Challenges and Opportunities in Clinical Applications of Blood-Based Proteomics in Cancer




Simple Summary
Abstract
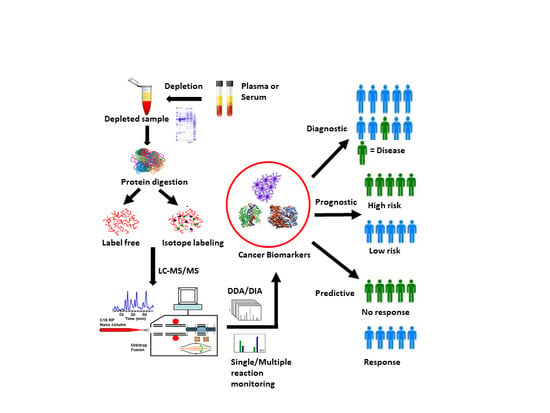
1. Introduction
2. General Considerations for Serum/Plasma Proteomics-Based Biomarker Discovery
2.1. Choice of Blood Plasma or Serum for Proteomics Studies
2.2. Important Factors for Designing Proteomic Studies for Cancer Biomarker Discovery
3. Pre-Analytic Challenges and Opportunities in Serum/Plasma Proteomics Cancer Biomarker Development
4. Experimental Design and Statistical Considerations in Serum/Plasma Proteomics
5. Global Comparative Proteomics versus Targeted Quantitative Proteomics for Serum/Plasma Cancer Biomarker Discovery
6. Clinical Serum/Plasma Proteomics and Molecular Signatures of Cancers
6.1. Lung Cancer
6.2. Breast Cancer
6.3. Ovarian Cancer
6.4. Prostate Cancer
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P. Clinical consequences of clonal hematopoiesis of indeterminate potential. Blood Adv. 2018, 2, 3404–3410. [Google Scholar] [CrossRef]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The evolutionary history of 2658 cancers. Nature 2020, 578, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.H.; Sun, H.; Yan, G.L.; Han, Y.; Wang, X.J. Serum proteomics in biomedical research: A systematic review. Appl. Biochem. Biotechnol. 2013, 170, 774–786. [Google Scholar] [CrossRef]
- Zhao, Y.; Lee, W.N.; Xiao, G.G. Quantitative proteomics and biomarker discovery in human cancer. Expert Rev. Proteom. 2009, 6, 115–118. [Google Scholar] [CrossRef]
- Hu, S.; Loo, J.A.; Wong, D.T. Human body fluid proteome analysis. Proteomics 2006, 6, 6326–6353. [Google Scholar] [CrossRef]
- Piersma, S.R.; Fiedler, U.; Span, S.; Lingnau, A.; Pham, T.V.; Hoffmann, S.; Kubbutat, M.H.; Jimenez, C.R. Workflow comparison for label-free, quantitative secretome proteomics for cancer biomarker discovery: Method evaluation, differential analysis, and verification in serum. J. Proteome Res. 2010, 9, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Srivastava, R.G. Proteomics in the forefront of cancer biomarker discovery. J. Proteome Res. 2005, 4, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Bichsel, V.E.; Liotta, L.A.; Petricoin, E.F., 3rd. Cancer proteomics: From biomarker discovery to signal pathway profiling. Cancer J. 2001, 7, 69–78. [Google Scholar]
- Rai, A.J.; Chan, D.W. Cancer proteomics: Serum diagnostics for tumor marker discovery. Ann. N. Y. Acad. Sci. 2004, 1022, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Krueger, K.E. The potential of serum proteomics for detection of cancer: Promise or only hope? Onkologie 2006, 29, 498–499. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.W.; Lo, S.C. Discovery of diagnostic serum biomarkers of gastric cancer using proteomics. Proteom. Clin. Appl. 2008, 2, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.W.; Park, J.; Han, H.S.; Yun, Y.M.; Kang, J.W.; Choi, D.Y.; Lee, J.W.; Jung, J.H.; Lee, K.Y.; Kim, K.P. Discovery of gastric cancer specific biomarkers by the application of serum proteomics. Proteomics 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Harlan, R.; Zhang, H. Targeted proteomics: A bridge between discovery and validation. Expert Rev. Proteom. 2014, 11, 657–661. [Google Scholar] [CrossRef]
- Gu, W.K.; Wang, Y.J. Gene Discovery for Disease Models Preface. Gene Discov. Dis. Models 2011. [Google Scholar] [CrossRef]
- Wulfkuhle, J.D.; Liotta, L.A.; Petricoin, E.F. Proteomic applications for the early detection of cancer. Nat. Rev. Cancer 2003, 3, 267–275. [Google Scholar] [CrossRef]
- Wang, D.L.; Xiao, C.; Fu, G.; Wang, X.; Li, L. Identification of potential serum biomarkers for breast cancer using a functional proteomics technology. Biomark Res. 2017, 5, 11. [Google Scholar] [CrossRef]
- Sun, C.; Rosendahl, A.H.; Ansari, D.; Andersson, R. Proteome-based biomarkers in pancreatic cancer. World J. Gastroenterol. 2011, 17, 4845–4852. [Google Scholar] [CrossRef]
- Pietrowska, M.; Marczak, L.; Polanska, J.; Behrendt, K.; Nowicka, E.; Walaszczyk, A.; Chmura, A.; Deja, R.; Stobiecki, M.; Polanski, A.; et al. Mass spectrometry-based serum proteome pattern analysis in molecular diagnostics of early stage breast cancer. J. Transl. Med. 2009, 7, 60. [Google Scholar] [CrossRef]
- Rosenblatt, K.P.; Bryant-Greenwood, P.; Killian, J.K.; Mehta, A.; Geho, D.; Espina, V.; Petricoin, E.F., 3rd; Liotta, L.A. Serum proteomics in cancer diagnosis and management. Annu. Rev. Med. 2004, 55, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Kohli, M.; Oberg, A.L.; Mahoney, D.W.; Riska, S.M.; Sherwood, R.; Zhang, Y.; Zenka, R.M.; Sahasrabudhe, D.; Qin, R.; Zhang, S. Serum Proteomics on the Basis of Discovery of Predictive Biomarkers of Response to Androgen Deprivation Therapy in Advanced Prostate Cancer. Clin. Genitourin Cancer 2019, 17, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Veenstra, T.D. Proteomic analysis of serum, plasma, and lymph for the identification of biomarkers. Proteom. Clin. Appl. 2007, 1, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Rifai, N.; Gillette, M.A.; Carr, S.A. Protein biomarker discovery and validation: The long and uncertain path to clinical utility. Nat. Biotechnol. 2006, 24, 971–983. [Google Scholar] [CrossRef]
- Rai, A.J.; Gelfand, C.A.; Haywood, B.C.; Warunek, D.J.; Yi, J.; Schuchard, M.D.; Mehigh, R.J.; Cockrill, S.L.; Scott, G.B.; Tammen, H.; et al. HUPO Plasma Proteome Project specimen collection and handling: Towards the standardization of parameters for plasma proteome samples. Proteomics 2005, 5, 3262–3277. [Google Scholar] [CrossRef]
- Faulk, W.P.; Torry, D.S.; McIntyre, J.A. Effects of serum versus plasma on agglutination of antibody-coated indicator cells by human rheumatoid factors. Clin. Immunol. Immunopathol. 1988, 46, 169–176. [Google Scholar] [CrossRef]
- Spence, G.M.; Graham, A.N.; Mulholland, K.; McAllister, I.; Sloan, J.M.; Armstrong, M.A.; Campbell, F.C.; McGuigan, J.A. Vascular endothelial growth factor levels in serum and plasma following esophageal cancer resection--relationship to platelet count. Int. J. Biol. Markers 2002, 17, 119–124. [Google Scholar] [CrossRef]
- Roos, P.H.; Jakubowski, N. Methods for the discovery of low-abundance biomarkers for urinary bladder cancer in biological fluids. Bioanalysis 2010, 2, 295–309. [Google Scholar] [CrossRef]
- Rai, A.J.; Vitzthum, F. Effects of preanalytical variables on peptide and protein measurements in human serum and plasma: Implications for clinical proteomics. Expert Rev. Proteom. 2006, 3, 409–426. [Google Scholar] [CrossRef]
- Simon, R. Roadmap for developing and validating therapeutically relevant genomic classifiers. J. Clin. Oncol. 2005, 23, 7332–7341. [Google Scholar] [CrossRef]
- Young, M.R.; Wagner, P.D.; Ghosh, S.; Rinaudo, J.A.; Baker, S.G.; Zaret, K.S.; Goggins, M.; Srivastava, S. Validation of Biomarkers for Early Detection of Pancreatic Cancer: Summary of The Alliance of Pancreatic Cancer Consortia for Biomarkers for Early Detection Workshop. Pancreas 2018, 47, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Pepe, M.S.; Janes, H.; Li, C.I.; Bossuyt, P.M.; Feng, Z.; Hilden, J. Early-Phase Studies of Biomarkers: What Target Sensitivity and Specificity Values Might Confer Clinical Utility? Clin. Chem. 2016, 62, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, D.F. How to improve reliability and efficiency of research about molecular markers: Roles of phases, guidelines, and study design. J. Clin. Epidemiol. 2007, 60, 1205–1219. [Google Scholar] [CrossRef]
- Zhu, C.S.; Pinsky, P.F.; Cramer, D.W.; Ransohoff, D.F.; Hartge, P.; Pfeiffer, R.M.; Urban, N.; Mor, G.; Bast, R.C., Jr.; Moore, L.E.; et al. A framework for evaluating biomarkers for early detection: Validation of biomarker panels for ovarian cancer. Cancer Prev. Res. 2011, 4, 375–383. [Google Scholar] [CrossRef]
- Ransohoff, D.F. Rules of evidence for cancer molecular-marker discovery and validation. Nat. Rev. Cancer 2004, 4, 309–314. [Google Scholar] [CrossRef] [PubMed]
- McShane, L.M.; Polley, M.Y. Development of omics-based clinical tests for prognosis and therapy selection: The challenge of achieving statistical robustness and clinical utility. Clin. Trials 2013, 10, 653–665. [Google Scholar] [CrossRef]
- Jacobs, I.; Menon, U. The sine qua non of discovering novel biomarkers for early detection of ovarian cancer: Carefully selected preclinical samples. Cancer Prev. Res. 2011, 4, 299–302. [Google Scholar] [CrossRef]
- Chari, S.T.; Leibson, C.L.; Rabe, K.G.; Ransom, J.; de Andrade, M.; Petersen, G.M. Probability of pancreatic cancer following diabetes: A population-based study. Gastroenterology 2005, 129, 504–511. [Google Scholar] [CrossRef]
- Sharma, A.; Kandlakunta, H.; Nagpal, S.J.S.; Feng, Z.; Hoos, W.; Petersen, G.M.; Chari, S.T. Model to Determine Risk of Pancreatic Cancer in Patients With New-Onset Diabetes. Gastroenterology 2018, 155, 730–739.e3. [Google Scholar] [CrossRef]
- Ransohoff, D.F. Bias as a threat to the validity of cancer molecular-marker research. Nat. Rev. Cancer 2005, 5, 142–149. [Google Scholar] [CrossRef]
- Schully, S.D.; Carrick, D.M.; Mechanic, L.E.; Srivastava, S.; Anderson, G.L.; Baron, J.A.; Berg, C.D.; Cullen, J.; Diamandis, E.P.; Doria-Rose, V.P.; et al. Leveraging biospecimen resources for discovery or validation of markers for early cancer detection. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- McShane, L.M. Biomarker Validation: Context and Complexities. J. Law Med. Ethics 2019, 47, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.M.; Paik, S.; Hayes, D.F. Use of archived specimens in evaluation of prognostic and predictive biomarkers. J. Natl. Cancer Inst. 2009, 101, 1446–1452. [Google Scholar] [CrossRef]
- Pepe, M.S.; Feng, Z.; Janes, H.; Bossuyt, P.M.; Potter, J.D. Pivotal evaluation of the accuracy of a biomarker used for classification or prediction: Standards for study design. J. Natl. Cancer Inst. 2008, 100, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Kim, J.W.; Jeon, S.Y.; Park, B.K.; Han, B.G. Proteomic analysis of the effect of storage temperature on human serum. Ann. Clin. Lab. Sci. 2010, 40, 61–70. [Google Scholar] [PubMed]
- Puangpila, C.; Mayadunne, E.; El Rassi, Z. Liquid phase based separation systems for depletion, prefractionation, and enrichment of proteins in biological fluids and matrices for in-depth proteomics analysis-An update covering the period 2011–2014. Electrophoresis 2015, 36, 238–252. [Google Scholar] [CrossRef]
- Qian, W.J.; Kaleta, D.T.; Petritis, B.O.; Jiang, H.; Liu, T.; Zhang, X.; Mottaz, H.M.; Varnum, S.M.; Camp, D.G., 2nd; Huang, L.; et al. Enhanced detection of low abundance human plasma proteins using a tandem IgY12-SuperMix immunoaffinity separation strategy. Mol. Cell Proteom. 2008, 7, 1963–1973. [Google Scholar] [CrossRef]
- Xu, R.; Gamst, A. Re: Lessons from controversy: Ovarian cancer screening and serum proteomics. J. Natl. Cancer Inst. 2005, 97, 1226. [Google Scholar] [CrossRef]
- Cheng, Y.; Liu, C.; Zhang, N.; Wang, S.; Zhang, Z. Proteomics analysis for finding serum markers of ovarian cancer. Biomed Res. Int. 2014, 2014, 179040. [Google Scholar] [CrossRef]
- Ransohoff, D.F. Lessons from controversy: Ovarian cancer screening and serum proteomics. J. Natl. Cancer Inst. 2005, 97, 315–319. [Google Scholar] [CrossRef]
- Skates, S.J.; Gillette, M.A.; LaBaer, J.; Carr, S.A.; Anderson, L.; Liebler, D.C.; Ransohoff, D.; Rifai, N.; Kondratovich, M.; Tezak, Z.; et al. Statistical design for biospecimen cohort size in proteomics-based biomarker discovery and verification studies. J. Proteome Res. 2013, 12, 5383–5394. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Picotti, P.; Huttenhain, R.; Heinzelmann-Schwarz, V.; Jovanovic, M.; Aebersold, R.; Vitek, O. Protein significance analysis in selected reaction monitoring (SRM) measurements. Mol. Cell Proteom. 2012, 11, M111.014662. [Google Scholar] [CrossRef] [PubMed]
- Huttenhain, R.; Choi, M.; Martin de la Fuente, L.; Oehl, K.; Chang, C.V.; Zimmermann, A.K.; Malander, S.; Olsson, H.; Surinova, S.; Clough, T.; et al. A Targeted Mass Spectrometry Strategy for Developing Proteomic Biomarkers: A Case Study of Epithelial Ovarian Cancer. Mol. Cell Proteom. 2019, 18, 1836–1850. [Google Scholar] [CrossRef] [PubMed]
- Clough, T.; Thaminy, S.; Ragg, S.; Aebersold, R.; Vitek, O. Statistical protein quantification and significance analysis in label-free LC-MS experiments with complex designs. BMC Bioinform. 2012, 13, S6. [Google Scholar] [CrossRef]
- Muntel, J.; Kirkpatrick, J.; Bruderer, R.; Huang, T.; Vitek, O.; Ori, A.; Reiter, L. Comparison of Protein Quantification in a Complex Background by DIA and TMT Workflows with Fixed Instrument Time. J. Proteome Res. 2019, 18, 1340–1351. [Google Scholar] [CrossRef]
- Tabb, D.L.; Wang, X.; Carr, S.A.; Clauser, K.R.; Mertins, P.; Chambers, M.C.; Holman, J.D.; Wang, J.; Zhang, B.; Zimmerman, L.J.; et al. Reproducibility of Differential Proteomic Technologies in CPTAC Fractionated Xenografts. J. Proteome Res. 2016, 15, 691–706. [Google Scholar] [CrossRef]
- Surinova, S.; Huttenhain, R.; Chang, C.Y.; Espona, L.; Vitek, O.; Aebersold, R. Automated selected reaction monitoring data analysis workflow for large-scale targeted proteomic studies. Nat. Protoc. 2013, 8, 1602–1619. [Google Scholar] [CrossRef]
- Borras, E.; Canto, E.; Choi, M.; Maria Villar, L.; Alvarez-Cermeno, J.C.; Chiva, C.; Montalban, X.; Vitek, O.; Comabella, M.; Sabido, E. Protein-Based Classifier to Predict Conversion from Clinically Isolated Syndrome to Multiple Sclerosis. Mol. Cell Proteom. 2016, 15, 318–328. [Google Scholar] [CrossRef]
- Oberg, A.L.; Vitek, O. Statistical design of quantitative mass spectrometry-based proteomic experiments. J. Proteome Res. 2009, 8, 2144–2156. [Google Scholar] [CrossRef]
- Oberg, A.L.; Mahoney, D.W. Statistical methods for quantitative mass spectrometry proteomic experiments with labeling. BMC Bioinform. 2012, 13, S7. [Google Scholar] [CrossRef]
- Oberg, A.L.; Mahoney, D.W.; Eckel-Passow, J.E.; Malone, C.J.; Wolfinger, R.D.; Hill, E.G.; Cooper, L.T.; Onuma, O.K.; Spiro, C.; Therneau, T.M.; et al. Statistical analysis of relative labeled mass spectrometry data from complex samples using ANOVA. J. Proteome Res. 2008, 7, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef]
- Kutner, M.H.; Neter, J.; Nachtsheim, C.J.; Li, W. Applied Linear Statistical Models, 5th ed.; McGraw-Hill/Irwin: New York, NY, USA, 2005. [Google Scholar]
- Choi, M.; Eren-Dogu, Z.F.; Colangelo, C.; Cottrell, J.; Hoopmann, M.R.; Kapp, E.A.; Kim, S.; Lam, H.; Neubert, T.A.; Palmblad, M.; et al. ABRF Proteome Informatics Research Group (iPRG) 2015 Study: Detection of Differentially Abundant Proteins in Label-Free Quantitative LC-MS/MS Experiments. J. Proteome Res. 2017, 16, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Clough, T.; Key, M.; Ott, I.; Ragg, S.; Schadow, G.; Vitek, O. Protein quantification in label-free LC-MS experiments. J. Proteome Res. 2009, 8, 5275–5284. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Chang, C.Y.; Clough, T.; Broudy, D.; Killeen, T.; MacLean, B.; Vitek, O. MSstats: An R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 2014, 30, 2524–2526. [Google Scholar] [CrossRef]
- James, G. An Introduction to Statistical Learning: With Applications in R; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Hastie, T.; Tibshirani, R.; Friedman, J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Witten, D.M.; Tibshirani, R.; Hastie, T. A penalized matrix decomposition, with applications to sparse principal components and canonical correlation analysis. Biostatistics 2009, 10, 515–534. [Google Scholar] [CrossRef] [PubMed]
- Dobbin, K.K.; Simon, R.M. Sample size planning for developing classifiers using high-dimensional DNA microarray data. Biostatistics 2007, 8, 101–117. [Google Scholar] [CrossRef]
- Dobbin, K.K.; Zhao, Y.; Simon, R.M. How large a training set is needed to develop a classifier for microarray data? Clin. Cancer Res. 2008, 14, 108–114. [Google Scholar] [CrossRef]
- Steyerberg, E.W. Clinical Prediction Models: A Practical Approach to Development, Validation, and Updating, 2nd ed.; Springer International Publishing: Cham, Switzerland, 2019. [Google Scholar]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. 2005, 67, 301–320. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Ransohoff, D.F.; Gourlay, M.L. Sources of bias in specimens for research about molecular markers for cancer. J. Clin. Oncol. 2010, 28, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Steyerberg, E.W.; Harrell, F.E., Jr. Prediction models need appropriate internal, internal-external, and external validation. J. Clin. Epidemiol. 2016, 69, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, J.; Simon, R. An evaluation of resampling methods for assessment of survival risk prediction in high-dimensional settings. Stat. Med. 2011, 30, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.M.; Subramanian, J.; Li, M.C.; Menezes, S. Using cross-validation to evaluate predictive accuracy of survival risk classifiers based on high-dimensional data. Brief. Bioinform. 2011, 12, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M.; Ankerst, D.P.; Andridge, R.R. Validation of biomarker-based risk prediction models. Clin. Cancer Res. 2008, 14, 5977–5983. [Google Scholar] [CrossRef]
- Cramer, D.W.; Bast, R.C., Jr.; Berg, C.D.; Diamandis, E.P.; Godwin, A.K.; Hartge, P.; Lokshin, A.E.; Lu, K.H.; McIntosh, M.W.; Mor, G.; et al. Ovarian cancer biomarker performance in prostate, lung, colorectal, and ovarian cancer screening trial specimens. Cancer Prev. Res. 2011, 4, 365–374. [Google Scholar] [CrossRef]
- Sjostrom, M.; Ossola, R.; Breslin, T.; Rinner, O.; Malmstrom, L.; Schmidt, A.; Aebersold, R.; Malmstrom, J.; Nimeus, E. A Combined Shotgun and Targeted Mass Spectrometry Strategy for Breast Cancer Biomarker Discovery. J. Proteome Res. 2015, 14, 2807–2818. [Google Scholar] [CrossRef]
- Moons, K.G.; Altman, D.G.; Reitsma, J.B.; Ioannidis, J.P.; Macaskill, P.; Steyerberg, E.W.; Vickers, A.J.; Ransohoff, D.F.; Collins, G.S. Transparent Reporting of a multivariable prediction model for Individual Prognosis or Diagnosis (TRIPOD): Explanation and elaboration. Ann. Intern. Med. 2015, 162, W1-73. [Google Scholar] [CrossRef]
- Collins, G.S.; Reitsma, J.B.; Altman, D.G.; Moons, K.G. Transparent Reporting of a multivariable prediction model for Individual Prognosis Or Diagnosis (TRIPOD). Ann. Intern. Med. 2015, 162, 735–736. [Google Scholar] [CrossRef]
- Altman, D.G.; McShane, L.M.; Sauerbrei, W.; Taube, S.E. Reporting recommendations for tumor marker prognostic studies (REMARK): Explanation and elaboration. BMC Med. 2012, 10, 51. [Google Scholar] [CrossRef]
- McShane, L.M.; Altman, D.G.; Sauerbrei, W.; Taube, S.E.; Gion, M.; Clark, G.M.; Statistics Subcommittee of the NCI-EORTC Working Group on Cancer Diagnostics. Reporting recommendations for tumor marker prognostic studies (REMARK). J. Natl. Cancer Inst. 2005, 97, 1180–1184. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Chen, B.; Min, S.; Gao, Y.; Zhang, Y.; Xu, P.; Li, C.; Chen, J.; Luo, G.; Liu, C. iTRAQ-based Comparative Serum Proteomic Analysis of Prostate Cancer Patients with or without Bone Metastasis. J. Cancer 2019, 10, 4165–4177. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Huang, J.; Rabii, B.; Rabii, R.; Hu, S. Quantitative proteomic analysis of serum proteins from oral cancer patients: Comparison of two analytical methods. Int. J. Mol. Sci 2014, 15, 14386–14395. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.G.; Recker, R.R.; Deng, H.W. Recent advances in proteomics and cancer biomarker discovery. Clin. Med. Oncol. 2008, 2, 63–72. [Google Scholar] [CrossRef]
- Higgs, R.E.; Knierman, M.D.; Gelfanova, V.; Butler, J.P.; Hale, J.E. Comprehensive label-free method for the relative quantification of proteins from biological samples. J. Proteome Res. 2005, 4, 1442–1450. [Google Scholar] [CrossRef]
- Levin, Y.; Schwarz, E.; Wang, L.; Leweke, F.M.; Bahn, S. Label-free LC-MS/MS quantitative proteomics for large-scale biomarker discovery in complex samples. J. Sep. Sci. 2007, 30, 2198–2203. [Google Scholar] [CrossRef]
- Domon, B.; Aebersold, R. Mass spectrometry and protein analysis. Science 2006, 312, 212–217. [Google Scholar] [CrossRef]
- Zhu, W.; Smith, J.W.; Huang, C.M. Mass spectrometry-based label-free quantitative proteomics. J. Biomed. Biotechnol. 2010, 2010, 840518. [Google Scholar] [CrossRef]
- Saraswat, M.; Joenvaara, S.; Seppanen, H.; Mustonen, H.; Haglund, C.; Renkonen, R. Comparative proteomic profiling of the serum differentiates pancreatic cancer from chronic pancreatitis. Cancer Med. 2017, 6, 1738–1751. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Siegel, E.R.; Petersen, G.M.; Chari, S.T.; Suva, L.J.; Haun, R.S. Diagnosis of pancreatic cancer using serum proteomic profiling. Neoplasia 2004, 6, 674–686. [Google Scholar] [CrossRef]
- Saraswat, M.; Makitie, A.; Agarwal, R.; Joenvaara, S.; Renkonen, S. Oral squamous cell carcinoma patients can be differentiated from healthy individuals with label-free serum proteomics. Br. J. Cancer 2017, 117, 376–384. [Google Scholar] [CrossRef]
- Schubert, O.T.; Rost, H.L.; Collins, B.C.; Rosenberger, G.; Aebersold, R. Quantitative proteomics: Challenges and opportunities in basic and applied research. Nat. Protoc. 2017, 12, 1289–1294. [Google Scholar] [CrossRef] [PubMed]
- Sechi, S.; Oda, Y. Quantitative proteomics using mass spectrometry. Curr. Opin. Chem. Biol. 2003, 7, 70–77. [Google Scholar] [CrossRef]
- Thompson, A.; Wolmer, N.; Koncarevic, S.; Selzer, S.; Bohm, G.; Legner, H.; Schmid, P.; Kienle, S.; Penning, P.; Hohle, C.; et al. TMTpro: Design, Synthesis, and Initial Evaluation of a Proline-Based Isobaric 16-Plex Tandem Mass Tag Reagent Set. Anal. Chem. 2019, 91, 15941–15950. [Google Scholar] [CrossRef] [PubMed]
- Sogawa, K.; Takano, S.; Iida, F.; Satoh, M.; Tsuchida, S.; Kawashima, Y.; Yoshitomi, H.; Sanda, A.; Kodera, Y.; Takizawa, H.; et al. Identification of a novel serum biomarker for pancreatic cancer, C4b-binding protein alpha-chain (C4BPA) by quantitative proteomic analysis using tandem mass tags. Br. J. Cancer 2016, 115, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shi, T.; Qian, W.J.; Liu, T.; Kagan, J.; Srivastava, S.; Smith, R.D.; Rodland, K.D.; Camp, D.G., 2nd. The clinical impact of recent advances in LC-MS for cancer biomarker discovery and verification. Expert Rev. Proteom. 2016, 13, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Vidova, V.; Spacil, Z. A review on mass spectrometry-based quantitative proteomics: Targeted and data independent acquisition. Anal. Chim. Acta 2017, 964, 7–23. [Google Scholar] [CrossRef]
- Holman, S.W.; Sims, P.F.; Eyers, C.E. The use of selected reaction monitoring in quantitative proteomics. Bioanalysis 2012, 4, 1763–1786. [Google Scholar] [CrossRef]
- Gillette, M.A.; Carr, S.A. Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nat. Methods 2013, 10, 28–34. [Google Scholar] [CrossRef]
- Schiess, R.; Wollscheid, B.; Aebersold, R. Targeted proteomic strategy for clinical biomarker discovery. Mol. Oncol. 2009, 3, 33–44. [Google Scholar] [CrossRef]
- Krueger, K.E.; Srivastava, S. Posttranslational protein modifications: Current implications for cancer detection, prevention, and therapeutics. Mol. Cell Proteom. 2006, 5, 1799–1810. [Google Scholar] [CrossRef]
- Hitosugi, T.; Chen, J. Post-translational modifications and the Warburg effect. Oncogene 2014, 33, 4279–4285. [Google Scholar] [CrossRef]
- Beach, A.; Zhang, H.G.; Ratajczak, M.Z.; Kakar, S.S. Exosomes: An overview of biogenesis, composition and role in ovarian cancer. J. Ovarian Res. 2014, 7, 14. [Google Scholar] [CrossRef]
- Sinha, A.; Ignatchenko, V.; Ignatchenko, A.; Mejia-Guerrero, S.; Kislinger, T. In-depth proteomic analyses of ovarian cancer cell line exosomes reveals differential enrichment of functional categories compared to the NCI 60 proteome. Biochem. Biophys. Res. Commun. 2014, 445, 694–701. [Google Scholar] [CrossRef]
- Sajic, T.; Liu, Y.; Aebersold, R. Using data-independent, high-resolution mass spectrometry in protein biomarker research: Perspectives and clinical applications. Proteom. Clin. Appl. 2015, 9, 307–321. [Google Scholar] [CrossRef]
- Egertson, J.D.; Kuehn, A.; Merrihew, G.E.; Bateman, N.W.; MacLean, B.X.; Ting, Y.S.; Canterbury, J.D.; Marsh, D.M.; Kellmann, M.; Zabrouskov, V.; et al. Multiplexed MS/MS for improved data-independent acquisition. Nat. Methods 2013, 10, 744–746. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Rost, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell Proteom. 2012, 11, O111.016717. [Google Scholar] [CrossRef]
- Bruderer, R.; Sondermann, J.; Tsou, C.C.; Barrantes-Freer, A.; Stadelmann, C.; Nesvizhskii, A.I.; Schmidt, M.; Reiter, L.; Gomez-Varela, D. New targeted approaches for the quantification of data-independent acquisition mass spectrometry. Proteomics 2017, 17. [Google Scholar] [CrossRef]
- Nigjeh, E.N.; Chen, R.; Brand, R.E.; Petersen, G.M.; Chari, S.T.; von Haller, P.D.; Eng, J.K.; Feng, Z.; Yan, Q.; Brentnall, T.A.; et al. Quantitative Proteomics Based on Optimized Data-Independent Acquisition in Plasma Analysis. J. Proteome Res. 2017, 16, 665–676. [Google Scholar] [CrossRef]
- Aebersold, R.; Bensimon, A.; Collins, B.C.; Ludwig, C.; Sabido, E. Applications and Developments in Targeted Proteomics: From SRM to DIA/SWATH. Proteomics 2016, 16, 2065–2067. [Google Scholar] [CrossRef]
- Song, Y.; Zhong, L.; Zhou, J.; Lu, M.; Xing, T.; Ma, L.; Shen, J. Data-Independent Acquisition-Based Quantitative Proteomic Analysis Reveals Potential Biomarkers of Kidney Cancer. Proteom. Clin. Appl. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Bouchal, P.; Schubert, O.T.; Faktor, J.; Capkova, L.; Imrichova, H.; Zoufalova, K.; Paralova, V.; Hrstka, R.; Liu, Y.; Ebhardt, H.A.; et al. Breast Cancer Classification Based on Proteotypes Obtained by SWATH Mass Spectrometry. Cell Rep. 2019, 28, 832–843.e837. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Asmis, R.; Hawkins, G.A.; Howard, T.D.; Cox, L.A. The Need for Multi-Omics Biomarker Signatures in Precision Medicine. Int. J. Mol. Sci. 2019, 20, 4781. [Google Scholar] [CrossRef]
- Gibson, G. On the utilization of polygenic risk scores for therapeutic targeting. PLoS Genet. 2019, 15, e1008060. [Google Scholar] [CrossRef]
- Sanhueza, C.; Kohli, M. Clinical and Novel Biomarkers in the Management of Prostate Cancer. Curr. Treat. Options Oncol. 2018, 19, 8. [Google Scholar] [CrossRef]
- Sestak, I.; Buus, R.; Cuzick, J.; Dubsky, P.; Kronenwett, R.; Denkert, C.; Ferree, S.; Sgroi, D.; Schnabel, C.; Baehner, F.L.; et al. Comparison of the Performance of 6 Prognostic Signatures for Estrogen Receptor-Positive Breast Cancer: A Secondary Analysis of a Randomized Clinical Trial. JAMA Oncol. 2018, 4, 545–553. [Google Scholar] [CrossRef]
- Buttigliero, C.; Shepherd, F.A.; Barlesi, F.; Schwartz, B.; Orlov, S.; Favaretto, A.G.; Santoro, A.; Hirsh, V.; Ramlau, R.; Blackler, A.R.; et al. Retrospective Assessment of a Serum Proteomic Test in a Phase III Study Comparing Erlotinib plus Placebo with Erlotinib plus Tivantinib (MARQUEE) in Previously Treated Patients with Advanced Non-Small Cell Lung Cancer. Oncologist 2019, 24, e251–e259. [Google Scholar] [CrossRef]
- Kao, S.C.; Kirschner, M.B.; Cooper, W.A.; Tran, T.; Burgers, S.; Wright, C.; Korse, T.; van den Broek, D.; Edelman, J.; Vallely, M.; et al. A proteomics-based approach identifies secreted protein acidic and rich in cysteine as a prognostic biomarker in malignant pleural mesothelioma. Br. J. Cancer 2016, 114, 524–531. [Google Scholar] [CrossRef]
- Khalil, F.K.; Altiok, S. Advances in EGFR as a Predictive Marker in Lung Adenocarcinoma. Cancer Control 2015, 22, 193–199. [Google Scholar] [CrossRef]
- Kang, S.M.; Sung, H.J.; Ahn, J.M.; Park, J.Y.; Lee, S.Y.; Park, C.S.; Cho, J.Y. The Haptoglobin beta chain as a supportive biomarker for human lung cancers. Mol. Biosyst. 2011, 7, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Kuo, S.W.; Hsu, L.M.; Huang, S.C.; Wang, C.H.; Tsai, P.R.; Chen, Y.S.; Jou, T.S.; Ko, W.J. Peroxiredoxin 1 induces inflammatory cytokine response and predicts outcome of cardiogenic shock patients necessitating extracorporeal membrane oxygenation: An observational cohort study and translational approach. J. Transl. Med. 2016, 14, 114. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Taib, N.A.; Ashrafzadeh, A.; Fadzli, F.; Harun, F.; Rahmat, K.; Hoong, S.M.; Abdul-Rahman, P.S.; Hashim, O.H. Unmasking Heavily O-Glycosylated Serum Proteins Using Perchloric Acid: Identification of Serum Proteoglycan 4 and Protease C1 Inhibitor as Molecular Indicators for Screening of Breast Cancer. PLoS ONE 2016, 11, e0149551. [Google Scholar] [CrossRef] [PubMed]
- Majidzadeh, A.K.; Gharechahi, J. Plasma proteomics analysis of tamoxifen resistance in breast cancer. Med. Oncol. 2013, 30, 753. [Google Scholar] [CrossRef]
- Suh, E.J.; Kabir, M.H.; Kang, U.B.; Lee, J.W.; Yu, J.; Noh, D.Y.; Lee, C. Comparative profiling of plasma proteome from breast cancer patients reveals thrombospondin-1 and BRWD3 as serological biomarkers. Exp. Mol. Med. 2012, 44, 36–44. [Google Scholar] [CrossRef]
- Bhardwaj, M.; Gies, A.; Weigl, K.; Tikk, K.; Benner, A.; Schrotz-King, P.; Borchers, C.H.; Brenner, H. Evaluation and Validation of Plasma Proteins Using Two Different Protein Detection Methods for Early Detection of Colorectal Cancer. Cancers 2019, 11, 1426. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Alborn, W.E.; Slebos, R.J.; Liebler, D.C. Comparison of protein immunoprecipitation-multiple reaction monitoring with ELISA for assay of biomarker candidates in plasma. J. Proteome Res. 2013, 12, 5996–6003. [Google Scholar] [CrossRef]
- Bhardwaj, M.; Weigl, K.; Tikk, K.; Holland-Letz, T.; Schrotz-King, P.; Borchers, C.H.; Brenner, H. Multiplex quantitation of 270 plasma protein markers to identify a signature for early detection of colorectal cancer. Eur. J. Cancer 2020, 127, 30–40. [Google Scholar] [CrossRef]
- Yepes, D.; Costina, V.; Pilz, L.R.; Hofheinz, R.; Neumaier, M.; Findeisen, P. Multiplex profiling of tumor-associated proteolytic activity in serum of colorectal cancer patients. Proteom. Clin. Appl. 2014, 8, 308–316. [Google Scholar] [CrossRef]
- Alvarez-Chaver, P.; De Chiara, L.; Martinez-Zorzano, V.S. Proteomic Profiling for Colorectal Cancer Biomarker Discovery. Methods Mol. Biol. 2018, 1765, 241–269. [Google Scholar] [CrossRef]
- Briggs, M.T.; Condina, M.R.; Klingler-Hoffmann, M.; Arentz, G.; Everest-Dass, A.V.; Kaur, G.; Oehler, M.K.; Packer, N.H.; Hoffmann, P. Translating N-Glycan Analytical Applications into Clinical Strategies for Ovarian Cancer. Proteom. Clin. Appl. 2019, 13, e1800099. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.R.; Walker, M.J.; Williamson, A.J.; Gentry-Maharaj, A.; Ryan, A.; Kalsi, J.; Skates, S.; D’Amato, A.; Dive, C.; Pernemalm, M.; et al. Protein Z: A putative novel biomarker for early detection of ovarian cancer. Int. J. Cancer 2016, 138, 2984–2992. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.Y.; Beer, L.A.; Tanyi, J.L.; Zhang, R.; Liu, Q.; Speicher, D.W. Protein isoform-specific validation defines multiple chloride intracellular channel and tropomyosin isoforms as serological biomarkers of ovarian cancer. J. Proteom. 2013, 89, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Tsaur, I.; Thurn, K.; Juengel, E.; Gust, K.M.; Borgmann, H.; Mager, R.; Bartsch, G.; Oppermann, E.; Ackermann, H.; Nelson, K.; et al. sE-cadherin serves as a diagnostic and predictive parameter in prostate cancer patients. J. Exp. Clin. Cancer Res. 2015, 34, 43. [Google Scholar] [CrossRef]
- Larkin, S.E.; Johnston, H.E.; Jackson, T.R.; Jamieson, D.G.; Roumeliotis, T.I.; Mockridge, C.I.; Michael, A.; Manousopoulou, A.; Papachristou, E.K.; Brown, M.D.; et al. Detection of candidate biomarkers of prostate cancer progression in serum: A depletion-free 3D LC/MS quantitative proteomics pilot study. Br. J. Cancer 2016, 115, 1078–1086. [Google Scholar] [CrossRef]
- Takakura, M.; Yokomizo, A.; Tanaka, Y.; Kobayashi, M.; Jung, G.; Banno, M.; Sakuma, T.; Imada, K.; Oda, Y.; Kamita, M.; et al. Carbonic anhydrase I as a new plasma biomarker for prostate cancer. ISRN Oncol. 2012, 2012, 768190. [Google Scholar] [CrossRef]
- Xie, Y.; Chen, L.; Lv, X.; Hou, G.; Wang, Y.; Jiang, C.; Zhu, H.; Xu, N.; Wu, L.; Lou, X.; et al. The levels of serine proteases in colon tissue interstitial fluid and serum serve as an indicator of colorectal cancer progression. Oncotarget 2016, 7, 32592–32606. [Google Scholar] [CrossRef]
- Wang, Y.; Shan, Q.; Hou, G.; Zhang, J.; Bai, J.; Lv, X.; Xie, Y.; Zhu, H.; Su, S.; Li, Y.; et al. Discovery of potential colorectal cancer serum biomarkers through quantitative proteomics on the colonic tissue interstitial fluids from the AOM-DSS mouse model. J. Proteom. 2016, 132, 31–40. [Google Scholar] [CrossRef]
- Sole, X.; Crous-Bou, M.; Cordero, D.; Olivares, D.; Guino, E.; Sanz-Pamplona, R.; Rodriguez-Moranta, F.; Sanjuan, X.; de Oca, J.; Salazar, R.; et al. Discovery and validation of new potential biomarkers for early detection of colon cancer. PLoS ONE 2014, 9, e106748. [Google Scholar] [CrossRef]
- Vase, M.O.; Ludvigsen, M.; Bendix, K.; Hamilton-Dutoit, S.; Mller, M.B.; Pedersen, C.; Pedersen, G.; Obel, N.; Larsen, C.S.; d’Amore, F.; et al. Proteomic profiling of pretreatment serum from HIV-infected patients identifies candidate markers predictive of lymphoma development. AIDS 2016, 30, 1889–1898. [Google Scholar] [CrossRef]
- Ludvigsen, M.; Hamilton-Dutoit, S.J.; d’Amore, F.; Honore, B. Proteomic approaches to the study of malignant lymphoma: Analyses on patient samples. Proteom. Clin. Appl. 2015, 9, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhai, J.; Wu, X.; Xie, G.; Shen, L. Serum proteome profiling reveals SOX3 as a candidate prognostic marker for gastric cancer. J. Cell Mol. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Polom, K.; Williams, C.; de Oliveira, F.M.S.; Guergova-Kuras, M.; Lisacek, F.; Karlsson, N.G.; Roviello, F.; Kamali-Moghaddam, M. A targeted proteomics approach reveals a serum protein signature as diagnostic biomarker for resectable gastric cancer. EBioMedicine 2019, 44, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Yang, H.; Zhang, X.; Wang, B.; Mao, J.; Li, X.; Wang, M.; Zhang, B.; Sun, Z.; Qian, H.; et al. Exosomal TRIM3 is a novel marker and therapy target for gastric cancer. J. Exp. Clin. Cancer Res. 2018, 37, 162. [Google Scholar] [CrossRef]
- Loei, H.; Tan, H.T.; Lim, T.K.; Lim, K.H.; So, J.B.; Yeoh, K.G.; Chung, M.C. Mining the gastric cancer secretome: Identification of GRN as a potential diagnostic marker for early gastric cancer. J. Proteome Res. 2012, 11, 1759–1772. [Google Scholar] [CrossRef]
- Tseng, C.W.; Yang, J.C.; Chen, C.N.; Huang, H.C.; Chuang, K.N.; Lin, C.C.; Lai, H.S.; Lee, P.H.; Chang, K.J.; Juan, H.F. Identification of 14-3-3beta in human gastric cancer cells and its potency as a diagnostic and prognostic biomarker. Proteomics 2011, 11, 2423–2439. [Google Scholar] [CrossRef]
- Liu, P.; Kong, L.; Liang, K.; Wu, Y.; Jin, H.; Song, B.; Tan, X. Identification of dissociation factors in pancreatic Cancer using a mass spectrometry-based proteomic approach. BMC Cancer 2020, 20, 45. [Google Scholar] [CrossRef]
- Duan, B.; Hu, X.; Fan, M.; Xiong, X.; Han, L.; Wang, Z.; Tong, D.; Liu, L.; Wang, X.; Li, W.; et al. RNA-Binding Motif Protein 6 is a Candidate Serum Biomarker for Pancreatic Cancer. Proteom. Clin. Appl. 2019, 13, e1900048. [Google Scholar] [CrossRef]
- Kosanam, H.; Prassas, I.; Chrystoja, C.C.; Soleas, I.; Chan, A.; Dimitromanolakis, A.; Blasutig, I.M.; Ruckert, F.; Gruetzmann, R.; Pilarsky, C.; et al. Laminin, gamma 2 (LAMC2): A promising new putative pancreatic cancer biomarker identified by proteomic analysis of pancreatic adenocarcinoma tissues. Mol. Cell Proteom. 2013, 12, 2820–2832. [Google Scholar] [CrossRef]
- Park, J.; Lee, E.; Park, K.J.; Park, H.D.; Kim, J.W.; Woo, H.I.; Lee, K.H.; Lee, K.T.; Lee, J.K.; Park, J.O.; et al. Large-scale clinical validation of biomarkers for pancreatic cancer using a mass spectrometry-based proteomics approach. Oncotarget 2017, 8, 42761–42771. [Google Scholar] [CrossRef]
- Jenkinson, C.; Elliott, V.; Menon, U.; Apostolidou, S.; Fourkala, O.E.; Gentry-Maharaj, A.; Pereira, S.P.; Jacobs, I.; Cox, T.F.; Greenhalf, W.; et al. Evaluation in pre-diagnosis samples discounts ICAM-1 and TIMP-1 as biomarkers for earlier diagnosis of pancreatic cancer. J. Proteom. 2015, 113, 400–402. [Google Scholar] [CrossRef]
- Qiu, F.; Chen, F.; Liu, D.; Xu, J.; He, J.; Xiao, J.; Cao, L.; Huang, X. LC-MS/MS-based screening of new protein biomarkers for cervical precancerous lesions and cervical cancer. Nan Fang Yi Ke Da Xue Xue Bao 2019, 39, 13–22. [Google Scholar] [CrossRef]
- Barba de la Rosa, A.P.; Lugo-Melchor, O.Y.; Briones-Cerecero, E.P.; Chagolla-Lopez, A.; De Leon-Rodriguez, A.; Santos, L.; Vazquez-Ortiz, G.; Salcedo, M. Analysis of human serum from women affected by cervical lesions. J. Exp. Ther. Oncol. 2008, 7, 65–72. [Google Scholar]
- Liu, Y.; Wei, F.; Wang, F.; Li, C.; Meng, G.; Duan, H.; Ma, Q.; Zhang, W. Serum peptidome profiling analysis for the identification of potential biomarkers in cervical intraepithelial neoplasia patients. Biochem. Biophys. Res. Commun. 2015, 465, 476–480. [Google Scholar] [CrossRef]
- Dytfeld, D.; Luczak, M.; Wrobel, T.; Usnarska-Zubkiewicz, L.; Brzezniakiewicz, K.; Jamroziak, K.; Giannopoulos, K.; Przybylowicz-Chalecka, A.; Ratajczak, B.; Czerwinska-Rybak, J.; et al. Comparative proteomic profiling of refractory/relapsed multiple myeloma reveals biomarkers involved in resistance to bortezomib-based therapy. Oncotarget 2016, 7, 56726–56736. [Google Scholar] [CrossRef]
- Zhang, H.T.; Tian, E.B.; Chen, Y.L.; Deng, H.T.; Wang, Q.T. Proteomic analysis for finding serum pathogenic factors and potential biomarkers in multiple myeloma. Chin. Med. J. (Engl.) 2015, 128, 1108–1113. [Google Scholar] [CrossRef]
- Harshman, S.W.; Canella, A.; Ciarlariello, P.D.; Agarwal, K.; Branson, O.E.; Rocci, A.; Cordero, H.; Phelps, M.A.; Hade, E.M.; Dubovsky, J.A.; et al. Proteomic characterization of circulating extracellular vesicles identifies novel serum myeloma associated markers. J. Proteom. 2016, 136, 89–98. [Google Scholar] [CrossRef]
- Barcelo, F.; Gomila, R.; de Paul, I.; Gili, X.; Segura, J.; Perez-Montana, A.; Jimenez-Marco, T.; Sampol, A.; Portugal, J. MALDI-TOF analysis of blood serum proteome can predict the presence of monoclonal gammopathy of undetermined significance. PLoS ONE 2018, 13, e0201793. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Hoseok, I.; Cho, J.Y. Lung Cancer Biomarkers. Adv. Clin. Chem. 2015, 72, 107–170. [Google Scholar] [CrossRef]
- Cagle, P.T.; Allen, T.C.; Olsen, R.J. Lung cancer biomarkers: Present status and future developments. Arch. Pathol. Lab. Med. 2013, 137, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Gallien, S.; El-Khoury, V.; Goswami, P.; Sertamo, K.; Schlesser, M.; Berchem, G.; Domon, B. Quantification of SAA1 and SAA2 in lung cancer plasma using the isotype-specific PRM assays. Proteomics 2015, 15, 3116–3125. [Google Scholar] [CrossRef] [PubMed]
- Bonotti, A.; Foddis, R.; Landi, S.; Melaiu, O.; De Santi, C.; Giusti, L.; Donadio, E.; Ciregia, F.; Mazzoni, M.R.; Lucacchini, A.; et al. A Novel Panel of Serum Biomarkers for MPM Diagnosis. Dis. Markers 2017, 2017, 3510984. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.J.; Cho, J.Y. Biomarkers for the lung cancer diagnosis and their advances in proteomics. BMB Rep. 2008, 41, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Youlden, D.R.; Cramb, S.M.; Dunn, N.A.; Muller, J.M.; Pyke, C.M.; Baade, P.D. The descriptive epidemiology of female breast cancer: An international comparison of screening, incidence, survival and mortality. Cancer Epidemiol. 2012, 36, 237–248. [Google Scholar] [CrossRef]
- Dos Anjos Pultz, B.; da Luz, F.A.; de Faria, P.R.; Oliveira, A.P.; de Araujo, R.A.; Silva, M.J. Far beyond the usual biomarkers in breast cancer: A review. J. Cancer 2014, 5, 559–571. [Google Scholar] [CrossRef]
- Yang, J.; Yang, J.; Gao, Y.; Zhao, L.; Liu, L.; Qin, Y.; Wang, X.; Song, T.; Huang, C. Identification of potential serum proteomic biomarkers for clear cell renal cell carcinoma. PLoS ONE 2014, 9, e111364. [Google Scholar] [CrossRef]
- Weigel, M.T.; Dowsett, M. Current and emerging biomarkers in breast cancer: Prognosis and prediction. Endocr. Relat. Cancer 2010, 17, R245–R262. [Google Scholar] [CrossRef]
- Molina, R.; Auge, J.M.; Escudero, J.M.; Filella, X.; Zanon, G.; Pahisa, J.; Farrus, B.; Munoz, M.; Velasco, M. Evaluation of tumor markers (HER-2/neu oncoprotein, CEA, and CA 15.3) in patients with locoregional breast cancer: Prognostic value. Tumour. Biol. 2010, 31, 171–180. [Google Scholar] [CrossRef]
- Di Gioia, D.; Dresse, M.; Mayr, D.; Nagel, D.; Heinemann, V.; Stieber, P. Serum HER2 in combination with CA 15-3 as a parameter for prognosis in patients with early breast cancer. Clin. Chim. Acta 2015, 440, 16–22. [Google Scholar] [CrossRef]
- Kazarian, A.; Blyuss, O.; Metodieva, G.; Gentry-Maharaj, A.; Ryan, A.; Kiseleva, E.M.; Prytomanova, O.M.; Jacobs, I.J.; Widschwendter, M.; Menon, U.; et al. Testing breast cancer serum biomarkers for early detection and prognosis in pre-diagnosis samples. Br. J. Cancer 2017, 116, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Opstal-van Winden, A.W.; Rodenburg, W.; Pennings, J.L.; van Oostrom, C.T.; Beijnen, J.H.; Peeters, P.H.; van Gils, C.H.; de Vries, A. A bead-based multiplexed immunoassay to evaluate breast cancer biomarkers for early detection in pre-diagnostic serum. Int. J. Mol. Sci. 2012, 13, 13587–13604. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.C.; Hollingsworth, A.B.; Gordon, K.; Silver, M.; Mulpuri, R.; Letsios, E.; Reese, D.E. Integration of Serum Protein Biomarker and Tumor Associated Autoantibody Expression Data Increases the Ability of a Blood-Based Proteomic Assay to Identify Breast Cancer. PLoS ONE 2016, 11, e0157692. [Google Scholar] [CrossRef] [PubMed]
- Giussani, M.; Landoni, E.; Merlino, G.; Turdo, F.; Veneroni, S.; Paolini, B.; Cappelletti, V.; Miceli, R.; Orlandi, R.; Triulzi, T.; et al. Extracellular matrix proteins as diagnostic markers of breast carcinoma. J. Cell Physiol. 2018, 233, 6280–6290. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Gehin, T.; Chevolot, Y.; Souteyrand, E.; Mange, A.; Solassol, J.; Laurenceau, E. Anti-heat shock protein autoantibody profiling in breast cancer using customized protein microarray. Anal. Bioanal. Chem. 2016, 408, 1497–1506. [Google Scholar] [CrossRef]
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [CrossRef]
- Zhang, B.; Barekati, Z.; Kohler, C.; Radpour, R.; Asadollahi, R.; Holzgreve, W.; Zhong, X.Y. Proteomics and biomarkers for ovarian cancer diagnosis. Ann. Clin. Lab. Sci. 2010, 40, 218–225. [Google Scholar] [CrossRef]
- Toss, A.; De Matteis, E.; Rossi, E.; Casa, L.D.; Iannone, A.; Federico, M.; Cortesi, L. Ovarian cancer: Can proteomics give new insights for therapy and diagnosis? Int. J. Mol. Sci. 2013, 14, 8271–8290. [Google Scholar] [CrossRef]
- Lin, Y.W.; Lin, C.Y.; Lai, H.C.; Chiou, J.Y.; Chang, C.C.; Yu, M.H.; Chu, T.Y. Plasma proteomic pattern as biomarkers for ovarian cancer. Int. J. Gynecol. Cancer 2006, 16, 139–146. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, X.; Ma, Y.; Rao, M.; Glimm, J.; Kovach, J.S. Detection of cancer-specific markers amid massive mass spectral data. Proc. Natl. Acad. Sci. USA 2003, 100, 14666–14671. [Google Scholar] [CrossRef]
- Jacobs, I.J.; Menon, U. Progress and challenges in screening for early detection of ovarian cancer. Mol. Cell Proteom. 2004, 3, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Elzek, M.A.; Rodland, K.D. Proteomics of ovarian cancer: Functional insights and clinical applications. Cancer Metastasis Rev. 2015, 34, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Yin, B.W.; Lloyd, K.O. Molecular cloning of the CA125 ovarian cancer antigen: Identification as a new mucin, MUC16. J. Biol. Chem. 2001, 276, 27371–27375. [Google Scholar] [CrossRef] [PubMed]
- Bast, R.C., Jr.; Urban, N.; Shridhar, V.; Smith, D.; Zhang, Z.; Skates, S.; Lu, K.; Liu, J.; Fishman, D.; Mills, G. Early detection of ovarian cancer: Promise and reality. Cancer Treat. Res. 2002, 107, 61–97. [Google Scholar] [CrossRef]
- Moss, E.L.; Hollingworth, J.; Reynolds, T.M. The role of CA125 in clinical practice. J. Clin. Pathol. 2005, 58, 308–312. [Google Scholar] [CrossRef]
- Vaughan, S.; Coward, J.I.; Bast, R.C., Jr.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef]
- Diamandis, E.P. Point: Proteomic patterns in biological fluids: Do they represent the future of cancer diagnostics? Clin. Chem. 2003, 49, 1272–1275. [Google Scholar] [CrossRef]
- Timms, J.F.; Arslan-Low, E.; Kabir, M.; Worthington, J.; Camuzeaux, S.; Sinclair, J.; Szaub, J.; Afrough, B.; Podust, V.N.; Fourkala, E.O.; et al. Discovery of serum biomarkers of ovarian cancer using complementary proteomic profiling strategies. Proteom. Clin. Appl. 2014, 8, 982–993. [Google Scholar] [CrossRef]
- Bast, R.C., Jr.; Badgwell, D.; Lu, Z.; Marquez, R.; Rosen, D.; Liu, J.; Baggerly, K.A.; Atkinson, E.N.; Skates, S.; Zhang, Z.; et al. New tumor markers: CA125 and beyond. Int. J. Gynecol Cancer 2005, 15, 274–281. [Google Scholar] [CrossRef]
- Bonifacio, V.D.B. Ovarian Cancer Biomarkers: Moving Forward in Early Detection. Adv. Exp. Med. Biol. 2020, 1219, 355–363. [Google Scholar] [CrossRef]
- Kristjansdottir, B.; Levan, K.; Partheen, K.; Carlsohn, E.; Sundfeldt, K. Potential tumor biomarkers identified in ovarian cyst fluid by quantitative proteomic analysis, iTRAQ. Clin. Proteom. 2013, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Li, L.; Li, Z.; Hong, S.; Yang, Q.; Qu, X.; Kong, B. Alterations in the serum proteome profile during the development of ovarian cancer. Int. J. Oncol. 2014, 45, 2495–2501. [Google Scholar] [CrossRef] [PubMed]
- Kozak, K.R.; Su, F.; Whitelegge, J.P.; Faull, K.; Reddy, S.; Farias-Eisner, R. Characterization of serum biomarkers for detection of early stage ovarian cancer. Proteomics 2005, 5, 4589–4596. [Google Scholar] [CrossRef] [PubMed]
- Pietrowska, M.; Widlak, P. MALDI-MS-Based Profiling of Serum Proteome: Detection of Changes Related to Progression of Cancer and Response to Anticancer Treatment. Int. J. Proteom. 2012, 2012, 926427. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef]
- Huang, R.; Chen, Z.; He, L.; He, N.; Xi, Z.; Li, Z.; Deng, Y.; Zeng, X. Mass spectrometry-assisted gel-based proteomics in cancer biomarker discovery: Approaches and application. Theranostics 2017, 7, 3559–3572. [Google Scholar] [CrossRef]
- Araujo, A.; Cook, L.M.; Lynch, C.C.; Basanta, D. An integrated computational model of the bone microenvironment in bone-metastatic prostate cancer. Cancer Res. 2014, 74, 2391–2401. [Google Scholar] [CrossRef]
- Smith-Palmer, J.; Takizawa, C.; Valentine, W. Literature review of the burden of prostate cancer in Germany, France, the United Kingdom and Canada. BMC Urol. 2019, 19, 19. [Google Scholar] [CrossRef]
- Kohli, M.; Tindall, D.J. New developments in the medical management of prostate cancer. Mayo Clin. Proc. 2010, 85, 77–86. [Google Scholar] [CrossRef]
- Sun, C.; Song, C.; Ma, Z.; Xu, K.; Zhang, Y.; Jin, H.; Tong, S.; Ding, W.; Xia, G.; Ding, Q. Periostin identified as a potential biomarker of prostate cancer by iTRAQ-proteomics analysis of prostate biopsy. Proteome Sci. 2011, 9, 22. [Google Scholar] [CrossRef]
- Sugie, S.; Mukai, S.; Yamasaki, K.; Kamibeppu, T.; Tsukino, H.; Kamoto, T. Significant Association of Caveolin-1 and Caveolin-2 with Prostate Cancer Progression. Cancer Genom. Proteom. 2015, 12, 391–396. [Google Scholar]
- Qian, X.; Li, C.; Pang, B.; Xue, M.; Wang, J.; Zhou, J. Spondin-2 (SPON2), a more prostate-cancer-specific diagnostic biomarker. PLoS ONE 2012, 7, e37225. [Google Scholar] [CrossRef] [PubMed]
- Worst, T.S.; von Hardenberg, J.; Gross, J.C.; Erben, P.; Schnolzer, M.; Hausser, I.; Bugert, P.; Michel, M.S.; Boutros, M. Database-augmented Mass Spectrometry Analysis of Exosomes Identifies Claudin 3 as a Putative Prostate Cancer Biomarker. Mol. Cell Proteom. 2017, 16, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lee, B.Y.; Brown, D.A.; Molloy, M.P.; Marx, G.M.; Pavlakis, N.; Boyer, M.J.; Stockler, M.R.; Kaplan, W.; Breit, S.N.; et al. Identification of candidate biomarkers of therapeutic response to docetaxel by proteomic profiling. Cancer Res. 2009, 69, 7696–7703. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Study Design Step | Statistical Considerations |
|---|---|
| Study Goal | Define the population in which the protein marker will be used. Define the purpose of the protein marker—e.g., early detection, disease monitoring, etc. |
| Specimens | Select case and control specimens randomly. Select from a prospectively collected (prior to knowledge of disease status) specimen biobank. Select specimens from the relevant time point in the disease course. Avoid convenience samples. Avoid pooling of specimens. |
| Study Design | Plan sufficient sample size for discovery in light of realistic expected differences. Randomize specimens to assay run order. |
| Differential Abundance Detection | Assess protein difference signals relative to variation. Incorporate statistical design into the analysis model. Apply correction for multiple comparisons. |
| Panel or Signature Model Building | Finalize analysis plan in writing prior to beginning analyses. Employ optimism correction methods. Generate a fixed, locked-down algorithm. |
| Validation | Perform verification of initial protein marker identifications. Perform internal model validation in the discovery sample set. Perform external model validation in an independent sample set. |
| Tumor Type | (Serum/Plasma)-Proteomics Method | Biomarker Identified | Clinical Application in Cancer | Stage of Tumor | References |
|---|---|---|---|---|---|
| Lung | Plasma-MALDI-TOF, PRM | EGFR, SPRAC, SAA1, SAA2 | Screening/Prognostic | Advanced/Local | [122,123,124] |
| Serum-Shotgun | PTM profiling, (Hp) β chain | Screening | Advanced | [125] | |
| Plasma-Shotgun | Cytokines, Peroxiredoxins | Diagnosis/screening | local | [126] | |
| Breast | Serum–QTOF LC-MS | PRG4, C1-inh | Screening | local | [127] |
| Plasma-2Dgel, shotgun | CLU, SAA, SERPINB4, COL11A1 | Screening | Advanced | [128] | |
| Plasma-iTRAQ | THBS1, BRWD3, EGFR, CFHR3 | Prognostic | local | [129] | |
| Colorectal | Plasma-MRM-MS | MASP1, OPN, PON3, TFRC | Screening | local | [130,131] |
| Plasma-MRM-MS | 270 protein biomarkers | Diagnosis | local | [132] | |
| Serum-LC-MS/MS, 2Dgel | Reporter peptides, NDKA | Screening/prognostic | Advanced | [133,134] | |
| Ovarian | Plasma-SRM | CA125, IGHG2, LGALS3BP, DSG2, L1CAM, THBS1 | Diagnosis | Advanced | [53] |
| Serum-MALDI-TOF, LC-MS/MS | PTM profiling, RBP4 | Diagnosis | Local/Advanced | [49,135] | |
| Serum-iTRAQ, MRM | Protein Z, CA125, CLIC4, TPM | Screening, Diagnosis | Local | [136,137] | |
| Prostate | Serum-, iTRAQ | sE-cadherin, TSR1, SAA, KLK3 | Diagnosis | Local | [138,139] |
| Plasma-, Shotgun | CA1 | Diagnosis | Advanced | [140] | |
| Colon | Serum-iTRAQ, MRM | LCN-2, CELA1, CEL2A, CTRL, LRG1, TUBB5 | Prognostic/Screening | Local/Advanced | [126,141,142,143] |
| Plasma-, MRM | SLeA, TIMP1, COMP, THBS2, MMP9 | Prognostic/Screening | Local/Advanced | [126,131,141,142] | |
| Serum-Shotgun | IGJ, APOA1, PCOLCE, SAA2 | Prognostic | Local | [131,144,145] | |
| Lymphoma | Serum-Shotgun | SOX3, CA9, MSLN, CCL20, SCF, MMP-10, IGF-1, TRIM3 | Prognostic/Diagnosis | Local/advanced | [144,145,146,147,148] |
| Gastric | Serum-iTRAQ, MALDI-TOF | GRN, 14-3-3β | Prognostic/Diagnosis | Local | [146,147,148,149,150] |
| Serum-SEC-Shotgun, MALDI-TOF | YWHAG, RBM6, LAMC2 | Prognostic/Diagnosis | Local/Advanced | [149,150,151,152,153] | |
| Pancreatic | Serum-MRM | apoA-IV, apo-CIII, IGFBP-2 ICAM-1, TIMP-1 | Diagnosis | Advanced | [151,152,153,154,155] |
| Plasma-LC-MS/MS | F9, CFI, AFM, HPR, ORM2 | Prognostic | Local | [154,155,156] | |
| Cervical | Serum-2Dgel | MMP | Diagnosis | Local/Advanced | [156,157] |
| Serum-MALDI-TOF | Peptide profiling | Diagnosis | Local | [157,158] | |
| Plasma-iTRAQ, LFQ | TXN, TXNDC5 | Screening | Local | [158,159] | |
| Multiple Myeloma | Serum-LC-MS/MS | SAA2, KLKLB1, APOA1, CD44 | Diagnosis | Advanced | [159,160,161] |
| Serum-MALDI-TOF | Peptide Profiling | Diagnosis | Local | [160,161,162] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhawal, R.; Oberg, A.L.; Zhang, S.; Kohli, M. Challenges and Opportunities in Clinical Applications of Blood-Based Proteomics in Cancer. Cancers 2020, 12, 2428. https://doi.org/10.3390/cancers12092428
Bhawal R, Oberg AL, Zhang S, Kohli M. Challenges and Opportunities in Clinical Applications of Blood-Based Proteomics in Cancer. Cancers. 2020; 12(9):2428. https://doi.org/10.3390/cancers12092428
Chicago/Turabian StyleBhawal, Ruchika, Ann L. Oberg, Sheng Zhang, and Manish Kohli. 2020. "Challenges and Opportunities in Clinical Applications of Blood-Based Proteomics in Cancer" Cancers 12, no. 9: 2428. https://doi.org/10.3390/cancers12092428
APA StyleBhawal, R., Oberg, A. L., Zhang, S., & Kohli, M. (2020). Challenges and Opportunities in Clinical Applications of Blood-Based Proteomics in Cancer. Cancers, 12(9), 2428. https://doi.org/10.3390/cancers12092428