Vitamin D Effects on Cell Differentiation and Stemness in Cancer

 , ,
, ,  , and
, and {kind=link}
{kind=link}
Abstract
1. Introduction
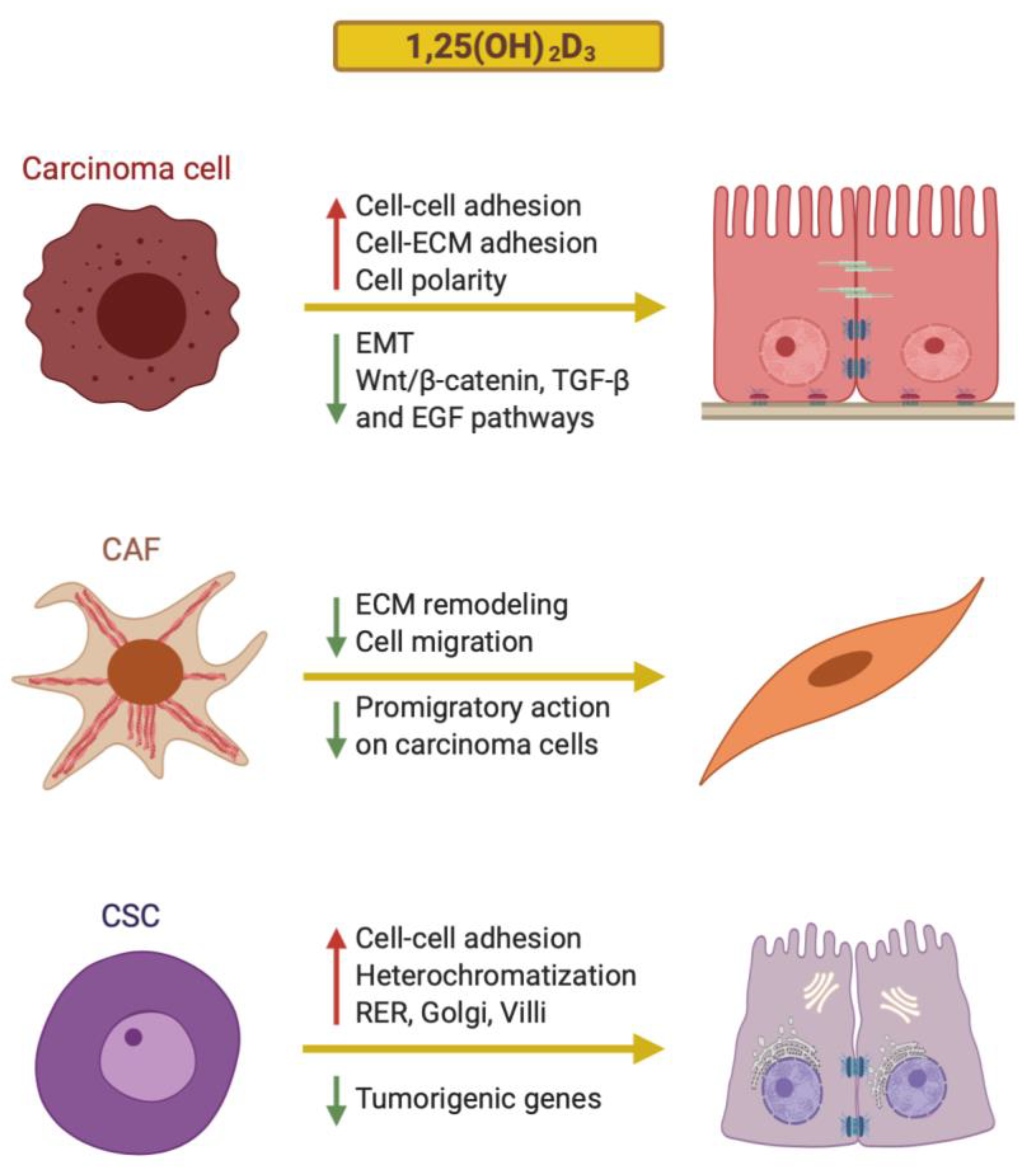
2. Effects of 1,25(OH)2D3 on Cancer Cell Differentiation
2.1. Carcinomas: The Epithelial–Mesenchymal Transition
2.2. Effects of 1,25(OH)2D3 on the Differentiation of Carcinoma Cells
2.2.1. Colon Cancer
1,25(OH)2D3 Induces Epithelial Differentiation and Inhibits EMT
Antagonism of the Wnt/β -Catenin Signaling Pathway by 1,25(OH)2D3
Other 1,25(OH)2D3 Target Genes Are Involved in Colon Cancer Cell Differentiation
Crosstalk between 1,25(OH)2D3 and EMT
2.2.2. Breast Cancer
2.2.3. Other Solid Cancers
2.3. Effects of 1,25(OH)2D3 on the Differentiation of Hematological Cancer Cells
3. Effects of 1,25(OH)2D3 on the Differentiation of Tumor Stromal Fibroblasts
4. Effects of 1,25(OH)2D3 on Cancer Stem Cells
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Fleet, J.C.; DeSmet, M.; Johnson, R.; Li, Y. Vitamin D and cancer: A review of molecular mechanisms. Biochem. J. 2012, 441, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.; Krishnan, A.V.; Swami, S.; Giovannucci, E.; Feldman, B.J. The role of vitamin D in reducing cancer risk and progression. Nat. Rev. Cancer 2014, 14, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Christakos, S.; Dhawan, P.; Verstuyf, A.; Verlinden, L.; Carmeliet, G. Vitamin D: Metabolism, Molecular Mechanism of Action, and Pleiotropic Effects. Physiol. Rev. 2016, 96, 365–408. [Google Scholar] [CrossRef] [PubMed]
- Tuoresmaki, P.; Vaisanen, S.; Neme, A.; Heikkinen, S.; Carlberg, C. Patterns of genome-wide VDR locations. PLoS ONE 2014, 9, e96105. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.J. Vitamin D and the RNA transcriptome: More than mRNA regulation. Front. Physiol. 2014, 5, 181. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C.; Munoz, A. An update on vitamin D signaling and cancer. Semin. Cancer Biol. 2020, in press. [Google Scholar] [CrossRef]
- Hanel, A.; Carlberg, C. Vitamin D and evolution: Pharmacologic implications. Biochem. Pharmacol. 2020, 173, 113595. [Google Scholar] [CrossRef]
- Abe, E.; Miyaura, C.; Sakagami, H.; Takeda, M.; Konno, K.; Yamazaki, T.; Yoshiki, S.; Suda, T. Differentiation of mouse myeloid leukemia cells induced by 1 alpha,25-dihydroxyvitamin D3. Proc. Natl. Acad. Sci. USA 1981, 78, 4990–4994. [Google Scholar] [CrossRef]
- Colston, K.; Colston, M.J.; Feldman, D. 1,25-dihydroxyvitamin D3 and malignant melanoma: The presence of receptors and inhibition of cell growth in culture. Endocrinology 1981, 108, 1083–1086. [Google Scholar] [CrossRef]
- Nieto, M.A. Context-specific roles of EMT programmes in cancer cell dissemination. Nat. Cell Biol. 2017, 19, 416–418. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Grant, W.B. Review of Recent Advances in Understanding the Role of Vitamin D in Reducing Cancer Risk: Breast, Colorectal, Prostate, and Overall Cancer. Anticancer Res. 2020, 40, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Barbachano, A.; Fernandez-Barral, A.; Ferrer-Mayorga, G.; Costales-Carrera, A.; Larriba, M.J.; Munoz, A. The endocrine vitamin D system in the gut. Mol. Cell. Endocrinol. 2017, 453, 79–87. [Google Scholar] [CrossRef]
- Christakos, S.; Li, S.; De La Cruz, J.; Shroyer, N.F.; Criss, Z.K.; Verzi, M.P.; Fleet, J.C. Vitamin D and the intestine: Review and update. J. Steroid Biochem. Mol. Biol. 2020, 196, 105501. [Google Scholar] [CrossRef]
- Palmer, H.G.; Gonzalez-Sancho, J.M.; Espada, J.; Berciano, M.T.; Puig, I.; Baulida, J.; Quintanilla, M.; Cano, A.; de Herreros, A.G.; Lafarga, M.; et al. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J. Cell Biol. 2001, 154, 369–387. [Google Scholar] [CrossRef]
- Ordonez-Moran, P.; Larriba, M.J.; Palmer, H.G.; Valero, R.A.; Barbachano, A.; Dunach, M.; de Herreros, A.G.; Villalobos, C.; Berciano, M.T.; Lafarga, M.; et al. RhoA—ROCK and p38MAPK-MSK1 mediate vitamin D effects on gene expression, phenotype, and Wnt pathway in colon cancer cells. J. Cell Biol. 2008, 183, 697–710. [Google Scholar] [CrossRef]
- Palmer, H.G.; Sanchez-Carbayo, M.; Ordonez-Moran, P.; Larriba, M.J.; Cordon-Cardo, C.; Munoz, A. Genetic signatures of differentiation induced by 1alpha,25-dihydroxyvitamin D3 in human colon cancer cells. Cancer Res. 2003, 63, 7799–7806. [Google Scholar]
- Fetahu, I.S.; Hummel, D.M.; Manhardt, T.; Aggarwal, A.; Baumgartner-Parzer, S.; Kallay, E. Regulation of the calcium-sensing receptor expression by 1,25-dihydroxyvitamin D3, interleukin-6, and tumor necrosis factor alpha in colon cancer cells. J. Steroid Biochem. Mol. Biol. 2014, 144, 228–231. [Google Scholar] [CrossRef]
- Aggarwal, A.; Hobaus, J.; Tennakoon, S.; Prinz-Wohlgenannt, M.; Graca, J.; Price, S.A.; Heffeter, P.; Berger, W.; Baumgartner-Parzer, S.; Kallay, E. Active vitamin D potentiates the anti-neoplastic effects of calcium in the colon: A cross talk through the calcium-sensing receptor. J. Steroid Biochem. Mol. Biol. 2016, 155, 231–238. [Google Scholar] [CrossRef]
- Aggarwal, A.; Kallay, E. Cross Talk between the Calcium-Sensing Receptor and the Vitamin D System in Prevention of Cancer. Front. Physiol. 2016, 7, 451. [Google Scholar] [CrossRef]
- Tennakoon, S.; Aggarwal, A.; Kallay, E. The calcium-sensing receptor and the hallmarks of cancer. Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1863, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Liu, L.; Masugi, Y.; Qian, Z.R.; Nishihara, R.; Keum, N.; Wu, K.; Smith-Warner, S.; Ma, Y.; Nowak, J.A.; et al. Calcium intake and risk of colorectal cancer according to expression status of calcium-sensing receptor (CASR). Gut 2018, 67, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, N.I.; Palmer, H.G.; Garcia, M.; Gonzalez-Martin, A.; del Rio, M.; Barettino, D.; Volpert, O.; Munoz, A.; Jimenez, B. 1alpha,25-Dihydroxyvitamin D3 regulates the expression of Id1 and Id2 genes and the angiogenic phenotype of human colon carcinoma cells. Oncogene 2005, 24, 6533–6544. [Google Scholar] [CrossRef]
- Kouchi, Z.; Fujiwara, Y.; Yamaguchi, H.; Nakamura, Y.; Fukami, K. Phosphatidylinositol 5-phosphate 4-kinase type II beta is required for vitamin D receptor-dependent E-cadherin expression in SW480 cells. Biochem. Biophys. Res. Commun. 2011, 408, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Ricca, C.; Aillon, A.; Viano, M.; Bergandi, L.; Aldieri, E.; Silvagno, F. Vitamin D inhibits the epithelial-mesenchymal transition by a negative feedback regulation of TGF-beta activity. J. Steroid Biochem. Mol. Biol. 2019, 187, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhu, J.; Zuo, S.; Ma, J.; Zhang, J.; Chen, G.; Wang, X.; Pan, Y.; Liu, Y.; Wang, P. 1,25(OH)2D3 attenuates TGF-beta1/beta2-induced increased migration and invasion via inhibiting epithelial-mesenchymal transition in colon cancer cells. Biochem. Biophys. Res. Commun. 2015, 468, 130–135. [Google Scholar] [CrossRef]
- Li, T.; Zhu, J.; Zuo, S.; Chen, S.; Ma, J.; Ma, Y.; Guo, S.; Wang, P.; Liu, Y. 1,25(OH)2D3 Attenuates IL-1beta-Induced Epithelial-to-Mesenchymal Transition Through Inhibiting the Expression of lncTCF7. Oncol. Res. 2019, 27, 739–750. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e123. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Merlos-Suarez, A.; Barriga, F.M.; Jung, P.; Iglesias, M.; Cespedes, M.V.; Rossell, D.; Sevillano, M.; Hernando-Momblona, X.; da Silva-Diz, V.; Munoz, P.; et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 2011, 8, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Krausova, M.; Korinek, V. Wnt signaling in adult intestinal stem cells and cancer. Cell Signal. 2014, 26, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Larriba, M.J.; Gonzalez-Sancho, J.M.; Barbachano, A.; Niell, N.; Ferrer-Mayorga, G.; Munoz, A. Vitamin D Is a Multilevel Repressor of Wnt/b-Catenin Signaling in Cancer Cells. Cancers 2013, 5, 1242–1260. [Google Scholar] [CrossRef]
- Shah, S.; Islam, M.N.; Dakshanamurthy, S.; Rizvi, I.; Rao, M.; Herrell, R.; Zinser, G.; Valrance, M.; Aranda, A.; Moras, D.; et al. The molecular basis of vitamin D receptor and beta-catenin crossregulation. Mol. Cell 2006, 21, 799–809. [Google Scholar] [CrossRef]
- Aguilera, O.; Pena, C.; Garcia, J.M.; Larriba, M.J.; Ordonez-Moran, P.; Navarro, D.; Barbachano, A.; Lopez de Silanes, I.; Ballestar, E.; Fraga, M.F.; et al. The Wnt antagonist DICKKOPF-1 gene is induced by 1alpha,25-dihydroxyvitamin D3 associated to the differentiation of human colon cancer cells. Carcinogenesis 2007, 28, 1877–1884. [Google Scholar] [CrossRef]
- Beildeck, M.E.; Islam, M.; Shah, S.; Welsh, J.; Byers, S.W. Control of TCF-4 expression by VDR and vitamin D in the mouse mammary gland and colorectal cancer cell lines. PLoS ONE 2009, 4, e7872. [Google Scholar] [CrossRef]
- Kaler, P.; Augenlicht, L.; Klampfer, L. Macrophage-derived IL-1beta stimulates Wnt signaling and growth of colon cancer cells: A crosstalk interrupted by vitamin D3. Oncogene 2009, 28, 3892–3902. [Google Scholar] [CrossRef]
- Groschel, C.; Aggarwal, A.; Tennakoon, S.; Hobaus, J.; Prinz-Wohlgenannt, M.; Marian, B.; Heffeter, P.; Berger, W.; Kallay, E. Effect of 1,25-dihydroxyvitamin D3 on the Wnt pathway in non-malignant colonic cells. J. Steroid Biochem. Mol. Biol. 2016, 155, 224–230. [Google Scholar] [CrossRef]
- Larriba, M.J.; Ordonez-Moran, P.; Chicote, I.; Martin-Fernandez, G.; Puig, I.; Munoz, A.; Palmer, H.G. Vitamin D receptor deficiency enhances Wnt/beta-catenin signaling and tumor burden in colon cancer. PLoS ONE 2011, 6, e23524. [Google Scholar] [CrossRef]
- Zheng, W.; Wong, K.E.; Zhang, Z.; Dougherty, U.; Mustafi, R.; Kong, J.; Deb, D.K.; Zheng, H.; Bissonnette, M.; Li, Y.C. Inactivation of the vitamin D receptor in APC(min/+) mice reveals a critical role for the vitamin D receptor in intestinal tumor growth. Int. J. Cancer 2012, 130, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Bostick, R.M. Effects of supplemental vitamin D and calcium on normal colon tissue and circulating biomarkers of risk for colorectal neoplasms. J. Steroid Biochem. Mol. Biol. 2015, 148, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Fedirko, V.; Bostick, R.M.; Flanders, W.D.; Long, Q.; Sidelnikov, E.; Shaukat, A.; Daniel, C.R.; Rutherford, R.E.; Woodard, J.J. Effects of vitamin d and calcium on proliferation and differentiation in normal colon mucosa: A randomized clinical trial. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2933–2941. [Google Scholar] [CrossRef] [PubMed]
- Ahearn, T.U.; Shaukat, A.; Flanders, W.D.; Rutherford, R.E.; Bostick, R.M. A randomized clinical trial of the effects of supplemental calcium and vitamin D3 on the APC/beta-catenin pathway in the normal mucosa of colorectal adenoma patients. Cancer Prev. Res. 2012, 5, 1247–1256. [Google Scholar] [CrossRef]
- Ferrer-Mayorga, G.; Alvarez-Diaz, S.; Valle, N.; De Las Rivas, J.; Mendes, M.; Barderas, R.; Canals, F.; Tapia, O.; Casal, J.I.; Lafarga, M.; et al. Cystatin D locates in the nucleus at sites of active transcription and modulates gene and protein expression. J. Biol. Chem. 2015, 290, 26533–26548. [Google Scholar] [CrossRef]
- Alvarez-Diaz, S.; Valle, N.; Garcia, J.M.; Pena, C.; Freije, J.M.; Quesada, V.; Astudillo, A.; Bonilla, F.; Lopez-Otin, C.; Munoz, A. Cystatin D is a candidate tumor suppressor gene induced by vitamin D in human colon cancer cells. J. Clin. Investig. 2009, 119, 2343–2358. [Google Scholar] [CrossRef]
- Alvarez-Diaz, S.; Valle, N.; Ferrer-Mayorga, G.; Lombardia, L.; Herrera, M.; Dominguez, O.; Segura, M.F.; Bonilla, F.; Hernando, E.; Munoz, A. MicroRNA-22 is induced by vitamin D and contributes to its antiproliferative, antimigratory and gene regulatory effects in colon cancer cells. Hum. Mol. Genet. 2012, 21, 2157–2165. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, X.; Cheng, R.; Yang, F.; Yu, M.; Wang, C.; Cui, S.; Hong, Y.; Liang, H.; Liu, M.; et al. The Jun/miR-22/HuR regulatory axis contributes to tumourigenesis in colorectal cancer. Mol. Cancer 2018, 17, 11. [Google Scholar] [CrossRef]
- Xu, M.; Li, J.; Wang, X.; Meng, S.; Shen, J.; Wang, S.; Xu, X.; Xie, B.; Liu, B.; Xie, L. MiR-22 suppresses epithelial-mesenchymal transition in bladder cancer by inhibiting Snail and MAPK1/Slug/vimentin feedback loop. Cell Death Dis. 2018, 9, 209. [Google Scholar] [CrossRef]
- Pereira, F.; Barbachano, A.; Silva, J.; Bonilla, F.; Campbell, M.J.; Munoz, A.; Larriba, M.J. KDM6B/JMJD3 histone demethylase is induced by vitamin D and modulates its effects in colon cancer cells. Hum. Mol. Genet. 2011, 20, 4655–4665. [Google Scholar] [CrossRef]
- Pereira, F.; Barbachano, A.; Singh, P.K.; Campbell, M.J.; Munoz, A.; Larriba, M.J. Vitamin D has wide regulatory effects on histone demethylase genes. Cell Cycle 2012, 11, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Barbachano, A.; Ordonez-Moran, P.; Garcia, J.M.; Sanchez, A.; Pereira, F.; Larriba, M.J.; Martinez, N.; Hernandez, J.; Landolfi, S.; Bonilla, F.; et al. SPROUTY-2 and E-cadherin regulate reciprocally and dictate colon cancer cell tumourigenicity. Oncogene 2010, 29, 4800–4813. [Google Scholar] [CrossRef] [PubMed]
- Barbachano, A.; Fernandez-Barral, A.; Pereira, F.; Segura, M.F.; Ordonez-Moran, P.; Pau, E.C.D.S.; Gonzalez-Sancho, J.M.; Hanniford, D.; Martinez, N.; Costales-Carrera, A.; et al. SPROUTY-2 represses the epithelial phenotype of colon carcinoma cells via upregulation of ZEB1 mediated by ETS1 and miR-200/miR-150. Oncogene 2016, 35, 2991–3003. [Google Scholar] [CrossRef] [PubMed]
- Ordonez-Moran, P.; Irmisch, A.; Barbachano, A.; Chicote, I.; Tenbaum, S.; Landolfi, S.; Tabernero, J.; Huelsken, J.; Munoz, A.; Palmer, H.G. SPROUTY2 is a beta-catenin and FOXO3a target gene indicative of poor prognosis in colon cancer. Oncogene 2014, 33, 1975–1985. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Palmer, H.G.; Larriba, M.J.; Garcia, J.M.; Ordonez-Moran, P.; Pena, C.; Peiro, S.; Puig, I.; Rodriguez, R.; de la Fuente, R.; Bernad, A.; et al. The transcription factor SNAIL represses vitamin D receptor expression and responsiveness in human colon cancer. Nat. Med. 2004, 10, 917–919. [Google Scholar] [CrossRef] [PubMed]
- Larriba, M.J.; Valle, N.; Palmer, H.G.; Ordonez-Moran, P.; Alvarez-Diaz, S.; Becker, K.F.; Gamallo, C.; de Herreros, A.G.; Gonzalez-Sancho, J.M.; Munoz, A. The inhibition of Wnt/beta-catenin signalling by 1alpha,25-dihydroxyvitamin D3 is abrogated by Snail1 in human colon cancer cells. Endocr. Relat. Cancer 2007, 14, 141–151. [Google Scholar] [CrossRef]
- Larriba, M.J.; Martin-Villar, E.; Garcia, J.M.; Pereira, F.; Pena, C.; de Herreros, A.G.; Bonilla, F.; Munoz, A. Snail2 cooperates with Snail1 in the repression of vitamin D receptor in colon cancer. Carcinogenesis 2009, 30, 1459–1468. [Google Scholar] [CrossRef]
- Pena, C.; Garcia, J.M.; Silva, J.; Garcia, V.; Rodriguez, R.; Alonso, I.; Millan, I.; Salas, C.; de Herreros, A.G.; Munoz, A.; et al. E-cadherin and vitamin D receptor regulation by SNAIL and ZEB1 in colon cancer: Clinicopathological correlations. Hum. Mol. Genet. 2005, 14, 3361–3370. [Google Scholar] [CrossRef]
- Pena, C.; Garcia, J.M.; Garcia, V.; Silva, J.; Dominguez, G.; Rodriguez, R.; Maximiano, C.; de Herreros, A.G.; Munoz, A.; Bonilla, F. The expression levels of the transcriptional regulators p300 and CtBP modulate the correlations between SNAIL, ZEB1, E-cadherin and vitamin D receptor in human colon carcinomas. Int. J. Cancer 2006, 119, 2098–2104. [Google Scholar] [CrossRef]
- Pena, C.; Garcia, J.M.; Larriba, M.J.; Barderas, R.; Gomez, I.; Herrera, M.; Garcia, V.; Silva, J.; Dominguez, G.; Rodriguez, R.; et al. SNAI1 expression in colon cancer related with CDH1 and VDR downregulation in normal adjacent tissue. Oncogene 2009, 28, 4375–4385. [Google Scholar] [CrossRef][Green Version]
- Larriba, M.J.; Gonzalez-Sancho, J.M.; Bonilla, F.; Munoz, A. Interaction of vitamin D with membrane-based signaling pathways. Front. Physiol. 2014, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Larriba, M.J.; de Herreros, A.G.; Munoz, A. Vitamin D and the Epithelial to Mesenchymal Transition. Stem. Cells Int. 2016, 2016, 6213872. [Google Scholar] [CrossRef] [PubMed]
- Andl, C.D.; Rustgi, A.K. No one-way street: Cross-talk between e-cadherin and receptor tyrosine kinase (RTK) signaling: A mechanism to regulate RTK activity. Cancer Biol. Ther. 2005, 4, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.M.; Kallay, E.; Hofer, H.; Hulla, W.; Manhardt, T.; Peterlik, M.; Cross, H.S. Growth regulation of human colon cancer cells by epidermal growth factor and 1,25-dihydroxyvitamin D3 is mediated by mutual modulation of receptor expression. Eur. J. Cancer 1998, 34, 2119–2125. [Google Scholar] [CrossRef]
- Tong, W.M.; Hofer, H.; Ellinger, A.; Peterlik, M.; Cross, H.S. Mechanism of antimitogenic action of vitamin D in human colon carcinoma cells: Relevance for suppression of epidermal growth factor-stimulated cell growth. Oncol. Res. 1999, 11, 77–84. [Google Scholar] [PubMed]
- Dougherty, U.; Mustafi, R.; Sadiq, F.; Almoghrabi, A.; Mustafi, D.; Kreisheh, M.; Sundaramurthy, S.; Liu, W.; Konda, V.J.; Pekow, J.; et al. The renin-angiotensin system mediates EGF receptor-vitamin d receptor cross-talk in colitis-associated colon cancer. Clin. Cancer Res. 2014, 20, 5848–5859. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Blasiak, J.; Pawlowska, E.; Chojnacki, J.; Szczepanska, J.; Fila, M.; Chojnacki, C. Vitamin D in Triple—Negative and BRCA1-Deficient Breast Cancer-Implications for Pathogenesis and Therapy. Int. J. Mol. Sci. 2020, 21, 3670. [Google Scholar] [CrossRef]
- Welsh, J. Function of the vitamin D endocrine system in mammary gland and breast cancer. Mol. Cell. Endocrinol. 2017, 453, 88–95. [Google Scholar] [CrossRef]
- Berger, U.; Wilson, P.; McClelland, R.A.; Colston, K.; Haussler, M.R.; Pike, J.W.; Coombes, R.C. Immunocytochemical detection of 1,25-dihydroxyvitamin D receptors in normal human tissues. J. Clin. Endocrinol. Metab. 1988, 67, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Pendas-Franco, N.; Gonzalez-Sancho, J.M.; Suarez, Y.; Aguilera, O.; Steinmeyer, A.; Gamallo, C.; Berciano, M.T.; Lafarga, M.; Munoz, A. Vitamin D regulates the phenotype of human breast cancer cells. Differentiation 2007, 75, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Lopes, N.; Sousa, B.; Martins, D.; Gomes, M.; Vieira, D.; Veronese, L.A.; Milanezi, F.; Paredes, J.; Costa, J.L.; Schmitt, F. Alterations in Vitamin D signalling and metabolic pathways in breast cancer progression: A study of VDR, CYP27B1 and CYP24A1 expression in benign and malignant breast lesions. BMC Cancer 2010, 10, 483. [Google Scholar] [CrossRef]
- Al-Azhri, J.; Zhang, Y.; Bshara, W.; Zirpoli, G.; McCann, S.E.; Khoury, T.; Morrison, C.D.; Edge, S.B.; Ambrosone, C.B.; Yao, S. Tumor Expression of Vitamin D Receptor and Breast Cancer Histopathological Characteristics and Prognosis. Clin. Cancer Res. 2017, 23, 97–103. [Google Scholar] [CrossRef]
- Gocek, E.; Studzinski, G.P. Vitamin D and differentiation in cancer. Crit. Rev. Clin. Lab. Sci. 2009, 46, 190–209. [Google Scholar] [CrossRef] [PubMed]
- Narvaez, C.J.; Matthews, D.; LaPorta, E.; Simmons, K.M.; Beaudin, S.; Welsh, J. The impact of vitamin D in breast cancer: Genomics, pathways, metabolism. Front. Physiol. 2014, 5, 213. [Google Scholar] [CrossRef]
- Welsh, J. Vitamin D and breast cancer: Past and present. J. Steroid Biochem. Mol. Biol. 2018, 177, 15–20. [Google Scholar] [CrossRef]
- Lopes, N.; Carvalho, J.; Duraes, C.; Sousa, B.; Gomes, M.; Costa, J.L.; Oliveira, C.; Paredes, J.; Schmitt, F. 1Alpha,25-dihydroxyvitamin D3 induces de novo E-cadherin expression in triple-negative breast cancer cells by CDH1-promoter demethylation. Anticancer Res. 2012, 32, 249–257. [Google Scholar]
- Gonzalez-Sancho, J.M.; Alvarez-Dolado, M.; Munoz, A. 1,25-Dihydroxyvitamin D3 inhibits tenascin-C expression in mammary epithelial cells. FEBS Lett. 1998, 426, 225–228. [Google Scholar] [CrossRef]
- Koli, K.; Keski-Oja, J. 1alpha,25-dihydroxyvitamin D3 and its analogues down-regulate cell invasion-associated proteases in cultured malignant cells. Cell Growth Differ. 2000, 11, 221–229. [Google Scholar]
- Sundaram, S.; Beckman, M.J.; Bajwa, A.; Wei, J.; Smith, K.M.; Posner, G.H.; Gewirtz, D.A. QW-1624F2-2, a synthetic analogue of 1,25-dihydroxyvitamin D3, enhances the response to other deltanoids and suppresses the invasiveness of human metastatic breast tumor cells. Mol. Cancer Ther. 2006, 5, 2806–2814. [Google Scholar] [CrossRef] [PubMed]
- LaPorta, E.; Welsh, J. Modeling vitamin D actions in triple negative/basal-like breast cancer. J. Steroid Biochem. Mol. Biol. 2014, 144, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Wilmanski, T.; Barnard, A.; Parikh, M.R.; Kirshner, J.; Buhman, K.; Burgess, J.; Teegarden, D. 1alpha,25-Dihydroxyvitamin D Inhibits the Metastatic Capability of MCF10CA1a and MDA-MB-231 Cells in an In Vitro Model of Breast to Bone Metastasis. Nutr. Cancer 2016, 68, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.M.; Frandsen, T.L.; Brunner, N.; Binderup, L. 1 alpha,25-Dihydroxyvitamin D3 inhibits the invasive potential of human breast cancer cells in vitro. Clin. Exp. Metastasis 1994, 12, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Vanoirbeek, E.; Eelen, G.; Verlinden, L.; Carmeliet, G.; Mathieu, C.; Bouillon, R.; O’Connor, R.; Xiao, G.; Verstuyf, A. PDLIM2 expression is driven by vitamin D and is involved in the pro-adhesion, and anti-migration and -invasion activity of vitamin D. Oncogene 2014, 33, 1904–1911. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.D.; Aggarwal, A.; Swami, S.; Krishnan, A.V.; Ji, L.; Albertelli, M.A.; Feldman, B.J. Tumor Autonomous Effects of Vitamin D Deficiency Promote Breast Cancer Metastasis. Endocrinology 2016, 157, 1341–1347. [Google Scholar] [CrossRef]
- Arensman, M.D.; Nguyen, P.; Kershaw, K.M.; Lay, A.R.; Ostertag-Hill, C.A.; Sherman, M.H.; Downes, M.; Liddle, C.; Evans, R.M.; Dawson, D.W. Calcipotriol Targets LRP6 to Inhibit Wnt Signaling in Pancreatic Cancer. Mol. Cancer Res. 2015, 13, 1509–1519. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, Z.H.; Fu, L.; Song, J.; Xie, D.D.; Yu, D.X.; Xu, D.X.; Sun, G.P. Calcitriol inhibits migration and invasion of renal cell carcinoma cells by suppressing Smad2/3-, STAT3- and beta-catenin-mediated epithelial-mesenchymal transition. Cancer Sci. 2020, 111, 59–71. [Google Scholar] [CrossRef]
- Palmer, H.G.; Anjos-Afonso, F.; Carmeliet, G.; Takeda, H.; Watt, F.M. The vitamin D receptor is a Wnt effector that controls hair follicle differentiation and specifies tumor type in adult epidermis. PLoS ONE 2008, 3, e1483. [Google Scholar] [CrossRef]
- Jiang, Y.J.; Teichert, A.E.; Fong, F.; Oda, Y.; Bikle, D.D. 1alpha,25(OH)2-dihydroxyvitamin D3/VDR protects the skin from UVB-induced tumor formation by interacting with the beta-catenin pathway. J. Steroid Biochem. Mol. Biol. 2013, 136, 229–232. [Google Scholar] [CrossRef]
- Tapia, C.; Suares, A.; De Genaro, P.; Gonzalez-Pardo, V. In vitro studies revealed a downregulation of Wnt/beta-catenin cascade by active vitamin D and TX 527 analog in a Kaposi’s sarcoma cellular model. Toxicol. In Vitro 2020, 63, 104748. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.C.; Kuo, S.F.; Chen, C.H.; Ng, S.; Lin, S.F.; Yeh, C.N.; Chen, L.W.; Takano, M.; Chen, T.C.; Juang, H.H.; et al. MART-10, the vitamin D analog, is a potent drug to inhibit anaplastic thyroid cancer cell metastatic potential. Cancer Lett. 2015, 369, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.F.; Gao, S.H.; Wang, P.; Zhang, H.M.; Liu, L.Z.; Ye, M.X.; Zhou, G.M.; Zhang, Z.L.; Li, B.Y. 1alpha,25(OH)(2)D(3) Suppresses the Migration of Ovarian Cancer SKOV-3 Cells through the Inhibition of Epithelial-Mesenchymal Transition. Int. J. Mol. Sci. 2016, 17, 1285. [Google Scholar] [CrossRef]
- Jiang, F.; Yang, Y.; Xue, L.; Li, B.; Zhang, Z. 1alpha,25-dihydroxyvitamin D3 Attenuates TGF-beta-Induced Pro-Fibrotic Effects in Human Lung Epithelial Cells through Inhibition of Epithelial-Mesenchymal Transition. Nutrients 2017, 9, 980. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Jeon, S.H.; Yi, J.Y.; Jin, Y.J.; Son, Y.S. Calcipotriol inhibits autocrine phosphorylation of EGF receptor in a calcium-dependent manner, a possible mechanism for its inhibition of cell proliferation and stimulation of cell differentiation. Biochem. Biophys. Res. Commun. 2001, 284, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Cordero, J.B.; Cozzolino, M.; Lu, Y.; Vidal, M.; Slatopolsky, E.; Stahl, P.D.; Barbieri, M.A.; Dusso, A. 1,25-Dihydroxyvitamin D down-regulates cell membrane growth- and nuclear growth-promoting signals by the epidermal growth factor receptor. J. Biol. Chem. 2002, 277, 38965–38971. [Google Scholar] [CrossRef]
- Boisseau-Garsaud, A.M.; Donatien, P.; Margerin, C.; Taieb, A. EGF receptor expression and growth of psoriatic and normal human keratinocytes are modulated by 1.25 (OH)2-vitamin D3 ex vivo. Arch. Dermatol. Res. 1996, 288, 453–457. [Google Scholar] [CrossRef]
- Kulling, P.M.; Olson, K.C.; Olson, T.L.; Feith, D.J.; Loughran, T.P., Jr. Vitamin D in hematological disorders and malignancies. Eur. J. Haematol. 2017, 98, 187–197. [Google Scholar] [CrossRef]
- Tanaka, H.; Abe, E.; Miyaura, C.; Shiina, Y.; Suda, T. 1 alpha,25-dihydroxyvitamin D3 induces differentiation of human promyelocytic leukemia cells (HL-60) into monocyte-macrophages, but not into granulocytes. Biochem. Biophys. Res. Commun. 1983, 117, 86–92. [Google Scholar] [CrossRef]
- Koeffler, H.P.; Amatruda, T.; Ikekawa, N.; Kobayashi, Y.; DeLuca, H.F. Induction of macrophage differentiation of human normal and leukemic myeloid stem cells by 1,25-dihydroxyvitamin D3 and its fluorinated analogues. Cancer Res. 1984, 44, 5624–5628. [Google Scholar]
- Liu, M.; Lee, M.H.; Cohen, M.; Bommakanti, M.; Freedman, L.P. Transcriptional activation of the Cdk inhibitor p21 by vitamin D3 leads to the induced differentiation of the myelomonocytic cell line U937. Genes Dev. 1996, 10, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Studzinski, G.P. Retinoblastoma protein and CCAAT/enhancer-binding protein beta are required for 1,25-dihydroxyvitamin D3-induced monocytic differentiation of HL60 cells. Cancer Res. 2004, 64, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Marchwicka, A.; Marcinkowska, E. Regulation of Expression of CEBP Genes by Variably Expressed Vitamin D Receptor and Retinoic Acid Receptor alpha in Human Acute Myeloid Leukemia Cell Lines. Int. J. Mol. Sci. 2018, 19, 1918. [Google Scholar] [CrossRef] [PubMed]
- Hmama, Z.; Nandan, D.; Sly, L.; Knutson, K.L.; Herrera-Velit, P.; Reiner, N.E. 1alpha,25-dihydroxyvitamin D(3)-induced myeloid cell differentiation is regulated by a vitamin D receptor-phosphatidylinositol 3-kinase signaling complex. J. Exp. Med. 1999, 190, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Studzinski, G.P.; Wang, X.; Ji, Y.; Wang, Q.; Zhang, Y.; Kutner, A.; Harrison, J.S. The rationale for deltanoids in therapy for myeloid leukemia: Role of KSR-MAPK-C/EBP pathway. J. Steroid Biochem. Mol. Biol. 2005, 97, 47–55. [Google Scholar] [CrossRef]
- Cao, H.; Xu, Y.; de Necochea-Campion, R.; Baylink, D.J.; Payne, K.J.; Tang, X.; Ratanatharathorn, C.; Ji, Y.; Mirshahidi, S.; Chen, C.S. Application of vitamin D and vitamin D analogs in acute myelogenous leukemia. Exp. Hematol. 2017, 50, 1–12. [Google Scholar] [CrossRef]
- Muto, A.; Kizaki, M.; Yamato, K.; Kawai, Y.; Kamata-Matsushita, M.; Ueno, H.; Ohguchi, M.; Nishihara, T.; Koeffler, H.P.; Ikeda, Y. 1,25-Dihydroxyvitamin D3 induces differentiation of a retinoic acid-resistant acute promyelocytic leukemia cell line (UF-1) associated with expression of p21(WAF1/CIP1) and p27(KIP1). Blood 1999, 93, 2225–2233. [Google Scholar] [CrossRef]
- Hickish, T.; Cunningham, D.; Colston, K.; Millar, B.C.; Sandle, J.; Mackay, A.G.; Soukop, M.; Sloane, J. The effect of 1,25-dihydroxyvitamin D3 on lymphoma cell lines and expression of vitamin D receptor in lymphoma. Br. J. Cancer 1993, 68, 668–672. [Google Scholar] [CrossRef]
- Ohlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Augsten, M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef]
- Ding, N.; Yu, R.T.; Subramaniam, N.; Sherman, M.H.; Wilson, C.; Rao, R.; Leblanc, M.; Coulter, S.; He, M.; Scott, C.; et al. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell 2013, 153, 601–613. [Google Scholar] [CrossRef]
- Duran, A.; Hernandez, E.D.; Reina-Campos, M.; Castilla, E.A.; Subramaniam, S.; Raghunandan, S.; Roberts, L.R.; Kisseleva, T.; Karin, M.; Diaz-Meco, M.T.; et al. p62/SQSTM1 by Binding to Vitamin D Receptor Inhibits Hepatic Stellate Cell Activity, Fibrosis, and Liver Cancer. Cancer Cell 2016, 30, 595–609. [Google Scholar] [CrossRef]
- Campos, L.T.; Brentani, H.; Roela, R.A.; Katayama, M.L.; Lima, L.; Rolim, C.F.; Milani, C.; Folgueira, M.A.; Brentani, M.M. Differences in transcriptional effects of 1alpha,25 dihydroxyvitamin D3 on fibroblasts associated to breast carcinomas and from paired normal breast tissues. J. Steroid Biochem. Mol. Biol. 2013, 133, 12–24. [Google Scholar] [CrossRef]
- Ferrer-Mayorga, G.; Gomez-Lopez, G.; Barbachano, A.; Fernandez-Barral, A.; Pena, C.; Pisano, D.G.; Cantero, R.; Rojo, F.; Munoz, A.; Larriba, M.J. Vitamin D receptor expression and associated gene signature in tumour stromal fibroblasts predict clinical outcome in colorectal cancer. Gut 2017, 66, 1449–1462. [Google Scholar] [CrossRef]
- Kong, F.; Li, L.; Wang, G.; Deng, X.; Li, Z.; Kong, X. VDR signaling inhibits cancer-associated-fibroblasts’ release of exosomal miR-10a-5p and limits their supportive effects on pancreatic cancer cells. Gut 2019, 68, 950–951. [Google Scholar] [CrossRef]
- Niell, N.; Larriba, M.J.; Ferrer-Mayorga, G.; Sanchez-Perez, I.; Cantero, R.; Real, F.X.; Del Peso, L.; Munoz, A.; Gonzalez-Sancho, J.M. The human PKP2/plakophilin-2 gene is induced by Wnt/beta-catenin in normal and colon cancer-associated fibroblasts. Int. J. Cancer 2018, 142, 792–804. [Google Scholar] [CrossRef]
- Ferrer-Mayorga, G.; Niell, N.; Cantero, R.; Gonzalez-Sancho, J.M.; Del Peso, L.; Munoz, A.; Larriba, M.J. Vitamin D and Wnt3A have additive and partially overlapping modulatory effects on gene expression and phenotype in human colon fibroblasts. Sci. Rep. 2019, 9, 8085. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Hu, W.; Lu, L.; Zhao, Y.; Zhou, Y.; Xiao, Z.; Zhang, L.; Zhang, H.; Li, X.; Li, W.; et al. Repurposing vitamin D for treatment of human malignancies via targeting tumor microenvironment. Acta Pharm. Sin. B 2019, 9, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Goktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Murata, K.; Jadhav, U.; Madha, S.; van Es, J.; Dean, J.; Cavazza, A.; Wucherpfennig, K.; Michor, F.; Clevers, H.; Shivdasani, R.A. Ascl2-Dependent Cell Dedifferentiation Drives Regeneration of Ablated Intestinal Stem Cells. Cell Stem Cell 2020, 26, 377–390.e376. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef]
- Jeong, Y.; Swami, S.; Krishnan, A.V.; Williams, J.D.; Martin, S.; Horst, R.L.; Albertelli, M.A.; Feldman, B.J.; Feldman, D.; Diehn, M. Inhibition of Mouse Breast Tumor-Initiating Cells by Calcitriol and Dietary Vitamin D. Mol. Cancer Ther. 2015, 14, 1951–1961. [Google Scholar] [CrossRef]
- So, J.Y.; Wahler, J.; Das Gupta, S.; Salerno, D.M.; Maehr, H.; Uskokovic, M.; Suh, N. HES1-mediated inhibition of Notch1 signaling by a Gemini vitamin D analog leads to decreased CD44(+)/CD24(-/low) tumor-initiating subpopulation in basal-like breast cancer. J. Steroid Biochem. Mol. Biol. 2015, 148, 111–121. [Google Scholar] [CrossRef]
- Wahler, J.; So, J.Y.; Cheng, L.C.; Maehr, H.; Uskokovic, M.; Suh, N. Vitamin D compounds reduce mammosphere formation and decrease expression of putative stem cell markers in breast cancer. J. Steroid Biochem. Mol. Biol. 2015, 148, 148–155. [Google Scholar] [CrossRef]
- Shan, N.L.; Wahler, J.; Lee, H.J.; Bak, M.J.; Gupta, S.D.; Maehr, H.; Suh, N. Vitamin D compounds inhibit cancer stem-like cells and induce differentiation in triple negative breast cancer. J. Steroid Biochem. Mol. Biol. 2017, 173, 122–129. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Rizvi, A.; Cui, T.; Han, C.; Banerjee, A.; Naseem, I.; Zheng, Y.; Wani, A.A.; Wang, Q.E. Depleting ovarian cancer stem cells with calcitriol. Oncotarget 2018, 9, 14481–14491. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Liu, L.; Hou, Y.; Li, B. 1alpha,25Dihydroxyvitamin D3 restrains stem celllike properties of ovarian cancer cells by enhancing vitamin D receptor and suppressing CD44. Oncol. Rep. 2019, 41, 3393–3403. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.B.; Jorgensen, A.; Nielsen, J.E.; Steinmeyer, A.; Leffers, H.; Juul, A.; De Meyts, E.R. Vitamin D metabolism and effects on pluripotency genes and cell differentiation in testicular germ cell tumors in vitro and in vivo. Neoplasia 2012, 14, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Barral, A.; Costales-Carrera, A.; Buira, S.P.; Jung, P.; Ferrer-Mayorga, G.; Larriba, M.J.; Bustamante-Madrid, P.; Dominguez, O.; Real, F.X.; Guerra-Pastrian, L.; et al. Vitamin D differentially regulates colon stem cells in patient-derived normal and tumor organoids. FEBS J. 2020, 287, 53–72. [Google Scholar] [CrossRef]
- Costales-Carrera, A.; Fernandez-Barral, A.; Bustamante-Madrid, P.; Dominguez, O.; Guerra-Pastrian, L.; Cantero, R.; del Peso, L.; Burgos, A.; Barbachano, A.; Muñoz, A. Comparative study of organoids from patient-derived normal and tumor colon and rectal tissue. Cancers 2020, 12, 2302. [Google Scholar] [CrossRef] [PubMed]
- Peregrina, K.; Houston, M.; Daroqui, C.; Dhima, E.; Sellers, R.S.; Augenlicht, L.H. Vitamin D is a determinant of mouse intestinal Lgr5 stem cell functions. Carcinogenesis 2015, 36, 25–31. [Google Scholar] [CrossRef]
- Augenlicht, L.H. Environmental Impact on Intestinal Stem Cell Functions in Mucosal Homeostasis and Tumorigenesis. J. Cell. Biochem. 2017, 118, 943–952. [Google Scholar] [CrossRef][Green Version]
- Li, W.; Zimmerman, S.E.; Peregrina, K.; Houston, M.; Mayoral, J.; Zhang, J.; Maqbool, S.; Zhang, Z.; Cai, Y.; Ye, K.; et al. The nutritional environment determines which and how intestinal stem cells contribute to homeostasis and tumorigenesis. Carcinogenesis 2019, 40, 937–946. [Google Scholar] [CrossRef]
- Wang, L.Q.; Yu, P.; Li, B.; Guo, Y.H.; Liang, Z.R.; Zheng, L.L.; Yang, J.H.; Xu, H.; Liu, S.; Zheng, L.S.; et al. miR-372 and miR-373 enhance the stemness of colorectal cancer cells by repressing differentiation signaling pathways. Mol. Oncol. 2018, 12, 1949–1964. [Google Scholar] [CrossRef]
- Paubelle, E.; Zylbersztejn, F.; Maciel, T.T.; Carvalho, C.; Mupo, A.; Cheok, M.; Lieben, L.; Sujobert, P.; Decroocq, J.; Yokoyama, A.; et al. Vitamin D Receptor Controls Cell Stemness in Acute Myeloid Leukemia and in Normal Bone Marrow. Cell Rep. 2020, 30, 739–754.e734. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Barral, A.; Bustamante-Madrid, P.; Ferrer-Mayorga, G.; Barbáchano, A.; Larriba, M.J.; Muñoz, A. Vitamin D Effects on Cell Differentiation and Stemness in Cancer. Cancers 2020, 12, 2413. https://doi.org/10.3390/cancers12092413
Fernández-Barral A, Bustamante-Madrid P, Ferrer-Mayorga G, Barbáchano A, Larriba MJ, Muñoz A. Vitamin D Effects on Cell Differentiation and Stemness in Cancer. Cancers. 2020; 12(9):2413. https://doi.org/10.3390/cancers12092413
Chicago/Turabian StyleFernández-Barral, Asunción, Pilar Bustamante-Madrid, Gemma Ferrer-Mayorga, Antonio Barbáchano, María Jesús Larriba, and Alberto Muñoz. 2020. "Vitamin D Effects on Cell Differentiation and Stemness in Cancer" Cancers 12, no. 9: 2413. https://doi.org/10.3390/cancers12092413
APA StyleFernández-Barral, A., Bustamante-Madrid, P., Ferrer-Mayorga, G., Barbáchano, A., Larriba, M. J., & Muñoz, A. (2020). Vitamin D Effects on Cell Differentiation and Stemness in Cancer. Cancers, 12(9), 2413. https://doi.org/10.3390/cancers12092413