Intronic TP53 Polymorphisms Are Associated with Increased Δ133TP53 Transcript, Immune Infiltration and Cancer Risk

 , , , ,
, , , ,  , , , and
, , , and
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Intronic SNPs rs9895829 and rs2909430 Are Associated with Increased Cancer Risk
2.2. Association of TP53 Polymorphisms with Patient Survival
2.3. Association of TP53 Polymorphisms with Clinical Parameters of Cancer Patients
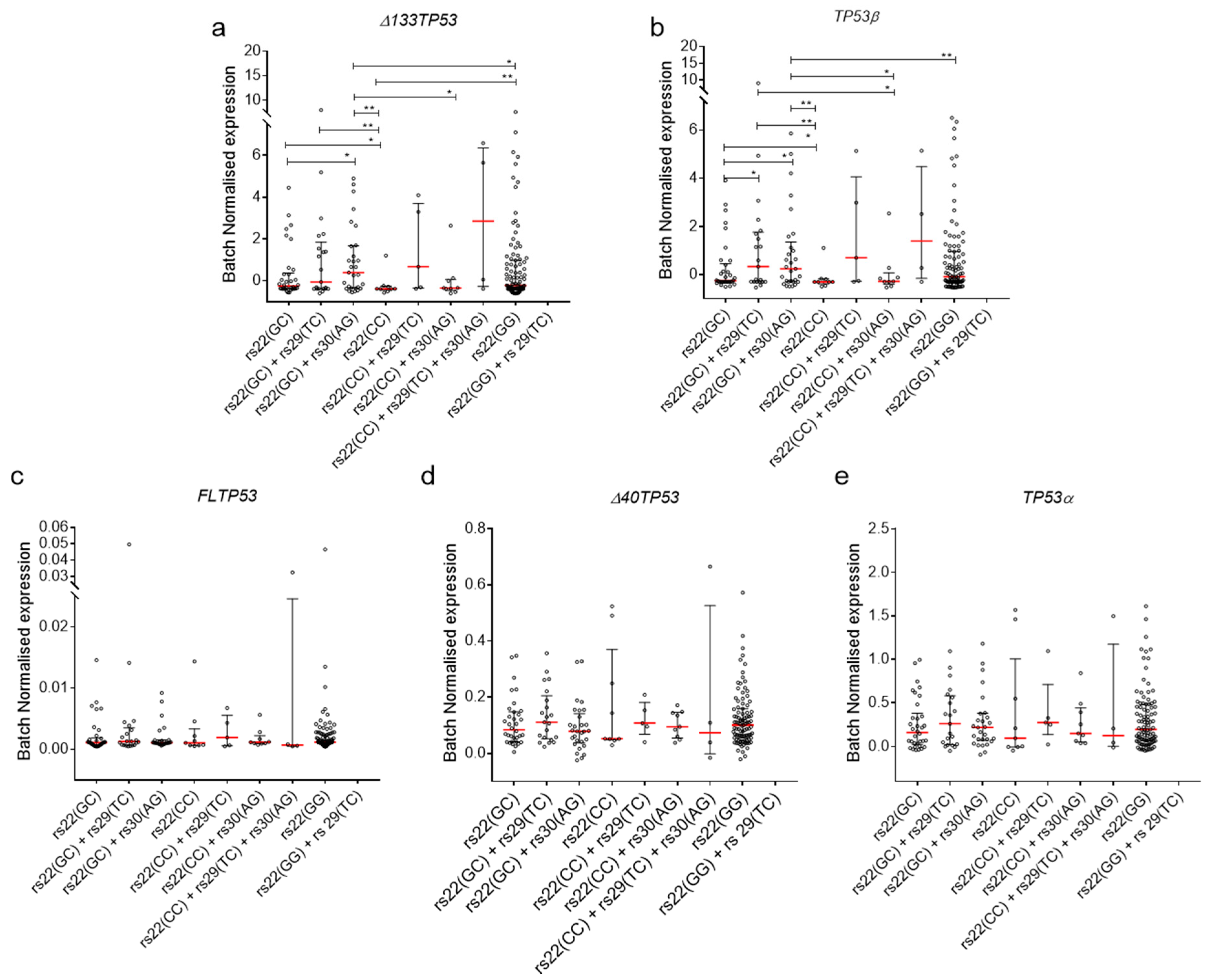
2.4. Association of TP53 Polymorphisms with Δ133TP53 and TP53β Isoform Expression in Cancers
2.5. Association of TP53 Polymorphisms with Δ133TP53 and TP53β Isoform Expression in Normal Cells
3. Discussion
4. Materials and Methods
4.1. Control Cohorts
4.2. Cancer Cohort
4.3. Cell Culture
4.4. Cloning Experiments
4.5. Genotyping
4.6. Preparation of RNA, cDNA and ddPCR for Analysis of p53 Isoforms
4.7. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B. Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 173, 371–385, e318. [Google Scholar] [CrossRef] [PubMed]
- Wasylishen, A.R.; Lozano, G. Attenuating the p53 pathway in human cancers: Many means to the same end. Cold Spring Harb. Perspect. Med. 2016, 6, a026211. [Google Scholar] [CrossRef] [PubMed]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef]
- Bourdon, J.-C. P53 and its isoforms in cancer. Br. J. Cancer 2007, 97, 277–282. [Google Scholar] [CrossRef]
- Bourdon, J.-C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. P53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef]
- Leroy, B.; Ballinger, M.L.; Baran-Marszak, F.; Bond, G.L.; Braithwaite, A.; Concin, N.; Donehower, L.A.; El-Deiry, W.S.; Fenaux, P.; Gaidano, G. Recommended guidelines for validation, quality control, and reporting of tp53 variants in clinical practice. Cancer Res. 2017, 77, 1250–1260. [Google Scholar] [CrossRef]
- Machado-Silva, A.; Perrier, S.; Bourdon, J.-C. p53 family members in cancer diagnosis and treatment. Semin. Cancer Biol. 2010, 20, 57–62. [Google Scholar] [CrossRef]
- Whibley, C.; Pharoah, P.D.; Hollstein, M. P53 polymorphisms: Cancer implications. Nat. Rev. Cancer 2009, 9, 95–107. [Google Scholar] [CrossRef]
- Zhu, F.; Dollé, M.E.; Berton, T.R.; Kuiper, R.V.; Capps, C.; Espejo, A.; McArthur, M.J.; Bedford, M.T.; Van Steeg, H.; De Vries, A. Mouse models for the p53 r72p polymorphism mimic human phenotypes. Cancer Res. 2010, 70, 5851–5859. [Google Scholar] [CrossRef]
- Thomas, M.; Kalita, A.; Labrecque, S.; Pim, D.; Banks, L.; Matlashewski, G. Two polymorphic variants of wild-type p53 differ biochemically and biologically. Mol. Cell. Biol. 1999, 19, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Dumont, P.; Leu, J.-J.; Della Pietra, A.C.; George, D.L.; Murphy, M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat. Genet. 2003, 33, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Banks, L. P53 polymorphic variants at codon 72 exert different effects on cell cycle progression. Int. J. Cancer 2004, 108, 196–199. [Google Scholar] [CrossRef]
- Siddique, M.; Sabapathy, K. Trp53-dependent DNA-repair is affected by the codon 72 polymorphism. Oncogene 2006, 25, 3489–3500. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Huang, R.; Pei, C.; Jia, X.; Jiang, C.; Ren, H. Tp53 codon 72 polymorphism and glioma risk: A meta-analysis. Oncol. Lett. 2012, 3, 599–606. [Google Scholar] [CrossRef]
- Jiang, P.; Liu, J.; Zeng, X.; Li, W.; Tang, J. Association of tp53 codon 72 polymorphism with cervical cancer risk in chinese women. Cancer Genet. Cytogenet. 2010, 197, 174–178. [Google Scholar] [CrossRef]
- Oliveira, S.; Sousa, H.; Santos, A.; Pinto, D.; Pinto-Correia, A.; Fontoura, D.; Moutinho, J.; Medeiros, R. The p53 r72p polymorphism does not influence cervical cancer development in a portuguese population: A study in exfoliated cervical cells. J. Med Virol. 2008, 80, 424–429. [Google Scholar] [CrossRef]
- Matakidou, A.; Eisen, T.; Houlston, R. Tp53 polymorphisms and lung cancer risk: A systematic review and meta-analysis. Mutagenesis 2003, 18, 377–385. [Google Scholar] [CrossRef]
- Ricks-Santi, L.; Mason, T.; Apprey, V.; Ahaghotu, C.; McLauchlin, A.; Josey, D.; Bonney, G.; Dunston, G. P53 pro72arg polymorphism and prostate cancer in men of african descent. Prostate 2010, 70, 1739–1745. [Google Scholar] [CrossRef]
- Biroš, E.; Kalina, I.; Salagovic, J.; Habalova, V.; ák Hriv, M.; Valanský, L. P53 single nucleotide polymorphisms and bladder cancer. Neoplasma 2000, 47, 303–306. [Google Scholar]
- Zhou, Y.; Li, N.; Zhuang, W.; Liu, G.J.; Wu, T.X.; Yao, X.; Du, L.; Wei, M.L.; Wu, X.T. P53 codon 72 polymorphism and gastric cancer: A meta-analysis of the literature. Int. J. Cancer 2007, 121, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, M.; Wu, D.; Wang, M.; Tong, N.; Tian, Y.; Zhang, Z. P53 codon 72 polymorphism contributes to breast cancer risk: A meta-analysis based on 39 case–control studies. Breast Cancer Res. Treat. 2010, 120, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Dahabreh, I.J.; Linardou, H.; Bouzika, P.; Varvarigou, V.; Murray, S. Tp53 arg72pro polymorphism and colorectal cancer risk: A systematic review and meta-analysis. Cancer Epidemiol. Biomarkers Prev. 2010, 19, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Marin, M.C.; Jost, C.A.; Brooks, L.A.; Irwin, M.S.; O’Nions, J.; Tidy, J.A.; James, N.; McGregor, J.M.; Harwood, C.A.; Yulug, I.G. A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nat. Genet. 2000, 25, 47–54. [Google Scholar] [CrossRef]
- Bergamaschi, D.; Gasco, M.; Hiller, L.; Sullivan, A.; Syed, N.; Trigiante, G.; Yulug, I.; Merlano, M.; Numico, G.; Comino, A. P53 polymorphism influences response in cancer chemotherapy via modulation of p73-dependent apoptosis. Cancer Cell 2003, 3, 387–402. [Google Scholar] [CrossRef]
- Langerød, A.; Bukholm, I.R.; Bregård, A.; Lønning, P.E.; Andersen, T.I.; Rognum, T.O.; Meling, G.I.; Lothe, R.A.; Børresen-Dale, A.-L. The tp53 codon 72 polymorphism may affect the function of tp53 mutations in breast carcinomas but not in colorectal carcinomas. Cancer Epidemiol. Biomarkers Prev. 2002, 11, 1684–1688. [Google Scholar]
- Burgdorf, K.S.; Grarup, N.; Justesen, J.M.; Harder, M.N.; Witte, D.R.; Jørgensen, T.; Sandbæk, A.; Lauritzen, T.; Madsbad, S.; Hansen, T. Studies of the association of arg72pro of tumor suppressor protein p53 with type 2 diabetes in a combined analysis of 55,521 europeans. PLoS ONE 2011, 6, e15813. [Google Scholar] [CrossRef]
- Bonfigli, A.R.; Sirolla, C.; Testa, R.; Cucchi, M.; Spazzafumo, L.; Salvioli, S.; Ceriello, A.; Olivieri, F.; Festa, R.; Procopio, A.D. The p53 codon 72 (arg72pro) polymorphism is associated with the degree of insulin resistance in type 2 diabetic subjects: A cross-sectional study. Acta Diabetol. 2013, 50, 429–436. [Google Scholar] [CrossRef]
- Assmann, G.; Voswinkel, J.; Mueller, M.; Bittenbring, J.; Koenig, J.; Menzel, A.; Pfreundschuh, M.; Roemer, K.; Melchers, I. Association of rheumatoid arthritis with mdm2 snp309 and genetic evidence for an allele-specific interaction between mdm2 and p53 p72r variants: A case control study. Clin. Exp. Rheumatol. 2009, 27, 615–619. [Google Scholar]
- Abderrahmane, R.; Moghtit, F.Z.; Boughrara, W.; Boubkeur, A.; Bnseddik, K.; Boudjema, A.; Benrrahal, F.; Aberkane, M.; Fodil, M.; Saidi-Mehtar, N. Association of mdm2 snp309 and tp53 arg72pro polymorphisms with risk of ra in the algerian population. J. Med. Sci. Clin. Res. 2014, 2, 1714–1723. [Google Scholar]
- Vietri, M.T.; Riegler, G.; Ursillo, A.; Caserta, L.; Cioffi, M.; Molinari, A.M. P53 codon 72 polymorphism in patients affected with ulcerative colitis. J. Gastroenterol. 2007, 42, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Vijayakumar, V.; Fernández-Cuesta, L.; Hafsi, H.; Sagne, C.; Hautefeuille, A.; Olivier, M.; Hainaut, P. P53 regulates the transcription of its delta133p53 isoform through specific response elements contained within the tp53 p2 internal promoter. Oncogene 2010, 29, 2691–2700. [Google Scholar] [CrossRef] [PubMed]
- Mechanic, L.E.; Bowman, E.D.; Welsh, J.A.; Khan, M.A.; Hagiwara, N.; Enewold, L.; Shields, P.G.; Burdette, L.; Chanock, S.; Harris, C.C. Common genetic variation in tp53 is associated with lung cancer risk and prognosis in african americans and somatic mutations in lung tumors. Cancer Epidemiol. Biomarkers Prev. 2007, 16, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Bellini, I.; Pitto, L.; Marini, M.G.; Porcu, L.; Moi, P.; Garritano, S.; Boldrini, L.; Rainaldi, G.; Fontanini, G.; Chiarugi, M.; et al. Deltan133p53 expression levels in relation to haplotypes of the tp53 internal promoter region. Hum. Mutat. 2010, 31, 456–465. [Google Scholar] [CrossRef]
- Kazantseva, M.; Mehta, S.; Eiholzer, R.A.; Hung, N.; Wiles, A.; Slatter, T.L.; Braithwaite, A.W. A mouse model of the δ133p53 isoform: Roles in cancer progression and inflammation. Mamm. Genome 2018, 29, 831–842. [Google Scholar] [CrossRef]
- Gadea, G.; Arsic, N.; Fernandes, K.; Diot, A.; Joruiz, S.M.; Abdallah, S.; Meuray, V.; Vinot, S.; Anguille, C.; Remenyi, J. Tp53 drives invasion through expression of its δ133p53β variant. Elife 2016, 5, e14734. [Google Scholar] [CrossRef]
- Avery-Kiejda, K.A.; Morten, B.; Wong-Brown, M.W.; Mathe, A.; Scott, R.J. The relative mrna expression of p53 isoforms in breast cancer is associated with clinical features and outcome. Carcinogenesis 2014, 35, 586–596. [Google Scholar] [CrossRef]
- Kazantseva, M.; Mehta, S.; Eiholzer, R.A.; Gimenez, G.; Bowie, S.; Campbell, H.; Reily-Bell, A.L.; Roth, I.; Ray, S.; Drummond, C.J.; et al. The δ133p53β isoform promotes an immunosuppressive environment leading to aggressive prostate cancer. Cell Death Dis. 2019, 10, 631. [Google Scholar] [CrossRef]
- Campbell, H.; Fleming, N.; Roth, I.; Mehta, S.; Wiles, A.; Williams, G.; Vennin, C.; Arsic, N.; Parkin, A.; Pajic, M.; et al. ∆133p53 isoform promotes tumour invasion and metastasis via interleukin-6 activation of jak-stat and rhoa-rock signalling. Nat. Commun. 2018, 9, 254. [Google Scholar] [CrossRef]
- Aoubala, M.; Murray-Zmijewski, F.; Khoury, M.; Fernandes, K.; Perrier, S.; Bernard, H.; Prats, A.; Lane, D.; Bourdon, J. P53 directly transactivates d133p53a, regulating cell fate outcome in response to DNA damage. Cell Death Differ. 2010, 21, 7. [Google Scholar]
- Consortium, G.P. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Hancox, R.J.; Poulton, R.; Welch, D.; Olova, N.; McLachlan, C.R.; Greene, J.M.; Sears, M.R.; Caspi, A.; Moffitt, T.E.; Robertson, S.P. Accelerated decline in lung function in cigarette smokers is associated with tp53/mdm2 polymorphisms. Hum. Genet. 2009, 126, 559. [Google Scholar] [CrossRef]
- Chen, Y.J.; Hakin-Smith, V.; Teo, M.; Xinarianos, G.E.; Jellinek, D.A.; Carroll, T.; McDowell, D.; MacFarlane, M.R.; Boet, R.; Baguley, B.C.; et al. Association of mutant tp53 with alternative lengthening of telomeres and favorable prognosis in glioma. Cancer Res. 2006, 66, 6473–6476. [Google Scholar] [CrossRef] [PubMed]
- Hakin-Smith, V.; Jellinek, D.; Levy, D.; Carroll, T.; Teo, M.; Timperley, W.; McKay, M.; Reddel, R.; Royds, J. Alternative lengthening of telomeres and survival in patients with glioblastoma multiforme. Lancet 2003, 361, 836–838. [Google Scholar] [CrossRef]
- Hung, N.A.; Eiholzer, R.A.; Kirs, S.; Zhou, J.; Ward-Hartstonge, K.; Wiles, A.K.; Frampton, C.M.; Taha, A.; Royds, J.A.; Slatter, T.L. Telomere profiles and tumor-associated macrophages with different immune signatures affect prognosis in glioblastoma. Mod. Pathol. 2016, 29, 212–226. [Google Scholar] [CrossRef]
- Zhou, J.; Reddy, M.V.; Wilson, B.K.J.; Blair, D.A.; Taha, A.; Frampton, C.M.; Eiholzer, R.A.; Gan, P.Y.C.; Ziad, F.; Thotathil, Z.; et al. Mr imaging characteristics associate with tumor-associated macrophages in glioblastoma and provide an improved signature for survival prognostication. Ajnr. Am. J. Neuroradiol. 2018, 39, 252–259. [Google Scholar] [CrossRef]
- Bryant, G.; Wang, L.; Mulholland, D.J. Overcoming oncogenic mediated tumor immunity in prostate cancer. Int. J. Mol. Sci. 2017, 18, 1542. [Google Scholar] [CrossRef]
- Ebelt, K.; Babaryka, G.; Frankenberger, B.; Stief, C.G.; Eisenmenger, W.; Kirchner, T.; Schendel, D.J.; Noessner, E. Prostate cancer lesions are surrounded by foxp3+, pd-1+ and b7-h1+ lymphocyte clusters. Eur. J. Cancer 2009, 45, 1664–1672. [Google Scholar] [CrossRef]
- Kazantseva, M.; Eiholzer, R.A.; Mehta, S.; Taha, A.; Bowie, S.; Roth, I.; Zhou, J.; Joruiz, S.M.; Royds, J.A.; Hung, N.A. Elevation of the tp53 isoform δ133p53β in glioblastomas: An alternative to mutant p53 in promoting tumor development. J. Pathol. 2018, 246, 77–88. [Google Scholar] [CrossRef]
- Mathews, D.H. Rna secondary structure analysis using rnastructure. Curr. Protoc. Bioinform. 2014, 46, 12–16. [Google Scholar] [CrossRef]
- Tsunoda, T.; Takagi, T. Estimating transcription factor bindability on DNA. Bioinformatics 1999, 15, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Saldi, T.; Cortazar, M.A.; Sheridan, R.M.; Bentley, D.L. Coupling of rna polymerase ii transcription elongation with pre-mrna splicing. J. Mol. Biol. 2016, 428, 2623–2635. [Google Scholar] [CrossRef] [PubMed]
- Mehta-Mujoo, P.; Cunliffe, H.; Hung, N.A.; Slatter, T.L. Long non-coding rna anril in the nucleus associates with periostin expression in breast cancer. Front. Oncol. 2019, 9, 885. [Google Scholar] [CrossRef] [PubMed]
- Royds, J.A.; Pilbrow, A.P.; Ahn, A.; Morrin, H.R.; Frampton, C.; Russell, I.A.; Moravec, C.S.; Sweet, W.E.; Tang, W.; Currie, M.J. The rs11515 polymorphism is more frequent and associated with aggressive breast tumors with increased anril and decreased p16ink4a expression. Front. Oncol. 2016, 5, 306. [Google Scholar] [CrossRef]
- Kazantseva, M.; Hung, N.A.; Mehta, S.; Roth, I.; Eiholzer, R.; Rich, A.M.; Seo, B.; Baird, M.A.; Braithwaite, A.W.; Slatter, T.L. Tumor protein 53 mutations are enriched in diffuse large b-cell lymphoma with irregular cd19 marker expression. Sci. Rep. 2017, 7, 1566. [Google Scholar] [CrossRef]
- Lasham, A.; Tsai, P.; Fitzgerald, S.J.; Mehta, S.Y.; Knowlton, N.S.; Braithwaite, A.W.; Print, C.G. Accessing a new dimension in tp53 biology: Multiplex long amplicon digital pcr to specifically detect and quantitate individual tp53 transcripts. Cancers 2020, 12, 769. [Google Scholar] [CrossRef]
- Mehta, S.; Tsai, P.; Lasham, A.; Campbell, H.; Reddel, R.; Braithwaite, A.; Print, C. A study of tp53 rna splicing illustrates pitfalls of rna-seq methodology. Cancer Res. 2016, 76, 7151–7159. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype (rs1042522) | Study Cohort-1 | Control Cohort 1 | χ2-Test p-Value | Control Cohort 2 | χ2-Test p-Value | Control Cohort 3 | χ2-Test p-Value |
| GG (REF) | 326 | 302 | 0.27 | 193 | 0.4223 | 373 | |
| CG/GC | 226 | 253 | 122 | 247 | 0.79 | ||
| CC (MA) | 48 | 44 | 20 | 48 | |||
| Genotype (rs9895829) | Study Cohort-1 | Control Cohort 1 | χ2-test p-value | Control Cohort 2 | χ2-test p-value | Control Cohort 3 | χ2-test p-value |
| TT (REF) | 291 | 535 | <0.0001 (****) | 304 | <0.0001 (****) | ||
| TC/CT | 302 | 62 | 21 | NA | |||
| CC (MA) | 7 | 2 | 0 | ||||
| Genotype (rs2909430) | Study Cohort-1 | Control Cohort 1 | χ2-test p-value | Control Cohort 2 | χ2-test p-value | Control Cohort 3 | χ2-test p-value |
| AA (REF) | 227 | 441 | <0.0001 (****) | 254 | <0.0001 (****) | ||
| AG/GA | 370 | 150 | 68 | NA | |||
| GG (MA) | 3 | 8 | 5 | ||||
| Genotype | Control Cohort 1 | Study Cohort | OR (95% CI), Fisher’s Exact Test p-Value | ||
|---|---|---|---|---|---|
| rs22(CC)+rs30(AG) | rs22(CC)+rs30(GG) | rs22(CC)+rs30(AA) | |||
| rs22(CC)+rs30(AA) | 17 | 18 | 1.34 (0.53–3.13), 0.82 | ||
| rs22(CC)+rs30(AG) | 19 | 27 | 0.26 (0.07–1.1), 0.09 | ||
| rs22(CC)+rs30(GG) | 8 | 3 | 0.35 (0.09–1.6), 0.18 | ||
| rs22(GG)+rs30(AG) | rs22(GG)+rs30(GG) | rs22(GG)+rs30(AA) | |||
| rs22(GG)+rs30(AA) | 301 | 144 | 380.4 (66.24–3826), 0.0001 | ||
| rs22(GG)+rs30(AG) | 1 | 182 | ND | ||
| rs22(GG)+rs30(GG) | 0 | 0 | ND | ||
| rs22(GC)+rs30(AG) | rs22(GC)+rs30(GG) | rs22(GC)+rs30(AA) | |||
| rs22(GC)+rs30(AA) | 123 | 65 | 2.34 (1.6–3.4), 0.0001 | ||
| rs22(GC)+rs30(AG) | 130 | 161 | ND | ||
| rs22(GC)+rs30(GG) | 0 | 0 | ND | ||
| rs22(CC)+rs29(TC) | rs22(CC)+rs29(CC) | rs22(CC)+rs29(TT) | |||
| rs22(CC)+rs29(TT) | 31 | 25 | 2.3 (0.95–5.9), 0.07 | ||
| rs22(CC)+rs29(TC) | 11 | 21 | 0.54(0.075–3.76), 0.6 | ||
| rs22(CC)+rs29(CC) | 2 | 2 | 1.24 (0.18–8.3), 0.99 | ||
| rs22(GG)+rs29(TC) | rs22(GG)+rs29(CC) | rs22(GG)+rs29(TT) | |||
| rs22(GG)+rs29(TT) | 302 | 172 | ∞ (75.89–∞), 0.0001 | ||
| rs22(GG)+rs29(TC) | 0 | 154 | ND | ||
| rs22(GG)+rs29(CC) | 0 | 0 | ND | ||
| rs22(GC)+rs29(TC) | rs22(GC)+rs29(CC) | rs22(GC)+rs29(TT) | |||
| rs22(GC)+rs29(TT) | 202 | 94 | 5.35 (3.53–8.01), 0.0001 | ||
| rs22(GC)+rs29(TC) | 51 | 127 | ∞ (0.57–∞), 0.32 | ||
| rs22(GC)+rs29(CC) | 0 | 5 | ∞ (3.1–∞), 0.0036 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eiholzer, R.A.; Mehta, S.; Kazantseva, M.; Drummond, C.J.; McKinney, C.; Young, K.; Slater, D.; Morten, B.C.; Avery-Kiejda, K.A.; Lasham, A.; et al. Intronic TP53 Polymorphisms Are Associated with Increased Δ133TP53 Transcript, Immune Infiltration and Cancer Risk. Cancers 2020, 12, 2472. https://doi.org/10.3390/cancers12092472
Eiholzer RA, Mehta S, Kazantseva M, Drummond CJ, McKinney C, Young K, Slater D, Morten BC, Avery-Kiejda KA, Lasham A, et al. Intronic TP53 Polymorphisms Are Associated with Increased Δ133TP53 Transcript, Immune Infiltration and Cancer Risk. Cancers. 2020; 12(9):2472. https://doi.org/10.3390/cancers12092472
Chicago/Turabian StyleEiholzer, Ramona A., Sunali Mehta, Marina Kazantseva, Catherine J. Drummond, Cushla McKinney, Katie Young, David Slater, Brianna C. Morten, Kelly A. Avery-Kiejda, Annette Lasham, and et al. 2020. "Intronic TP53 Polymorphisms Are Associated with Increased Δ133TP53 Transcript, Immune Infiltration and Cancer Risk" Cancers 12, no. 9: 2472. https://doi.org/10.3390/cancers12092472
APA StyleEiholzer, R. A., Mehta, S., Kazantseva, M., Drummond, C. J., McKinney, C., Young, K., Slater, D., Morten, B. C., Avery-Kiejda, K. A., Lasham, A., Fleming, N., Morrin, H. R., Reader, K., Royds, J. A., Landmann, M., Petrich, S., Reddel, R., Huschtscha, L., Taha, A., ... Braithwaite, A. W. (2020). Intronic TP53 Polymorphisms Are Associated with Increased Δ133TP53 Transcript, Immune Infiltration and Cancer Risk. Cancers, 12(9), 2472. https://doi.org/10.3390/cancers12092472