Sustained Inflammatory Signalling through Stat1/Stat2/IRF9 Is Associated with Amoeboid Phenotype of Melanoma Cells

 , , , and
, , , and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Data Analysis Reveals Upregulation of Inflammation-Associated Genes after MAT
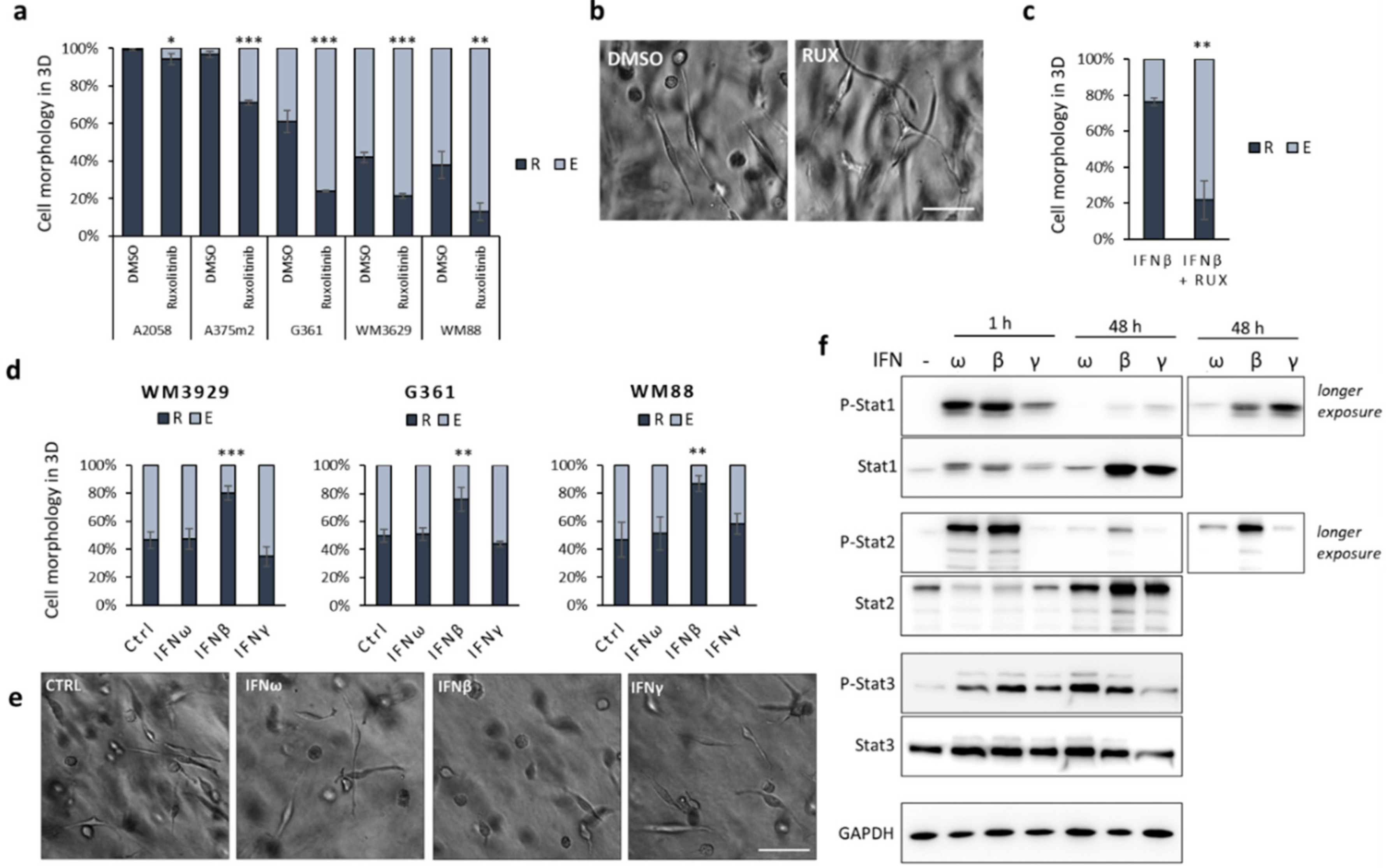
2.2. Inflammation-Associated Signalling Affects Invasion Plasticity in Melanoma Models
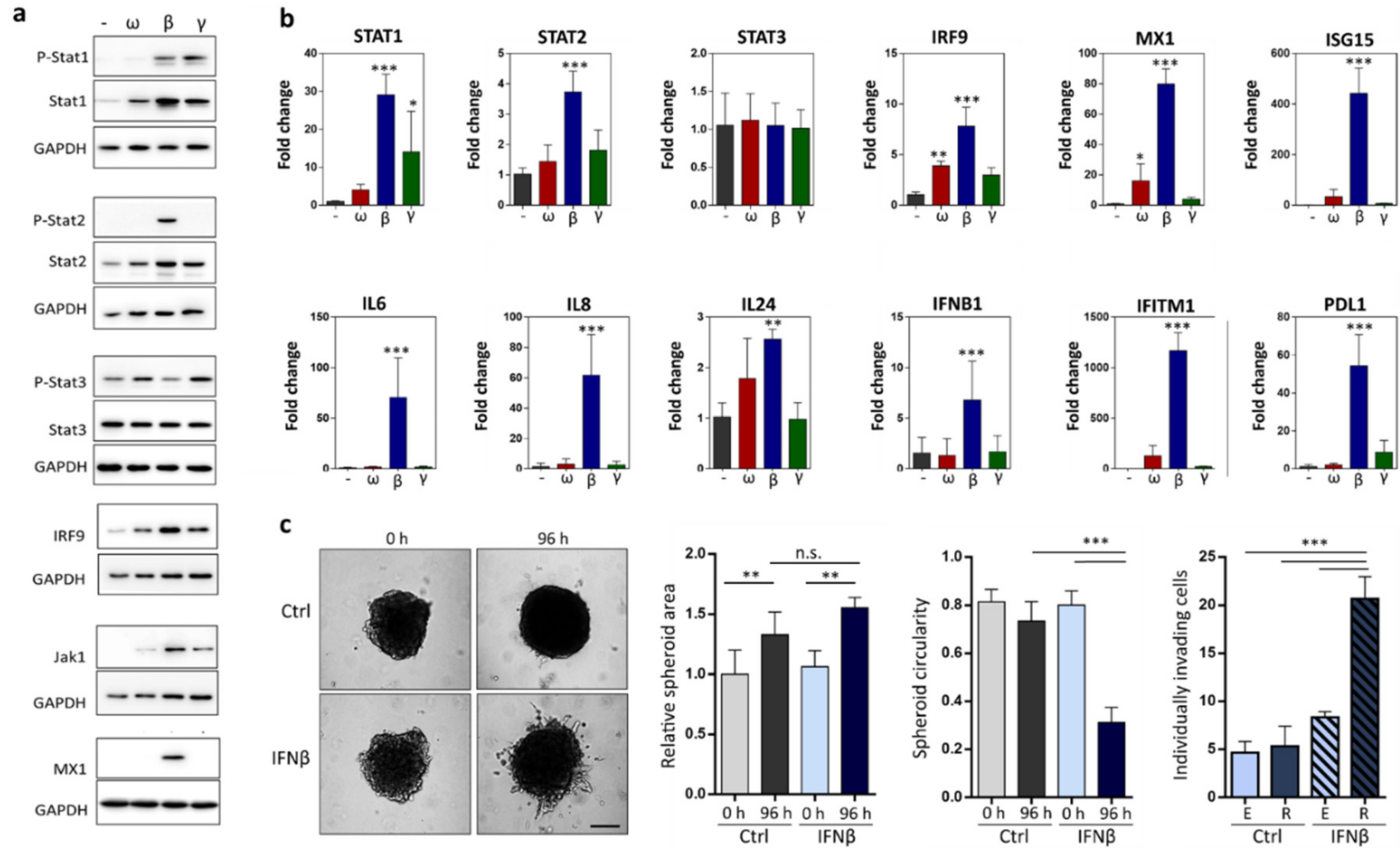
2.3. IFNβ Treated Cells Upregulate Expression of Pro-Invasive Cytokines and Increase Individual Invasion
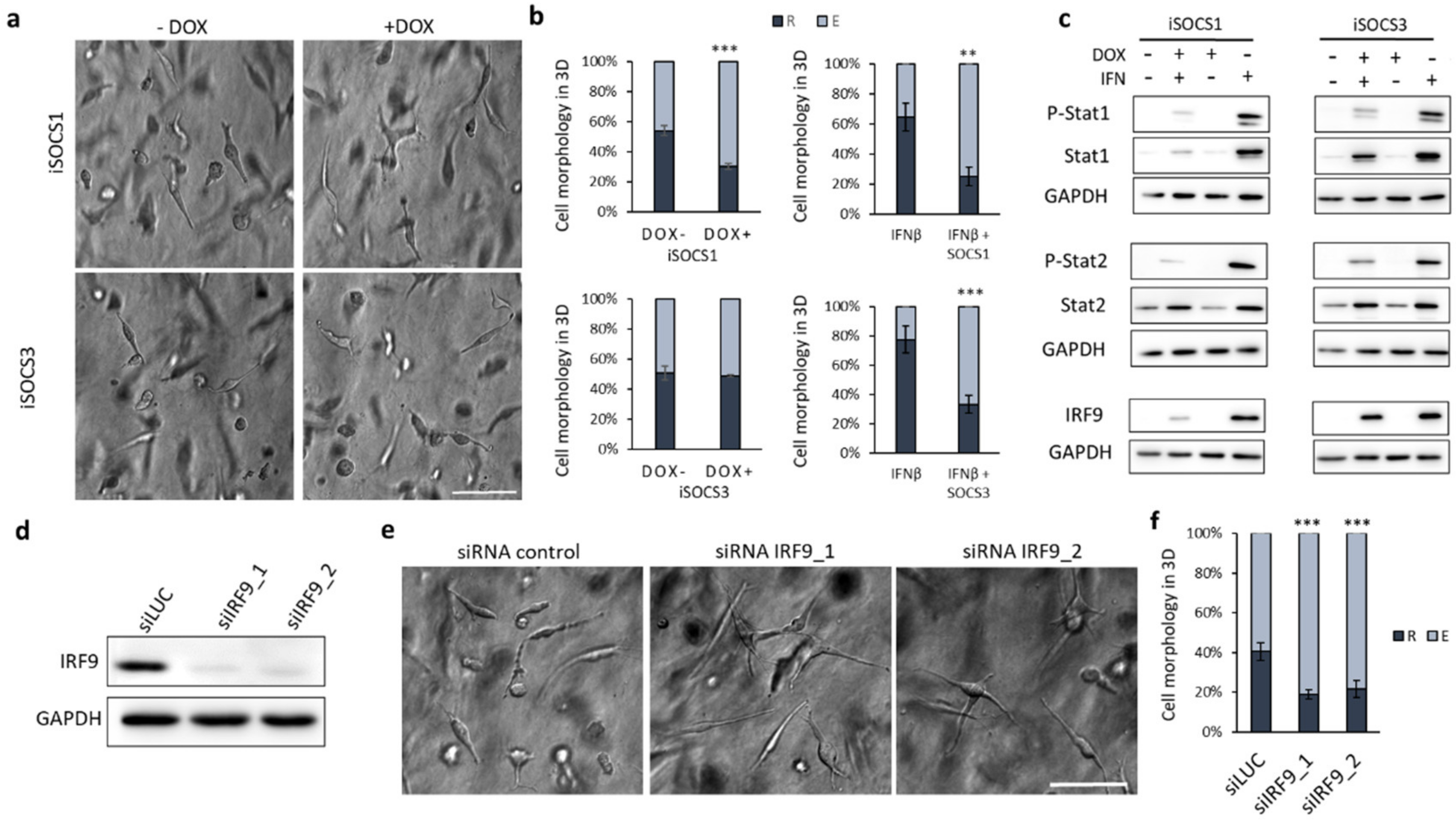
2.4. Suppression of IFN Signalling by SOCS1 Expression or IRF9 Knockdown Promotes the Mesenchymal Phenotype
3. Discussion
4. Materials and Methods
4.1. Data Analysis
4.2. Cell Lines, Constructs, and Transfection
4.3. Three-Dimensional Cell Culture and Morphology Analysis
4.4. Immunoblotting
4.5. Reverse Transcription–Quantitative Polymerase Chain Reaction (RT-qPCR)
4.6. Three-Dimensional Invasion Spheroid Assay
4.7. Proliferation Assay in 3D Collagen
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gandalovičová, A.; Rosel, D.; Fernandes, M.; Veselý, P.; Heneberg, P.; Čermák, V.; Petruželka, L.; Kumar, S.; Sanz-Moreno, V.; Brábek, J. Migrastatics—Anti-metastatic and Anti-invasion Drugs: Promises and Challenges. Trends Cancer 2017, 3, 391–406. [Google Scholar] [CrossRef] [Green Version]
- Rosel, D.; Fernandes, M.; Sanz-Moreno, V.; Brábek, J. Migrastatics: Redirecting R&D in Solid Cancer towards Metastasis? Trends Cancer 2019, 5, 755–756. [Google Scholar] [CrossRef]
- Odenthal, J.; Takes, R.; Friedl, P. Plasticity of tumor cell invasion: Governance by growth factors and cytokines. Carcinogenesis 2016. [Google Scholar] [CrossRef] [Green Version]
- Roizen, M. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Brábek, J.; Mierke, C.T.; Rosel, D.; Veselý, P.; Fabry, B. The role of the tissue microenvironment in the regulation of cancer cell motility and invasion. Cell Commun. Signal. 2010, 8, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parekh, A.; Weaver, A.M. Regulation of cancer invasiveness by the physical extracellular matrix environment. Cell Adhes. Migr. 2009, 3, 288–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedl, P.; Alexander, S. Cancer Invasion and the Microenvironment: Plasticity and Reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pánková, K.; Rosel, D.; Novotný, M.; Brábek, J. The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell. Mol. Life Sci. 2009, 67, 63–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedl, P.; Locker, J.; Sahai, E.; Segall, J.E. Classifying collective cancer cell invasion. Nat. Cell Biol. 2012, 14, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Tolde, O.; Gandalovičová, A.; Křížová, A.; Veselý, P.; Chmelík, R.; Rosel, D.; Brábek, J. Quantitative phase imaging unravels new insight into dynamics of mesenchymal and amoeboid cancer cell invasion. Sci. Rep. 2018, 8, 12020. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Wolf, K. Proteolytic interstitial cell migration: A five-step process. Cancer Metastasis Rev. 2009, 28, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyckoff, J.B.; Pinner, S.E.; Gschmeissner, S.; Condeelis, J.S.; Sahai, E. ROCK- and Myosin-Dependent Matrix Deformation Enables Protease-Independent Tumor-Cell Invasion In Vivo. Curr. Biol. 2006, 16, 1515–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lämmermann, T.; Sixt, M. Mechanical modes of ‘amoeboid’ cell migration. Curr. Opin. Cell Biol. 2009, 21, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Pandya, P.; Orgaz, J.L.; Sanz-Moreno, V. Modes of invasion during tumour dissemination. Mol. Oncol. 2016, 11, 5–27. [Google Scholar] [CrossRef] [Green Version]
- Gandalovičová, A.; Vomastek, T.; Rosel, D.; Brábek, J. Cell polarity signaling in the plasticity of cancer cell invasiveness. Oncotarget 2016, 7, 25022–25049. [Google Scholar] [CrossRef]
- Čermák, V.; Gandalovičová, A.; Merta, L.; Harant, K.; Rösel, D.; Brábek, J. High-throughput transcriptomic and proteomic profiling of mesenchymal-amoeboid transition in 3D collagen. Sci. Data 2020, 7, 1–11. [Google Scholar] [CrossRef]
- Boekhorst, V.T.; Friedl, P. Plasticity of Cancer Cell Invasion—Mechanisms and Implications for Therapy. Adv. Cancer Res. 2016, 132, 209–264. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gaggioli, C.; Yeo, M.; Albrengues, J.; Wallberg, F.; Virós, A.; Hooper, S.; Mitter, R.; Féral, C.C.; Cook, M.; et al. ROCK and JAK1 Signaling Cooperate to Control Actomyosin Contractility in Tumor Cells and Stroma. Cancer Cell 2011, 20, 229–245. [Google Scholar] [CrossRef] [Green Version]
- Georgouli, M.; Herraiz, C.; Crosas-Molist, E.; Fanshawe, B.; Maiques, O.; Perdrix, A.; Pandya, P.; Rodriguez-Hernandez, I.; Ilieva, K.M.; Cantelli, G.; et al. Regional Activation of Myosin II in Cancer Cells Drives Tumor Progression via a Secretory Cross-Talk with the Immune Microenvironment. Cell 2019, 176, 757–774. [Google Scholar] [CrossRef]
- Hölzel, M.; Tüting, T. Inflammation-Induced Plasticity in Melanoma Therapy and Metastasis. Trends Immunol. 2016, 37, 364–374. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2013, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Einav, U.; Tabach, Y.; Getz, G.; Yitzhaky, A.; Ozbek, U.; Amariglio, N.; Izraeli, S.; Rechavi, G.; Domany, E. Gene expression analysis reveals a strong signature of an interferon-induced pathway in childhood lymphoblastic leukemia as well as in breast and ovarian cancer. Oncogene 2005, 24, 6367–6375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichselbaum, R.R.; Ishwaran, H.; Yoon, T.; Nuyten, D.S.A.; Baker, S.W.; Khodarev, N.; Su, A.W.; Shaikh, A.Y.; Roach, P.; Kreike, B.; et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 18490–18495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyagi, S.; Hata, H.; Homma, E.; Shimizu, H. Sequential Local Injection of Low-Dose Interferon-Beta for Maintenance Therapy in Stage II and III Melanoma: A Single-Institution Matched Case-Control Study. Oncology 2012, 82, 139–146. [Google Scholar] [CrossRef]
- Fujimura, T.; Okuyama, R.; Ohtani, T.; Ito, Y.; Haga, T.; Hashimoto, A.; Aiba, S. Perilesional treatment of metastatic melanoma with interferon-β. Clin. Exp. Dermatol. 2009, 34, 793–799. [Google Scholar] [CrossRef]
- Uehara, J.; Ohkuri, T.; Kosaka, A.; Ishibashi, K.; Hirata, Y.; Ohara, K.; Nagato, T.; Oikawa, K.; Aoki, N.; Harabuchi, Y.; et al. Intratumoral injection of IFN-β induces chemokine production in melanoma and augments the therapeutic efficacy of anti-PD-L1 mAb. Biochem. Biophys. Res. Commun. 2017, 490, 521–527. [Google Scholar] [CrossRef] [Green Version]
- Kakizaki, A.; Fujimura, T.; Furudate, S.; Kambayashi, Y.; Yamauchi, T.; Yagita, H.; Aiba, S. Immunomodulatory effect of peritumorally administered interferon-beta on melanoma through tumor-associated macrophages. OncoImmunology 2015, 4, e1047584. [Google Scholar] [CrossRef]
- Čermák, V. Differential Expression Analyses Figshare Dataset. 2019. Available online: https://figshare.com/articles/Differential_expression_analyses/10329281/2 (accessed on 25 August 2020).
- Ge, S.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2019, 36, 2628–2629. [Google Scholar] [CrossRef]
- Cooper, S.; Sadok, A.; Bousgouni, V.; Bakal, C. Apolar and polar transitions drive the conversion between amoeboid and mesenchymal shapes in melanoma cells. Mol. Biol. Cell 2015, 26, 4163–4170. [Google Scholar] [CrossRef]
- Arozarena, I.; Wellbrock, C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazar-Molnar, E.; Hegyesi, H.; Tóth, S.; Falus, A. Autocrine and Paracrine Regulation by Cytokines and Growth Factors in Melanoma. Cytokine 2000, 12, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Michalska, A.; Blaszczyk, K.; Wesoly, J.; Bluyssen, H.A. A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and Type II IFN Responses. Front. Immunol. 2018, 9, 1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medrano, R.F.; Hunger, A.; Mendonça, S.A.; Barbuto, J.A.M.; Strauss, B.E. Immunomodulatory and antitumor effects of type I interferons and their application in cancer therapy. Oncotarget 2017, 8, 71249–71284. [Google Scholar] [CrossRef] [Green Version]
- Satomi, H.; Wang, B.; Fujisawa, H.; Otsuka, F. Interferon-β from melanoma cells suppresses the proliferations of melanoma cells in an autocrine manner. Cytokine 2002, 18, 108–115. [Google Scholar] [CrossRef]
- Tjandra, S.S.; Hsu, C.; Goh, I.; Gurung, A.; Poon, R.; Nadesan, P.; Alman, B.A. IFN- Signaling Positively Regulates Tumorigenesis in Aggressive Fibromatosis, Potentially by Modulating Mesenchymal Progenitors. Cancer Res. 2007, 67, 7124–7131. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, Z. STAT1 in cancer: Friend or foe? Discov. Med. 2017, 24, 19–29. [Google Scholar]
- Greenwood, C.; Metodieva, G.; Al-Janabi, K.; Lausen, B.; Alldridge, L.; Leng, L.; Bucala, R.; Fernández, N.; Metodiev, M.V. Stat1 and CD74 overexpression is co-dependent and linked to increased invasion and lymph node metastasis in triple-negative breast cancer. J. Proteom. 2012, 75, 3031–3040. [Google Scholar] [CrossRef]
- Emad, A.; Ray, T.; Jensen, T.W.; Parat, M.; Natrajan, R.; Sinha, S.; Ray, P.S. Superior breast cancer metastasis risk stratification using an epithelial-mesenchymal-amoeboid transition gene signature. Breast Cancer Res. 2020, 22, 1–13. [Google Scholar] [CrossRef]
- Cheon, H.; Borden, E.C.; Stark, G.R. Interferons and Their Stimulated Genes in the Tumor Microenvironment. Semin. Oncol. 2014, 41, 156–173. [Google Scholar] [CrossRef] [Green Version]
- Iriarte, A.R.; Arwert, E.; Milford, E.; Chakravarty, P.; Melcher, A.; Harrington, K.; Sahai, E. Interaction between cancer associated fibroblasts and cancer cells influence immune infiltrate and is modulated by therapeutic agents. Ann. Oncol. 2018, 29, viii657. [Google Scholar] [CrossRef]
- Lehmann, S.; Boekhorst, V.T.; Odenthal, J.; Bianchi, R.; Van Helvert, S.; Ikenberg, K.; Ilina, O.; Stoma, S.; Xandry, J.; Jiang, L.; et al. Hypoxia Induces a HIF-1-Dependent Transition from Collective-to-Amoeboid Dissemination in Epithelial Cancer Cells. Curr. Biol. 2017, 27, 392–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheon, H.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.C.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013, 32, 2751–2763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edsbäcker, E.; Serviss, J.T.; Kolosenko, I.; Palm-Apergi, C.; De Milito, A.; Tamm, K.P. STAT3 is activated in multicellular spheroids of colon carcinoma cells and mediates expression of IRF9 and interferon stimulated genes. Sci. Rep. 2019, 9, 536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolosenko, I.; Fryknäs, M.; Forsberg, S.; Johnsson, P.; Cheon, H.; Holvey-Bates, E.G.; Edsbäcker, E.; Pellegrini, P.; Rassoolzadeh, H.; Brnjic, S.; et al. Cell crowding induces interferon regulatory factor 9, which confers resistance to chemotherapeutic drugs. Int. J. Cancer 2014, 136, E51–E61. [Google Scholar] [CrossRef]
- E Luker, K.; Pica, C.M.; Schreiber, R.D.; Piwnica-Worms, D. Overexpression of IRF9 confers resistance to antimicrotubule agents in breast cancer cells. Cancer Res. 2001, 61, 6540–6547. [Google Scholar]
- Khodarev, N.N.; Beckett, M.; Labay, E.; Darga, T.; Roizman, B.; Weichselbaum, R.R. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc. Natl. Acad. Sci. USA 2004, 101, 1714–1719. [Google Scholar] [CrossRef] [Green Version]
- Silginer, M.; Nagy, S.; Happold, C.; Schneider, H.; Weller, M.; Roth, P. Autocrine activation of the IFN signaling pathway may promote immune escape in glioblastoma. Neuro Oncol. 2017, 19, 1338–1349. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Cardona, A.E. The myeloid cells of the central nervous system parenchyma. Nature 2010, 468, 253–262. [Google Scholar] [CrossRef]
- Scheu, S.; Ali, S.; Mann-Nüttel, R.; Richter, L.; Arolt, V.; Dannlowski, U.; Kuhlmann, T.; Klotz, L.; Alferink, J. Interferon β-Mediated Protective Functions of Microglia in Central Nervous System Autoimmunity. Int. J. Mol. Sci. 2019, 20, 190. [Google Scholar] [CrossRef] [Green Version]
- Ogony, J.; Choi, H.J.; Lui, A.; Cristofanilli, M.; Lewis-Wambi, J. Interferon-induced transmembrane protein 1 (IFITM1) overexpression enhances the aggressive phenotype of SUM149 inflammatory breast cancer cells in a signal transducer and activator of transcription 2 (STAT2)-dependent manner. Breast Cancer Res. 2016, 18, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, J.; Wang, Y.; Yang, J.; Stark, G.R. IRF9 and unphosphorylated STAT2 cooperate with NF-κB to drive IL6 expression. Proc. Natl. Acad. Sci. USA 2018, 115, 3906–3911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobe, N.P.; Rosel, D.; Dvořánková, B.; Kodet, O.; Lacina, L.; Mateu, R.; Smetana, K.; Brábek, J.; Smetana, K. Simultaneous blocking of IL-6 and IL-8 is sufficient to fully inhibit CAF-induced human melanoma cell invasiveness. Histochem. Cell Biol. 2016, 146, 205–217. [Google Scholar] [CrossRef]
- Croner, R.S.; Sturzl, M.; Rau, T.T.; Metodieva, G.; Geppert, C.I.; Naschberger, E.; Lausen, B.; Metodiev, M.V. Quantitative proteome profiling of lymph node-positive vs. -negative colorectal carcinomas pinpoints MX1 as a marker for lymph node metastasis. Int. J. Cancer 2014, 135, 2878–2886. [Google Scholar] [CrossRef] [Green Version]
- Desai, S.D.; E Reed, R.; Burks, J.; Wood, L.M.; Pullikuth, A.K.; Haas, A.L.; Liu, L.F.; Breslin, J.W.; Meiners, S.; Sankar, S. ISG15 disrupts cytoskeletal architecture and promotes motility in human breast cancer cells. Exp. Biol. Med. 2012, 237, 38–49. [Google Scholar] [CrossRef]
- Burks, J.; Reed, R.E.; Desai, S.D. ISGylation governs the oncogenic function of Ki-Ras in breast cancer. Oncogene 2013, 33, 794–803. [Google Scholar] [CrossRef]
- Cruz, A.C.T.; González, C.C.C.; Cruz-Ramos, E.; Jarquín, J.O.R.; Romero-Mandujano, A.K.; Sosa-Garrocho, M. Interplay between interferon-stimulated gene 15/ISGylation and interferon gamma signaling in breast cancer cells. Cell. Signal. 2019, 54, 91–101. [Google Scholar] [CrossRef]
- Cerikan, B.; Shaheen, R.; Colo, G.P.; Gläßer, C.; Hata, S.; Knobeloch, K.-P.; Alkuraya, F.S.; Fässler, R.; Schiebel, E. Cell-Intrinsic Adaptation Arising from Chronic Ablation of a Key Rho GTPase Regulator. Dev. Cell 2016, 39, 28–43. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Ramos, E.; Macías-Silva, M.; Sandoval-Hernández, A.; Tecalco-Cruz, A.C. Non-muscle myosin IIA is post-translationally modified by interferon-stimulated gene 15 in breast cancer cells. Int. J. Biochem. Cell Biol. 2019, 107, 14–26. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Choi, J.S.; Lee, J.Y.; Yu, K.R.; Kim, S.M.; Ka, S.H.; Oh, K.H.; Kim, K.I.; Zhang, D.-E.; Bang, O.S.; et al. ISG15 modification of filamin B negatively regulates the type I interferon-induced JNK signalling pathway. EMBO Rep. 2009, 10, 374–380. [Google Scholar] [CrossRef]
- Xie, B.; Zhao, J.; Kitagawa, M.; Durbin, J.; Madri, J.A.; Guan, J.-L.; Fu, X.-Y. Focal Adhesion Kinase Activates Stat1 in Integrin-mediated Cell Migration and Adhesion. J. Biol. Chem. 2001, 276, 19512–19523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zou, W. Inhibition of integrin β1 decreases the malignancy of ovarian cancer cells and potentiates anticancer therapy via the FAK/STAT1 signaling pathway. Mol. Med. Rep. 2015, 12, 7869–7876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, E.; Wang, Z.; Stanley, A.; Peyrollier, K.; Roesner, L.M.; Werfel, T.; Quondamatteo, F.; Brakebusch, C. RAC1 in keratinocytes regulates crosstalk to immune cells by Arp2/3-dependent control of STAT1. J. Cell Sci. 2012, 125, 5379–5390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, K.M.; Postovit, L.M. Investigating the utility of human melanoma cell lines as tumour models. Oncotarget 2017, 8, 10498–10509. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.M.; Baker, A.C.; Beroukhim, R.; Winckler, W.; Feng, W.; Marmion, J.M.; Laine, E.; Greulich, H.; Tseng, H.; Gates, C.; et al. Modeling Genomic Diversity and Tumor Dependency in Malignant Melanoma. Cancer Res. 2008, 68, 664–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoek, K.S.; Eichhoff, O.M.; Schlegel, N.C.; Döbbeling, U.; Kobert, N.; Schaerer, L.; Hemmi, S.; Dummer, R. In vivo Switching of Human Melanoma Cells between Proliferative and Invasive States. Cancer Res. 2008, 68, 650–656. [Google Scholar] [CrossRef] [Green Version]
- Merta, L.; Gandalovičová, A.; Cermak, V.; Dibus, M.; Gutschner, T.; Diederichs, S.; Rosel, D.; Brábek, J. Increased Level of Long Non-Coding RNA MALAT1 is a Common Feature of Amoeboid Invasion. Cancers 2020, 12, 1136. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.; Hellemans, J.; Huggett, J.F.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gandalovičová, A.; Šůchová, A.-M.; Čermák, V.; Merta, L.; Rösel, D.; Brábek, J. Sustained Inflammatory Signalling through Stat1/Stat2/IRF9 Is Associated with Amoeboid Phenotype of Melanoma Cells. Cancers 2020, 12, 2450. https://doi.org/10.3390/cancers12092450
Gandalovičová A, Šůchová A-M, Čermák V, Merta L, Rösel D, Brábek J. Sustained Inflammatory Signalling through Stat1/Stat2/IRF9 Is Associated with Amoeboid Phenotype of Melanoma Cells. Cancers. 2020; 12(9):2450. https://doi.org/10.3390/cancers12092450
Chicago/Turabian StyleGandalovičová, Aneta, Anna-Marie Šůchová, Vladimír Čermák, Ladislav Merta, Daniel Rösel, and Jan Brábek. 2020. "Sustained Inflammatory Signalling through Stat1/Stat2/IRF9 Is Associated with Amoeboid Phenotype of Melanoma Cells" Cancers 12, no. 9: 2450. https://doi.org/10.3390/cancers12092450
APA StyleGandalovičová, A., Šůchová, A.-M., Čermák, V., Merta, L., Rösel, D., & Brábek, J. (2020). Sustained Inflammatory Signalling through Stat1/Stat2/IRF9 Is Associated with Amoeboid Phenotype of Melanoma Cells. Cancers, 12(9), 2450. https://doi.org/10.3390/cancers12092450