Fibroblasts and Human Glioblastoma Cells (A172). Mol. Biol. Cell 1999, 10, 1093–1104. [Google Scholar] [CrossRef]
- Bell, H.S.; Whittle, I.R.; Walker, M.; Leaver, H.A.; Wharton, S.B. The development of necrosis and apoptosis in glioma: Experimental findings using spheroid culture systems. Neuropathol. Appl. Neurobiol. 2001, 27, 291–304. [Google Scholar] [CrossRef]
- Nirmala, C.; Rao, J.S.; Ruifrok, A.C.; Langford, L.A.; Obeyesekere, M. Growth characteristics of glioblastoma spheroids. Int. J. Oncol. 2001, 19, 1109–1115. [Google Scholar] [CrossRef]
- Madsen, S.J.; Sun, C.-H.; Tromberg, B.J.; Hirschberg, H. Repetitive 5-aminolevulinic acid-mediated photodynamic therapy on human glioma spheroids. J. Neuro-Oncol. 2003, 62, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Fedrigo, C.A.; Grivicich, I.; Schunemann, D.P.; Chemale, I.M.; Santos, D.D.; Jacovas, T.; Boschetti, P.S.; Jotz, G.P.; Filho, A.B.; da Rocha, A.B. Radioresistance of human glioma spheroids and expression of HSP70, p53 and EGFr. Radiat. Oncol 2011, 6, 156. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.N.; Graham, B.; Pacurari, M.; Leggett, S.S.; Tchounwou, P.B.; Ndebele, K. Di-Ethylhexylphthalate (DEHP) Modulates Cell Invasion, Migration and Anchorage Independent Growth through Targeting S100P in LN-229 Glioblastoma Cells. Int. J. Environ. Res. Public Health 2014, 11, 5006–5019. [Google Scholar] [CrossRef] [PubMed]
- Azzalin, A.; Moretti, E.; Arbustini, E.; Magrassi, L. Cell density modulates SHC3 expression and survival of human glioblastoma cells through Fak activation. J. Neurooncol. 2014, 120, 245–256. [Google Scholar] [CrossRef]
- Gdynia, G.; Grund, K.; Eckert, A.; Böck, B.C.; Funke, B.; Macher-Goeppinger, S.; Sieber, S.; Herold-Mende, C.; Wiestler, B.; Wiestler, O.D.; et al. Basal Caspase Activity Promotes Migration and Invasiveness in Glioblastoma Cells. Mol. Cancer Res. 2007, 5, 1232–1240. [Google Scholar] [CrossRef]
- Gordon, V.D.; Valentine, M.T.; Gardel, M.L.; Andor-Ardó, D.; Dennison, S.; Bogdanov, A.A.; Weitz, D.A.; Deisboeck, T.S. Measuring the mechanical stress induced by an expanding multicellular tumor system: A case study. Exp. Cell Res. 2003, 289, 58–66. [Google Scholar] [CrossRef]
- Haji-Karim, M.; Carisson, J. Proliferation and Viability in Cellular Spheroids of Human Origin. Cancer Res. 1978, 38, 1457–1464. [Google Scholar]
- Vinci, M.; Gowan, S.; Boxall, F.; Patterson, L.; Zimmermann, M.; Court, W.; Lomas, C.; Mendiola, M.; Hardisson, D.; Eccles, S.A. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol. 2012, 10, 29. [Google Scholar] [CrossRef]
- Del Duca, D.; Werbowetski, T.; Del Maestro, R.F. Spheroid preparation from hanging drops: Characterization of a model of brain tumor invasion. J. Neurooncol. 2004, 67, 295–303. [Google Scholar] [CrossRef]
- Jiguet Jiglaire, C.; Baeza-Kallee, N.; Denicolaï, E.; Barets, D.; Metellus, P.; Padovani, L.; Chinot, O.; Figarella-Branger, D.; Fernandez, C. Ex vivo cultures of glioblastoma in three-dimensional hydrogel maintain the original tumor growth behavior and are suitable for preclinical drug and radiation sensitivity screening. Exp. Cell Res. 2014, 321, 99–108. [Google Scholar] [CrossRef]
- Lv, D.; Wang, X.; Dong, J.; Zhuang, Y.; Huang, S.; Ma, B.; Chen, P.; Li, X.; Zhang, B.; Li, Z.; et al. Systematic characterization of lncRNAs’ cell-to-cell expression heterogeneity in glioblastoma cells. Oncotarget 2016, 7, 18403–18414. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, J.; Ebner, R.; Kunz-Schughart, L.A. Experimental anti-tumor therapy in 3-D: Spheroids—Old hat or new challenge? Int. J. Radiat. Biol. 2007, 83, 849–871. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.; Hsiao, A.Y.; Ingram, M.; Luker, G.D.; Takayama, S. Opportunities and Challenges for use of Tumor Spheroids as Models to Test Drug Delivery and Efficacy. J. Control. Release 2012, 164, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Nederman, T.; Norling, B.; Glimelius, B.; Carlsson, J.; Brunk, U. Demonstration of an Extracellular Matrix in Multicellular Tumor Spheroids. Cancer Res. 1984, 44, 3090–3097. [Google Scholar]
- Friedrich, J.; Seidel, C.; Ebner, R.; Kunz-Schughart, L.A. Spheroid-based drug screen: Considerations and practical approach. Nat. Protoc. 2009, 4, 309–324. [Google Scholar] [CrossRef]
- Berges, R.; Denicolai, E.; Tchoghandjian, A.; Baeza-Kallee, N.; Honore, S.; Figarella-Branger, D.; Braguer, D. Proscillaridin A exerts anti-tumor effects through GSK3β activation and alteration of microtubule dynamics in glioblastoma. Cell Death Dis. 2018, 9, 984. [Google Scholar] [CrossRef]
- Chaicharoenaudomrung, N.; Kunhorm, P.; Promjantuek, W.; Heebkaew, N.; Rujanapun, N.; Noisa, P. Fabrication of 3D calcium-alginate scaffolds for human glioblastoma modeling and anticancer drug response evaluation. J. Cell. Physiol. 2019, 234, 20085–20097. [Google Scholar] [CrossRef]
- Chaicharoenaudomrung, N.; Kunhorm, P.; Promjantuek, W.; Rujanapun, N.; Heebkaew, N.; Soraksa, N.; Noisa, P. Transcriptomic Profiling of 3D Glioblastoma Tumoroids for the Identification of Mechanisms Involved in Anticancer Drug Resistance. In Vivo 2020, 34, 199–211. [Google Scholar] [CrossRef]
- Ishiguro, T.; Ohata, H.; Sato, A.; Yamawaki, K.; Enomoto, T.; Okamoto, K. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci. 2017, 108, 283–289. [Google Scholar] [CrossRef]
- Li, A.; Walling, J.; Kotliarov, Y.; Center, A.; Steed, M.E.; Ahn, S.J.; Rosenblum, M.; Mikkelsen, T.; Zenklusen, J.C.; Fine, H.A. Genomic Changes and Gene Expression Profiles Reveal That Established Glioma Cell Lines Are Poorly Representative of Primary Human Gliomas. Mol. Cancer Res. 2008, 6, 21–30. [Google Scholar] [CrossRef]
- Allen, M.; Bjerke, M.; Edlund, H.; Nelander, S.; Westermark, B. Origin of the U87MG glioma cell line: Good news and bad news. Sci. Transl. Med. 2016, 8, 354re3. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, B.A.; Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 1992, 255, 1707–1710. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Ogden, A.T.; Waziri, A.E.; Lochhead, R.A.; Fusco, D.; Lopez, K.; Ellis, J.A.; Kang, J.; Assanah, M.; McKhann, G.M.; Sisti, M.B.; et al. Identification of A2B5+CD133- tumor-initiating cells in adult human gliomas. Neurosurgery 2008, 62, 505–514. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; Li, Z.; Sathornsumetee, S.; Wang, H.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Targeting Cancer Stem Cells through L1CAM Suppresses Glioma Growth. Cancer Res. 2008, 68, 6043–6048. [Google Scholar] [CrossRef]
- Lathia, J.D.; Gallagher, J.; Heddleston, J.M.; Wang, J.; Eyler, C.E.; Macswords, J.; Wu, Q.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell 2010, 6, 421–432. [Google Scholar] [CrossRef]
- Son, M.J.; Woolard, K.; Nam, D.-H.; Lee, J.; Fine, H.A. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef]
- Anido, J.; Sáez-Borderías, A.; Gonzàlez-Juncà, A.; Rodón, L.; Folch, G.; Carmona, M.A.; Prieto-Sánchez, R.M.; Barba, I.; Martínez-Sáez, E.; Prudkin, L.; et al. TGF-β Receptor Inhibitors Target the CD44high/Id1high Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell 2010, 18, 655–668. [Google Scholar] [CrossRef]
- Avril, T.; Vauleon, E.; Hamlat, A.; Saikali, S.; Etcheverry, A.; Delmas, C.; Diabira, S.; Mosser, J.; Quillien, V. Human Glioblastoma Stem-Like Cells are More Sensitive to Allogeneic NK and T Cell-Mediated Killing Compared with Serum-Cultured Glioblastoma Cells. Brain Pathol. 2012, 22, 159–174. [Google Scholar] [CrossRef]
- Eimer, S.; Dugay, F.; Airiau, K.; Avril, T.; Quillien, V.; Belaud-Rotureau, M.-A.; Belloc, F. Cyclopamine cooperates with EGFR inhibition to deplete stem-like cancer cells in glioblastoma-derived spheroid cultures. Neuro Oncol. 2012, 14, 1441–1451. [Google Scholar] [CrossRef]
- Ciccolini, F.; Svendsen, C.N. Fibroblast growth factor 2 (FGF-2) promotes acquisition of epidermal growth factor (EGF) responsiveness in mouse striatal precursor cells: Identification of neural precursors responding to both EGF and FGF-2. J. Neurosci. 1998, 18, 7869–7880. [Google Scholar] [CrossRef] [PubMed]
- Engebraaten, O.; Bjerkvig, R.; Pedersen, P.H.; Laerum, O.D. Effects of EGF, bFGF, NGF and PDGF(bb) on cell proliferative, migratory and invasive capacities of human brain-tumour biopsies in vitro. Int. J. Cancer 1993, 53, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Pastrana, E.; Silva-Vargas, V.; Doetsch, F. Eyes Wide Open: A Critical Review of Sphere-Formation as an Assay for Stem Cells. Cell Stem Cell 2011, 8, 486–498. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Soubéran, A.; Cappaï, J.; Chocry, M.; Nuccio, C.; Raujol, J.; Colin, C.; Lafitte, D.; Kovacic, H.; Quillien, V.; Baeza-Kallee, N.; et al. Inhibitor of Apoptosis Proteins Determines Glioblastoma Stem-Like Cell Fate in an Oxygen-Dependent Manner. Stem Cells 2019, 37, 731–742. [Google Scholar] [CrossRef]
- Tchoghandjian, A.; Jennewein, C.; Eckhardt, I.; Momma, S.; Figarella-Branger, D.; Fulda, S. Smac mimetic promotes glioblastoma cancer stem-like cell differentiation by activating NF-κB. Cell Death Differ. 2014, 21, 735–747. [Google Scholar] [CrossRef]
- Baeza-Kallee, N.; Bergès, R.; Soubéran, A.; Colin, C.; Denicolaï, E.; Appay, R.; Tchoghandjian, A.; Figarella-Branger, D. Glycolipids Recognized by A2B5 Antibody Promote Proliferation, Migration, and Clonogenicity in Glioblastoma Cells. Cancers 2019, 11, 1267. [Google Scholar] [CrossRef]
- Tchoghandjian, A.; Baeza-Kallee, N.; Beclin, C.; Metellus, P.; Colin, C.; Ducray, F.; Adélaïde, J.; Rougon, G.; Figarella-Branger, D. Cortical and subventricular zone glioblastoma-derived stem-like cells display different molecular profiles and differential in vitro and in vivo properties. Ann. Surg. Oncol. 2012, 19 (Suppl. 3), S608–S619. [Google Scholar] [CrossRef]
- Ohnishi, T.; Matsumura, H.; Izumoto, S.; Hiraga, S.; Hayakawa, T. A Novel Model of Glioma Cell Invasion Using Organotypic Brain Slice Culture. Cancer Res. 1998, 58, 2935–2940. [Google Scholar]
- Yamamoto, N.; Kurotani, T.; Toyama, K. Neural connections between the lateral geniculate nucleus and visual cortex in vitro. Science 1989, 245, 192–194. [Google Scholar] [CrossRef] [PubMed]
- Henrik Heiland, D.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strähle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef] [PubMed]
- Ravi, V.M.; Joseph, K.; Wurm, J.; Behringer, S.; Garrelfs, N.; d’Errico, P.; Naseri, Y.; Franco, P.; Meyer-Luehmann, M.; Sankowski, R.; et al. Human organotypic brain slice culture: A novel framework for environmental research in neuro-oncology. Life Sci. Alliance 2019, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Marques-Torrejon, M.A.; Gangoso, E.; Pollard, S.M. Modelling glioblastoma tumour-host cell interactions using adult brain organotypic slice co-culture. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Eisemann, T.; Costa, B.; Strelau, J.; Mittelbronn, M.; Angel, P.; Peterziel, H. An advanced glioma cell invasion assay based on organotypic brain slice cultures. BMC Cancer 2018, 18, 103. [Google Scholar] [CrossRef]
- Parker, J.J.; Lizarraga, M.; Waziri, A.; Foshay, K.M. A Human Glioblastoma Organotypic Slice Culture Model for Study of Tumor Cell Migration and Patient-specific Effects of Anti-Invasive Drugs. J. Vis. Exp. 2017. [Google Scholar] [CrossRef]
- Gritsenko, P.; Leenders, W.; Friedl, P. Recapitulating in vivo-like plasticity of glioma cell invasion along blood vessels and in astrocyte-rich stroma. Histochem. Cell Biol. 2017, 148, 395–406. [Google Scholar] [CrossRef]
- Gritsenko, P.G.; Friedl, P. Adaptive adhesion systems mediate glioma cell invasion in complex environments. J. Cell Sci. 2018, 131, jcs216382. [Google Scholar] [CrossRef]
- Sliwa, M.; Markovic, D.; Gabrusiewicz, K.; Synowitz, M.; Glass, R.; Zawadzka, M.; Wesolowska, A.; Kettenmann, H.; Kaminska, B. The invasion promoting effect of microglia on glioblastoma cells is inhibited by cyclosporin A. Brain 2007, 130, 476–489. [Google Scholar] [CrossRef]
- Hu, F.; Dzaye, O.D.A.; Hahn, A.; Yu, Y.; Scavetta, R.J.; Dittmar, G.; Kaczmarek, A.K.; Dunning, K.R.; Ricciardelli, C.; Rinnenthal, J.L.; et al. Glioma-derived versican promotes tumor expansion via glioma-associated microglial/macrophages Toll-like receptor 2 signaling. Neuro Oncol. 2015, 17, 200–210. [Google Scholar] [CrossRef]
- Wolf, K.J.; Shukla, P.; Springer, K.; Lee, S.; Coombes, J.D.; Choy, C.J.; Kenny, S.J.; Xu, K.; Kumar, S. A mode of cell adhesion and migration facilitated by CD44-dependent microtentacles. Proc. Natl. Acad. Sci. USA 2020, 117, 11432–11443. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Takahashi, H.; Inoue, A.; Harada, H.; Toshimori, S.; Kobayashi, Y.; Goto, K.; Sugimoto, K.; Yano, H.; Ohnishi, T.; et al. Oct-3/4 promotes migration and invasion of glioblastoma cells. J. Cell. Biochem. 2012, 113, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Ziemann, A.; Hess, S.; Bhuwania, R.; Linder, S.; Kloppenburg, P.; Noegel, A.A.; Clemen, C.S. CRN2 enhances the invasiveness of glioblastoma cells. Neuro Oncol. 2013, 15, 548–561. [Google Scholar] [CrossRef] [PubMed]
- Kurogi, R.; Nakamizo, A.; Suzuki, S.O.; Mizoguchi, M.; Yoshimoto, K.; Amano, T.; Amemiya, T.; Takagishi, S.; Iihara, K. Inhibition of glioblastoma cell invasion by hsa-miR-145-5p and hsa-miR-31-5p co-overexpression in human mesenchymal stem cells. J. Neurosurg. 2018, 130, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Jašprová, J.; Dal Ben, M.; Hurný, D.; Hwang, S.; Žížalová, K.; Kotek, J.; Wong, R.J.; Stevenson, D.K.; Gazzin, S.; Tiribelli, C.; et al. Neuro-inflammatory effects of photodegradative products of bilirubin. Sci Rep. 2018, 8, 7444. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.S.; Meyer, M.; Petterson, S.A.; Halle, B.; Rosager, A.M.; Aaberg-Jessen, C.; Thomassen, M.; Burton, M.; Kruse, T.A.; Kristensen, B.W. Establishment and Characterization of a Tumor Stem Cell-Based Glioblastoma Invasion Model. PLoS ONE 2016, 11, e0159746. [Google Scholar] [CrossRef]
- Humpel, C. Organotypic brain slice cultures: A review. Neuroscience 2015, 305, 86–98. [Google Scholar] [CrossRef]
- Davenport, R.D.; McKeever, P.E. DNA content and marker expression in human glioma explants. Acta Neuropathol. 1987, 74, 362–365. [Google Scholar] [CrossRef]
- Colin, C.; Baeza, N.; Tong, S.; Bouvier, C.; Quilichini, B.; Durbec, P.; Figarella-Branger, D. In vitro identification and functional characterization of glial precursor cells in human gliomas. Neuropathol. Appl. Neurobiol. 2006, 32, 189–202. [Google Scholar] [CrossRef]
- Asklund, T.; Appelskog, I.B.; Ammerpohl, O.; Langmoen, I.A.; Dilber, M.S.; Aints, A.; Ekström, T.J.; Almqvist, P.M. Gap junction-mediated bystander effect in primary cultures of human malignant gliomas with recombinant expression of the HSVtk gene. Exp. Cell Res. 2003, 284, 183–193. [Google Scholar] [CrossRef]
- Bayin, N.S.; Ma, L.; Placantonakis, D.G.; Barcellos-Hoff, M.H. Evaluation of radioresponse and radiosensitizers in glioblastoma organotypic cultures. Methods Mol. Biol. 2018, 1741, 171–182. [Google Scholar] [CrossRef]
- Manuelidis, E.E. Long-Term Lines of Tissue Cultures of Intracranial Tumors. J. Neurosurg. 1965, 22, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.; Sarkar, C.; Dinda, A.K.; Karak, A.K.; Mathur, M.; Roy, S. Morphological appearance, growth kinetics and glial fibrillary acidic protein (GFAP) expression in primary in vitro explant culture of astrocytic neoplasms. Acta Oncol. 1993, 32, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Sipe, J.C.; Herman, M.M.; Rubinstein, L.J. Electron Microscopic Observations on Human Glioblastomas and Astrocytomas Maintained in Organ Culture Systems. Am. J. Pathol. 1973, 73, 589–606. [Google Scholar]
- Escalona-Zapata, J.; Diez-Nau, M.D. The astrocytic nature of glioblastoma demonstrated by tissue culture. Acta Neuropathol. 1981, 53, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Tejero, R.; Huang, Y.; Katsyv, I.; Kluge, M.; Lin, J.-Y.; Tome-Garcia, J.; Daviaud, N.; Wang, Y.; Zhang, B.; Tsankova, N.M.; et al. Gene signatures of quiescent glioblastoma cells reveal mesenchymal shift and interactions with niche microenvironment. EBioMedicine 2019, 42, 252–269. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Generation of Cerebral Organoids from Human Pluripotent Stem Cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, B.; Zhou, C.; Li, Y.; Li, B.; Yu, M.; Luo, Y.; Gao, L.; Zhang, D.; Xue, Q.; et al. The comparison genomics analysis with glioblastoma multiforme (GBM) cells under 3D and 2D cell culture conditions. Colloids Surf. B: Biointerfaces 2018, 172, 665–673. [Google Scholar] [CrossRef]
- Abe, T.; Okamura, K.; Ono, M.; Kohno, K.; Mori, T.; Hori, S.; Kuwano, M. Induction of vascular endothelial tubular morphogenesis by human glioma cells. A model system for tumor angiogenesis. J. Clin. Investig. 1993, 92, 54–61. [Google Scholar] [CrossRef]
- Wei, Z.; Kale, S.; El Fatimy, R.; Rabinovsky, R.; Krichevsky, A.M. Co-cultures of Glioma Stem Cells and Primary Neurons, Astrocytes, Microglia, and Endothelial Cells for Investigation of Intercellular Communication in the Brain. Front. Neurosci. 2019, 13, 361. [Google Scholar] [CrossRef]
- Herting, C.J.; Chen, Z.; Maximov, V.; Duffy, A.; Szulzewsky, F.; Shayakhmetov, D.M.; Hambardzumyan, D. Tumour-associated macrophage-derived interleukin-1 mediates glioblastoma-associated cerebral oedema. Brain 2019, 142, 3834–3851. [Google Scholar] [CrossRef] [PubMed]
- Cosset, E.; Locatelli, M.; Marteyn, A.; Lescuyer, P.; Dall Antonia, F.; Mor, F.M.; Preynat-Seauve, O.; Stoppini, L.; Tieng, V. Human Neural Organoids for Studying Brain Cancer and Neurodegenerative Diseases. J. Vis. Exp. 2019, 148, e59682. [Google Scholar] [CrossRef] [PubMed]
- Krieger, T.G.; Tirier, S.M.; Park, J.; Jechow, K.; Eisemann, T.; Peterziel, H.; Angel, P.; Eils, R.; Conrad, C. Modeling glioblastoma invasion using human brain organoids and single-cell transcriptomics. Neuro Oncol. 2020. [Google Scholar] [CrossRef]
- Dai, X.; Ma, C.; Lan, Q.; Xu, T. 3D bioprinted glioma stem cells for brain tumor model and applications of drug susceptibility. Biofabrication 2016, 8, 045005. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.A.; Bansal, R.; Lammers, T.; Zhang, Y.S.; Schiffelers, R.M.; Prakash, J. 3D-Bioprinted Mini-Brain: A Glioblastoma Model to Study Cellular Interactions and Therapeutics. Adv. Mater. 2019, 31, 1806590. [Google Scholar] [CrossRef]
- Fan, Y.; Nguyen, D.T.; Akay, Y.; Xu, F.; Akay, M. Engineering a Brain Cancer Chip for High-throughput Drug Screening. Sci. Rep. 2016, 6, 25062. [Google Scholar] [CrossRef]
- Yi, H.-G.; Jeong, Y.H.; Kim, Y.; Choi, Y.-J.; Moon, H.E.; Park, S.H.; Kang, K.S.; Bae, M.; Jang, J.; Youn, H.; et al. A bioprinted human-glioblastoma-on-a-chip for the identification of patient-specific responses to chemoradiotherapy. Nat. Biomed. Eng. 2019, 3, 509–519. [Google Scholar] [CrossRef]
- Gomez-Roman, N.; Chalmers, A.J. Patient-specific 3D-printed glioblastomas. Nat. Biomed. Eng. 2019, 3, 498–499. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3D Models | Material | Alternative Names |
|---|---|---|
| Spheroids | Established glioblastoma cell lines | Spheroids [8] Neurospheroids [9] Multicellular tumor spheroids [7] Multicellular aggregates [10] |
| Tumorospheres | Primary glioblastoma stem-like cells | Spheres [11] Oncospheres [12] Neurosphere-like [13] |
| Organotypic slices | Grafted isolated cells or spheroids/tumorospheres on brain slices | - |
| Explants | Glioblastoma tissue | Tissue culture / Tissue particles [14] Organ culture [15] |
| Tumoroids | Glioblastoma tissue | Organoids [16] Glioblastoma organoids (GBO) [17] Spheroids [18] Multicellular tumor spheroids [19] Biopsy spheroids [20] Fragments spheroids [21] Primary spheroids [22] Patient-derived spheroids [23] Organotypic spheroids [24] Organotypic multicellular spheroids [25] |
| GBM-derived from cerebral organoids | Embryonic stem cells and induced pluripotent stem cells | Organoids [26] Organoid glioma (GLICO) [27] Neoplastic cerebral organoids (NeoCOR) [28] |
| Method | Procedure | Matrix | Advantages | Disadvantages | Ref. |
|---|---|---|---|---|---|
| Liquid overlay | Tumor cells are placed on tissue culture plastic covered with a thin layer of inert substrate. | Agar Agarose PolyHEMA | Easy-to-use protocol; Easily promotes the aggregation of cells to become spheroids; Co-culture ability; High reproducibility; Inexpensive; Easy to image. | Difficulty to monitor the number and size of spheroids; Heterogeneity of the cell lineage; Lack of interactions between cells and matrix. | [7,34,47] |
| Ultra–low attachment plates | Cells are seeded in an ultra-low attachment plate without coating as the polystyrene surface offers low adhesion properties. | - | Capability to produce one spheroid per well; Spheroids have a more compact structure than those on agar-coated plates; Easy to image. | Difficulty to monitor the number and size of the spheroids; Heterogeneity of the cell lineage; Lack of interactions between cells and matrix | [48] |
| Hanging drop method | Cells are dropped in a small volume in the petri dish lid. The lid is subsequently inverted, and aliquots of cell suspension turned into hanging drops without dripping due to surface tension. | - | Easy-to-use protocol; Consistent size and shape controlled by adjusting the density of cell seeding; High reproducibility; Inexpensive; Easy to image. | Heterogeneity of cell lineage; Lack of interactions between cells and matrix; Limited volume of the cell suspension; Difficulty in changing the culture medium. | [49] |
| Hydrogel embedding/Scaffold | Microcapsules with matrix /cells obtained from cells resuspended in hydrogel 3D structures that are constructed from a wide-range of materials and possess different porosities, permeabilities, surface chemistries, and mechanical characteristics. | Alginate Matrigel Methylcellulose Collagen Gelatin Silk Chitosan | Large variety of natural or synthetic materials; Customizable; Co-culture possible; Resemble natural extracellular matrix; Circulation of nutrients and cellular waste in and out of the hydrogels. | Deficiency in gelation kinetic control; Undefined composition in natural gels; May not be transparent; Difficulty to remove cells. | [50,51] |
| Spinner flask bioreactor | Cells are inserted into a chamber with continuous agitation (by gently stirring, rotating the chamber, or perfusing culture media through a scaffold using a pump system). Bioreactors are equipped with media-flowing systems to provide nutrient circulation, metabolic waste removal, and homogeneity of the physical and chemical factors within the bioreactors. | With or without matrix | Easy-to-use protocol; Great spheroid formation; Precise control system and guaranteed reproducibility; Motion of culture assists in nutrient transport; Large scale production. | No control of the cell number/size of spheroids; Cells possibly exposed to shear force in spinner flasks; Specialized equipment required. | [52] |
| Experimental Possibilities | Sphere-Based Models | Organotypic Models | Organoids | ||||
|---|---|---|---|---|---|---|---|
| Spheroids | Tumorospheres | Organotypic Slices | Explants | Tumoroids | GBM-Derived from Cerebral Organoids | ||
| CHARACTERISTICS | Success rate | 100% | 60% | n.s | 50% | 30 to 90% | n.s |
| Heterogeneity in tumor cells/in the peritumoral microenvironment cells | −/− | −/− | −/+ | +/− | +/− | −/− | |
| Genetic stability | − | + | − | + | + | + | |
| Cryoconservation | + | + | − | − | + | n.s | |
| Lifespan of the culture | Indefinitely | Indefinitely (1) | 4 weeks | <3 weeks | >1 year | n.s | |
| Standardization | + | + | − | − | − | − | |
| Patient specific | − | + | + (2) | + | + | + (3) | |
| CSLCs | − | + | + (2) | + | + | + | |
| PARAMETERS STUDIED | Tumor growth | + | + | + | + | + | + |
| Tumor invasion-migration model/can be used to study migration-invasion | −/+ | −/+ | +/+ | +/+ | −/+ | +/+ | |
| Stemness properties | − | + | + (4) | + | + | + (5) | |
| Environmental influence of the tumor/of the healthy surrounding tissue | −/− | −/− | −/+ | +/− | +/− | −/+ | |
| Drug testing | + | + | + | + | + | + | |
| Radiotherapy | + | + | + | + | + | n.s | |
| Mechanisms of drug resistance | + | + | − | − | − | + | |
| High throughput drug screening | + | + | − | − | − | − | |
| Personalized medicine | − | + | + (6) | + | + | − | |
| Immune response | − | − | + | + | + | − | |
| Gliomagenesis process | − | − | − | − | − | + | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soubéran, A.; Tchoghandjian, A. Practical Review on Preclinical Human 3D Glioblastoma Models: Advances and Challenges for Clinical Translation. Cancers 2020, 12, 2347. https://doi.org/10.3390/cancers12092347
Soubéran A, Tchoghandjian A. Practical Review on Preclinical Human 3D Glioblastoma Models: Advances and Challenges for Clinical Translation. Cancers. 2020; 12(9):2347. https://doi.org/10.3390/cancers12092347
Chicago/Turabian StyleSoubéran, Aurélie, and Aurélie Tchoghandjian. 2020. "Practical Review on Preclinical Human 3D Glioblastoma Models: Advances and Challenges for Clinical Translation" Cancers 12, no. 9: 2347. https://doi.org/10.3390/cancers12092347
APA StyleSoubéran, A., & Tchoghandjian, A. (2020). Practical Review on Preclinical Human 3D Glioblastoma Models: Advances and Challenges for Clinical Translation. Cancers, 12(9), 2347. https://doi.org/10.3390/cancers12092347