Novel Caffeic Acid Phenethyl Ester-Mortalin Antibody Nanoparticles Offer Enhanced Selective Cytotoxicity to Cancer Cells

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
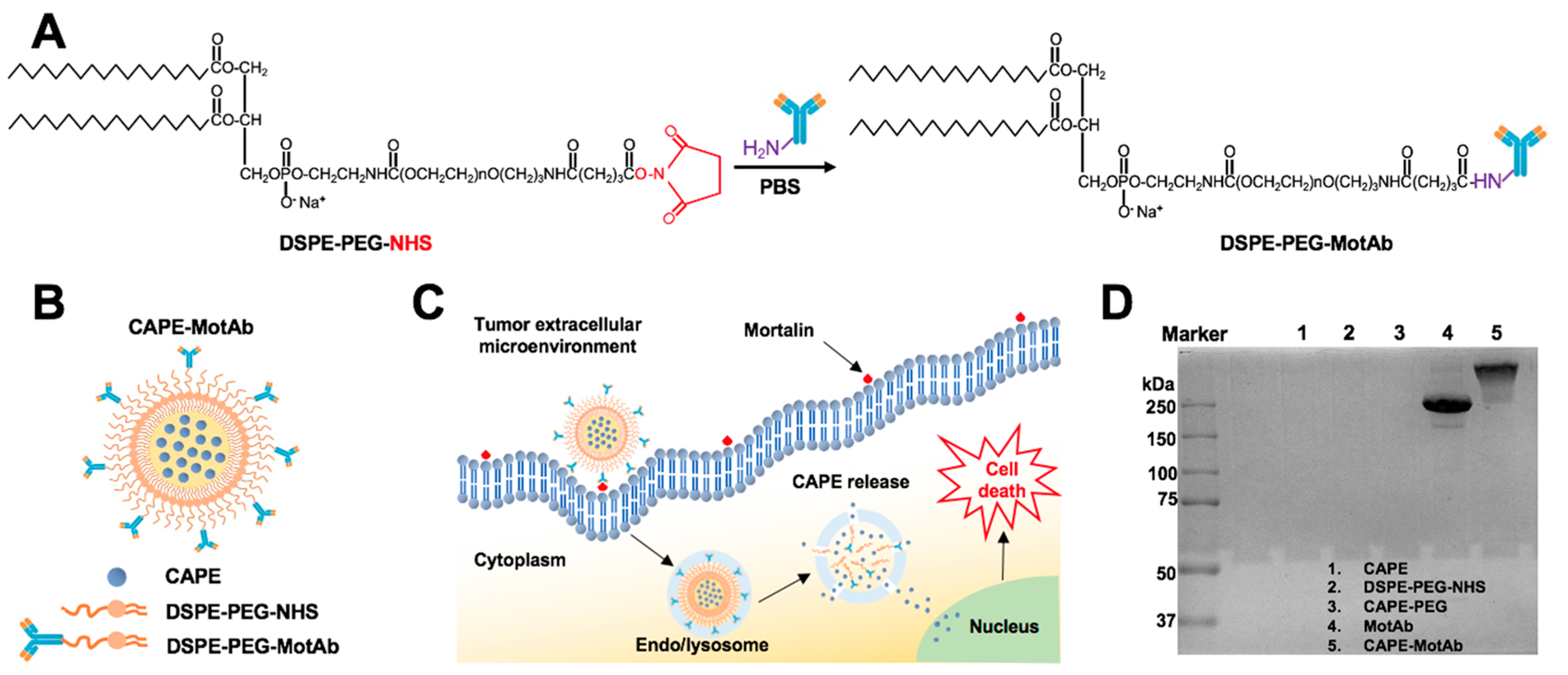
2.1. Preparation of CAPE-MotAb
2.2. Characterization of CAPE-MotAb
2.3. Encapsulation and Loading Efficiency of CAPE-MotAb
2.4. Cell Lines and Culture
2.5. Cell Viability and Morphological Observations
2.6. Colony Formation Assay
2.7. Flow Cytometry Analysis
2.8. Apoptosis Assay
2.9. Western Blotting
2.10. Fluorescence Microscopy
2.11. Cellular Uptake
2.12. Wound Scratch Assay
2.13. Cell Invasion Assay
2.14. In Vivo Xenograft Assay
2.15. Statistical Analysis
3. Results
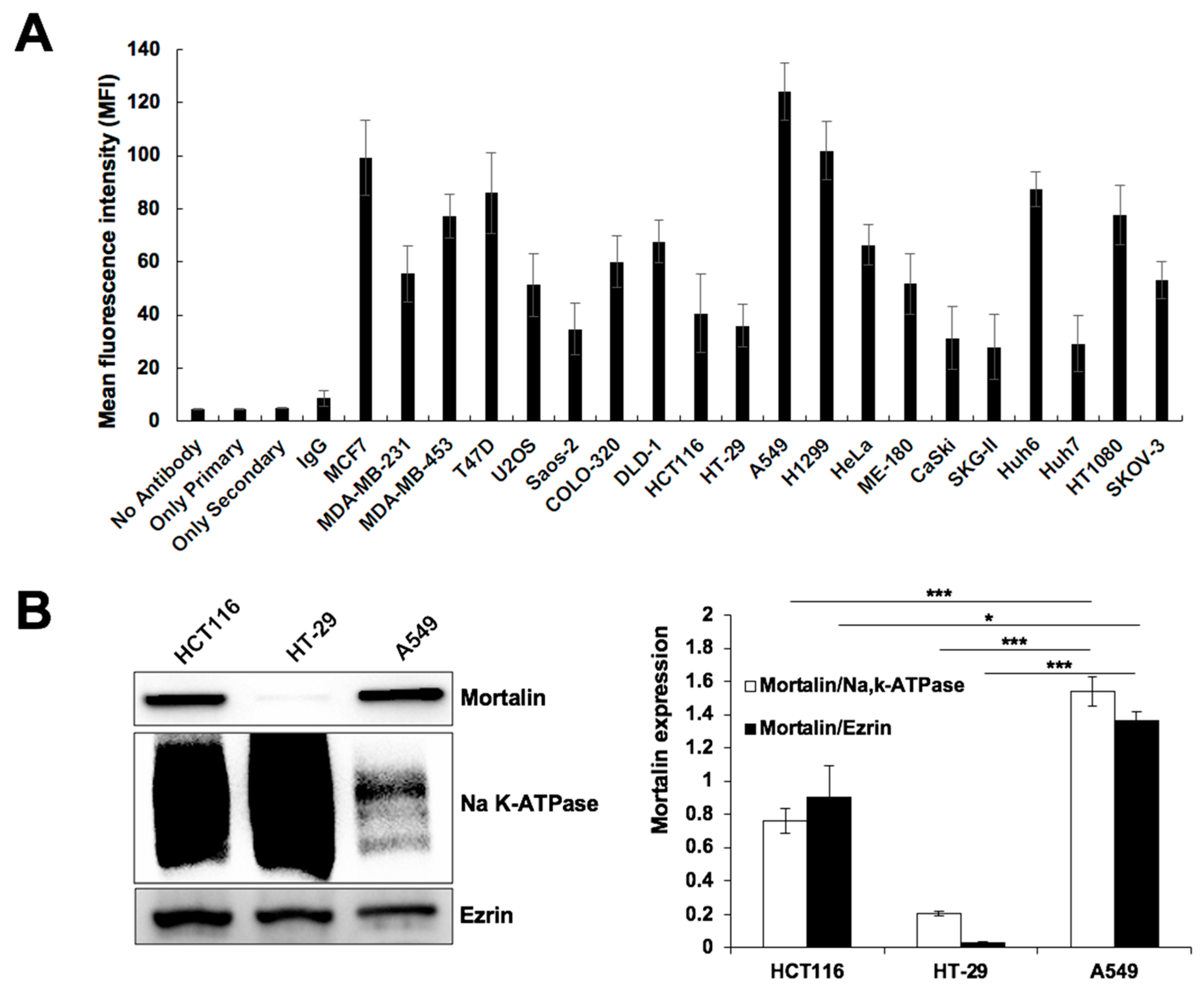
3.1. Generation and Characterization of CAPE-MotAb Nanoparticles
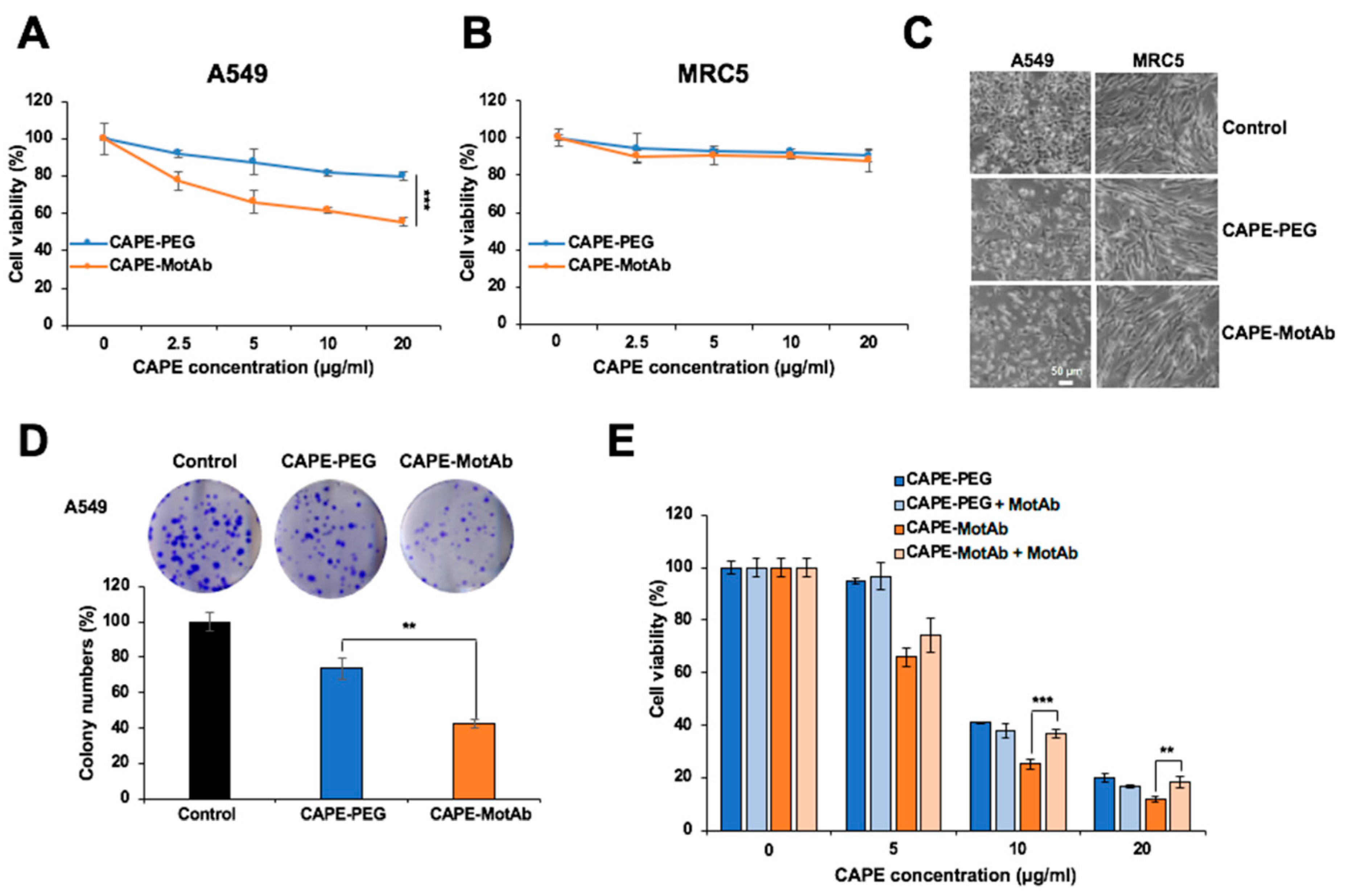
3.2. CAPE-MotAb Showed Enhanced Selective Cytotoxicity in Cancer Cells
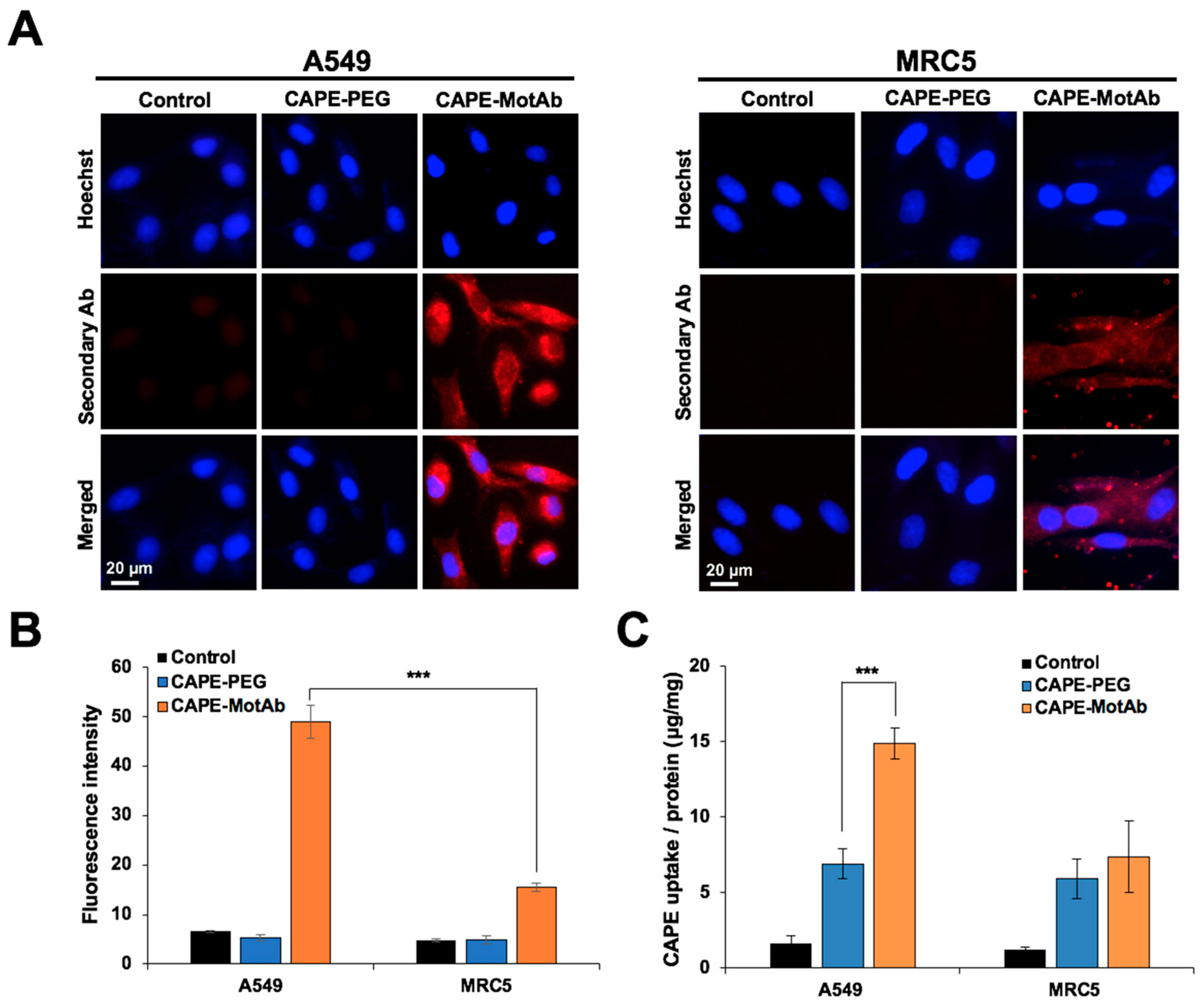
3.3. CAPE-MotAb Showed Selective Uptake in Cancer Cells
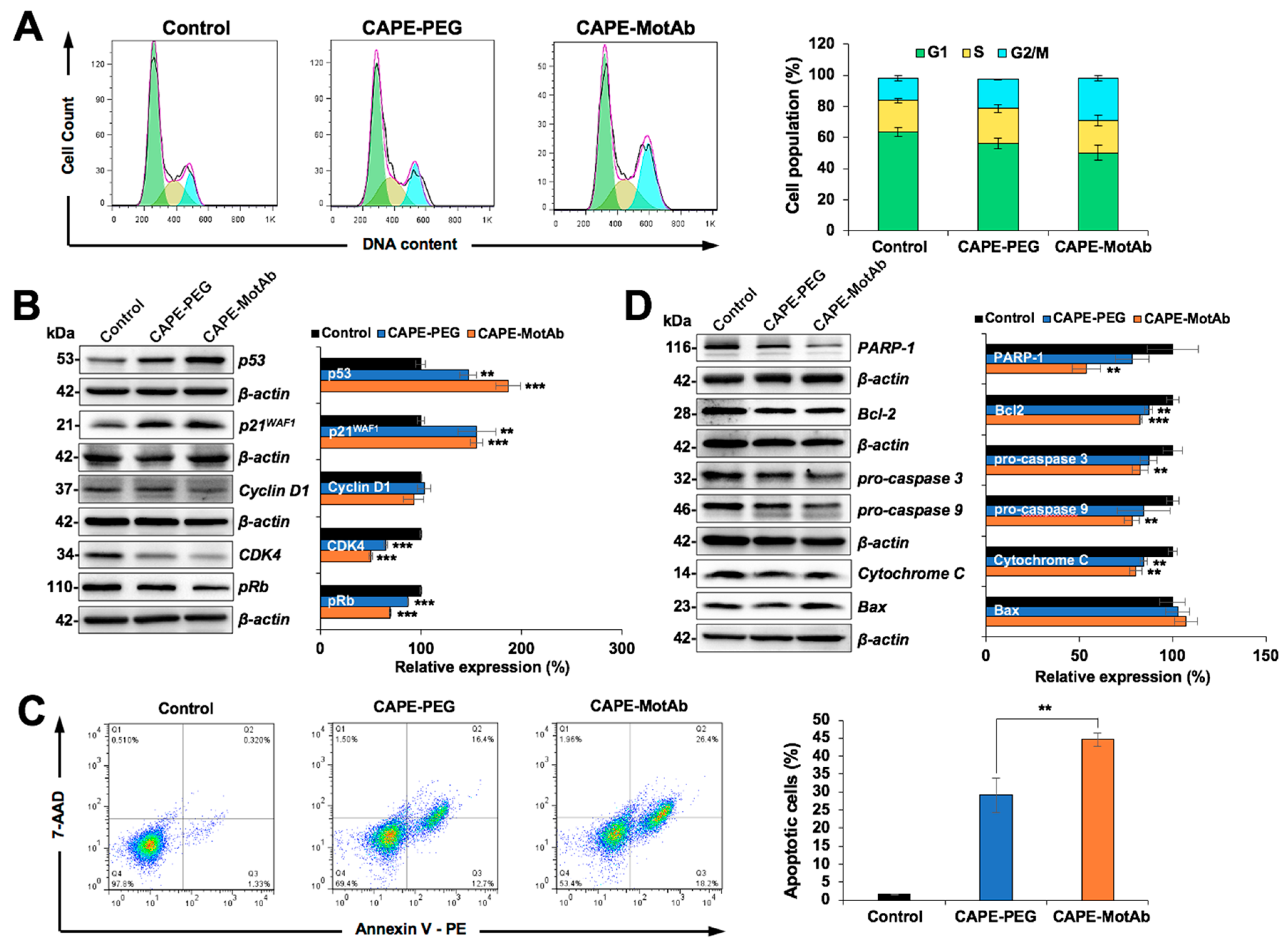
3.4. CAPE-MotAb Caused Stronger Cell Cycle Arrest and Apoptosis in Cancer Cells
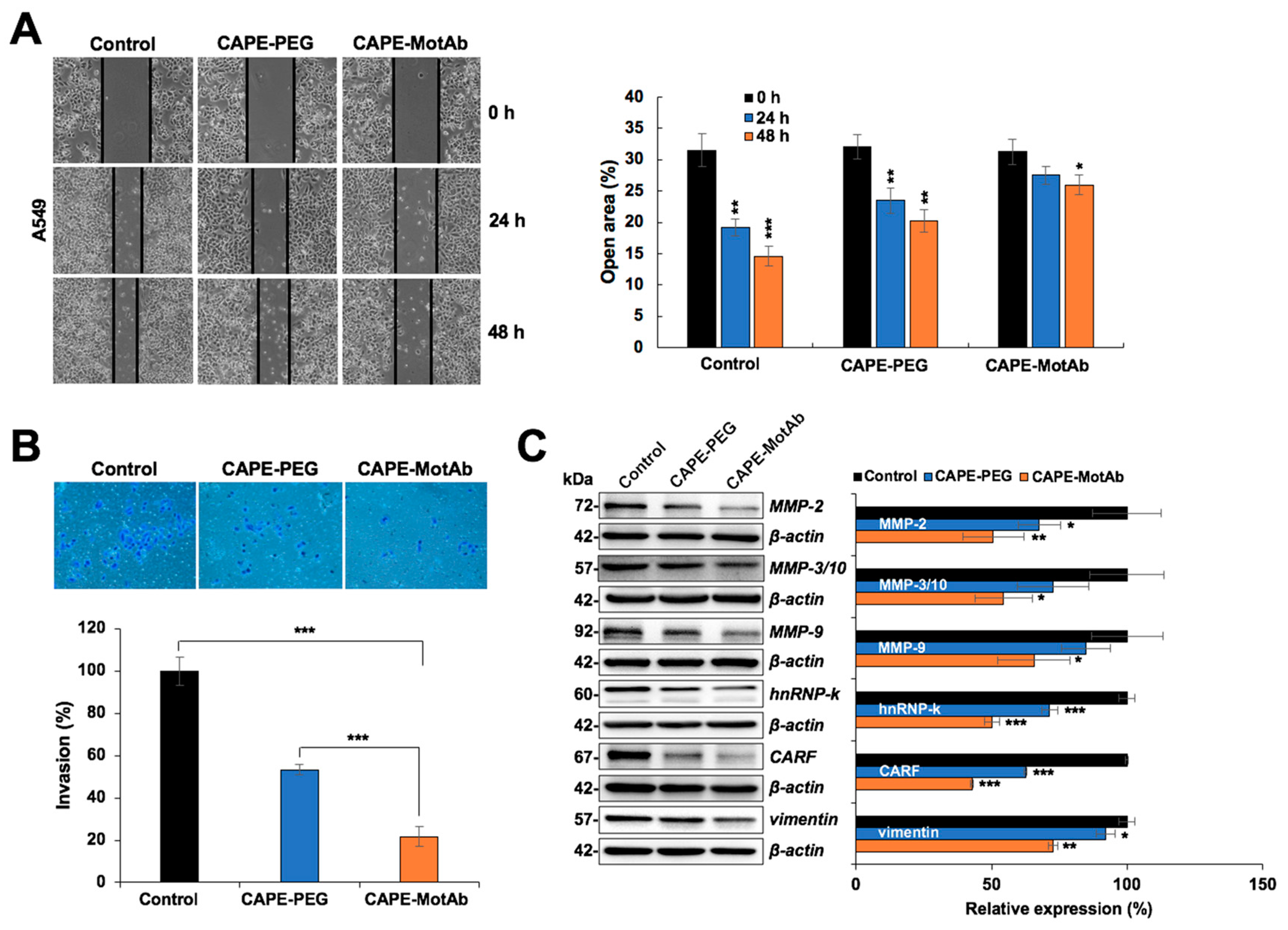
3.5. CAPE-MotAb Caused Enhanced Anti-Migration and Anti-Invasion Activities
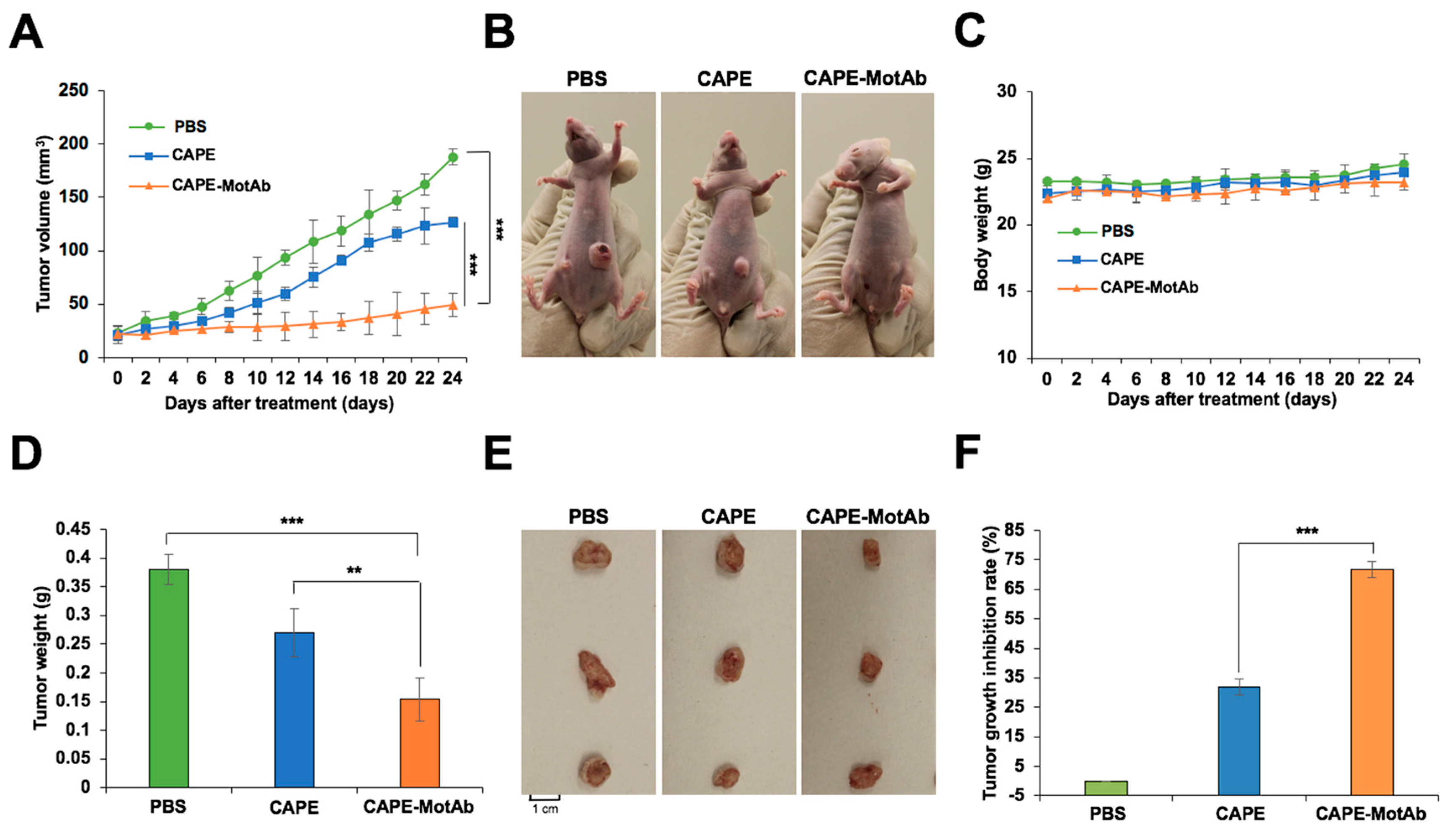
3.6. CAPE-MotAb Nanoparticles Caused Enhanced Tumor Suppression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Khayyal, M.T.; el-Ghazaly, M.A.; el-Khatib, A.S. Mechanisms involved in the antiinflammatory effect of propolis extract. Drugs Exp. Clin. Res. 1993, 19, 197–203. [Google Scholar]
- Przybylek, I.; Karpinski, T.M. Antibacterial Properties of Propolis. Molecules 2019, 24, 2047. [Google Scholar] [CrossRef]
- Schnitzler, P.; Neuner, A.; Nolkemper, S.; Zundel, C.; Nowack, H.; Sensch, K.H.; Reichling, J. Antiviral activity and mode of action of propolis extracts and selected compounds. Phytother. Res. 2010, 24 (Suppl. 1), S20–S28. [Google Scholar] [CrossRef]
- Banskota, A.H.; Nagaoka, T.; Sumioka, L.Y.; Tezuka, Y.; Awale, S.; Midorikawa, K.; Matsushige, K.; Kadota, S. Antiproliferative activity of the Netherlands propolis and its active principles in cancer cell lines. J. Ethnopharmacol. 2002, 80, 67–73. [Google Scholar] [CrossRef]
- Galeotti, F.; Maccari, F.; Fachini, A.; Volpi, N. Chemical Composition and Antioxidant Activity of Propolis Prepared in Different Forms and in Different Solvents Useful for Finished Products. Foods 2018, 7, 41. [Google Scholar] [CrossRef]
- Kujumgiev, A.; Tsvetkova, I.; Serkedjieva, Y.; Bankova, V.; Christov, R.; Popov, S. Antibacterial, antifungal and antiviral activity of propolis of different geographic origin. J. Ethnopharmacol. 1999, 64, 235–240. [Google Scholar] [CrossRef]
- Sforcin, J.M.; Bankova, V. Propolis: Is there a potential for the development of new drugs? J. Ethnopharmacol. 2011, 133, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Viuda-Martos, M.; Ruiz-Navajas, Y.; Fernandez-Lopez, J.; Perez-Alvarez, J.A. Functional properties of honey, propolis, and royal jelly. J. Food Sci. 2008, 73, R117–R124. [Google Scholar] [CrossRef] [PubMed]
- Bankova, V.; Popova, M.; Trusheva, B. Propolis volatile compounds: Chemical diversity and biological activity: A review. Chem. Cent. J. 2014, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Toreti, V.C.; Sato, H.H.; Pastore, G.M.; Park, Y.K. Recent progress of propolis for its biological and chemical compositions and its botanical origin. Evid. Based Complement Altern. Med. 2013, 2013, 697390. [Google Scholar] [CrossRef]
- Bhargava, P.; Grover, A.; Nigam, N.; Kaul, A.; Doi, M.; Ishida, Y.; Kakuta, H.; Kaul, S.C.; Terao, K.; Wadhwa, R. Anticancer activity of the supercritical extract of Brazilian green propolis and its active component, artepillin C: Bioinformatics and experimental analyses of its mechanisms of action. Int. J. Oncol. 2018, 52, 925–932. [Google Scholar] [PubMed]
- Ozturk, G.; Ginis, Z.; Akyol, S.; Erden, G.; Gurel, A.; Akyol, O. The anticancer mechanism of caffeic acid phenethyl ester (CAPE): Review of melanomas, lung and prostate cancers. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 2064–2068. [Google Scholar] [PubMed]
- Xiang, D.; Wang, D.; He, Y.; Xie, J.; Zhong, Z.; Li, Z.; Xie, J. Caffeic acid phenethyl ester induces growth arrest and apoptosis of colon cancer cells via the beta-catenin/T-cell factor signaling. Anticancer Drugs 2006, 17, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.F.; Wu, C.T.; Chen, Y.J.; Keng, P.C.; Chen, W.C. Cell killing and radiosensitization by caffeic acid phenethyl ester (CAPE) in lung cancer cells. J. Radiat Res. 2004, 45, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Omene, C.O.; Wu, J.; Frenkel, K. Caffeic Acid Phenethyl Ester (CAPE) derived from propolis, a honeybee product, inhibits growth of breast cancer stem cells. Investig. New Drugs 2012, 30, 1279–1288. [Google Scholar] [CrossRef]
- Motawi, T.K.; Abdelazim, S.A.; Darwish, H.A.; Elbaz, E.M.; Shouman, S.A. Modulation of Tamoxifen Cytotoxicity by Caffeic Acid Phenethyl Ester in MCF-7 Breast Cancer Cells. Oxid. Med. Cell. Longev. 2016, 2016, 3017108. [Google Scholar] [CrossRef]
- Kabala-Dzik, A.; Rzepecka-Stojko, A.; Kubina, R.; Jastrzebska-Stojko, Z.; Stojko, R.; Wojtyczka, R.D.; Stojko, J. Comparison of Two Components of Propolis: Caffeic Acid (CA) and Caffeic Acid Phenethyl Ester (CAPE) Induce Apoptosis and Cell Cycle Arrest of Breast Cancer Cells MDA-MB-231. Molecules 2017, 22, 1554. [Google Scholar] [CrossRef]
- Chen, M.J.; Chang, W.H.; Lin, C.C.; Liu, C.Y.; Wang, T.E.; Chu, C.H.; Shih, S.C.; Chen, Y.J. Caffeic acid phenethyl ester induces apoptosis of human pancreatic cancer cells involving caspase and mitochondrial dysfunction. Pancreatology 2008, 8, 566–576. [Google Scholar] [CrossRef]
- Rzepecka-Stojko, A.; Kabala-Dzik, A.; Mozdzierz, A.; Kubina, R.; Wojtyczka, R.D.; Stojko, R.; Dziedzic, A.; Jastrzebska-Stojko, Z.; Jurzak, M.; Buszman, E.; et al. Caffeic Acid phenethyl ester and ethanol extract of propolis induce the complementary cytotoxic effect on triple-negative breast cancer cell lines. Molecules 2015, 20, 9242–9262. [Google Scholar] [CrossRef]
- Kuo, Y.Y.; Lin, H.P.; Huo, C.; Su, L.C.; Yang, J.; Hsiao, P.H.; Chiang, H.C.; Chung, C.J.; Wang, H.D.; Chang, J.Y.; et al. Caffeic acid phenethyl ester suppresses proliferation and survival of TW2.6 human oral cancer cells via inhibition of Akt signaling. Int. J. Mol. Sci. 2013, 14, 8801–8817. [Google Scholar] [CrossRef]
- Chuu, C.P.; Lin, H.P.; Ciaccio, M.F.; Kokontis, J.M.; Hause, R.J., Jr.; Hiipakka, R.A.; Liao, S.; Jones, R.B. Caffeic acid phenethyl ester suppresses the proliferation of human prostate cancer cells through inhibition of p70S6K and Akt signaling networks. Cancer Prev. Res. 2012, 5, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.P.; Lin, C.Y.; Huo, C.; Hsiao, P.H.; Su, L.C.; Jiang, S.S.; Chan, T.M.; Chang, C.H.; Chen, L.T.; Kung, H.J.; et al. Caffeic acid phenethyl ester induced cell cycle arrest and growth inhibition in androgen-independent prostate cancer cells via regulation of Skp2, p53, p21Cip1 and p27Kip1. Oncotarget 2015, 6, 6684–6707. [Google Scholar] [CrossRef] [PubMed]
- Mirzoeva, O.K.; Yaqoob, P.; Knox, K.A.; Calder, P.C. Inhibition of ICE-family cysteine proteases rescues murine lymphocytes from lipoxygenase inhibitor-induced apoptosis. FEBS Lett. 1996, 396, 266–270. [Google Scholar] [CrossRef][Green Version]
- Watabe, M.; Hishikawa, K.; Takayanagi, A.; Shimizu, N.; Nakaki, T. Caffeic acid phenethyl ester induces apoptosis by inhibition of NFkappaB and activation of Fas in human breast cancer MCF-7 cells. J. Biol. Chem. 2004, 279, 6017–6026. [Google Scholar] [CrossRef]
- Natarajan, K.; Singh, S.; Burke, T.R., Jr.; Grunberger, D.; Aggarwal, B.B. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc. Natl. Acad. Sci. USA 1996, 93, 9090–9095. [Google Scholar] [CrossRef]
- Chiang, E.P.; Tsai, S.Y.; Kuo, Y.H.; Pai, M.H.; Chiu, H.L.; Rodriguez, R.L.; Tang, F.Y. Caffeic acid derivatives inhibit the growth of colon cancer: Involvement of the PI3-K/Akt and AMPK signaling pathways. PLoS ONE 2014, 9, e99631. [Google Scholar] [CrossRef]
- He, Y.J.; Liu, B.H.; Xiang, D.B.; Qiao, Z.Y.; Fu, T.; He, Y.H. Inhibitory effect of caffeic acid phenethyl ester on the growth of SW480 colorectal tumor cells involves beta-catenin associated signaling pathway down-regulation. World J. Gastroenterol. 2006, 12, 4981–4985. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kuo, H.C.; Chu, C.Y.; Wang, C.J.; Lin, W.C.; Tseng, T.H. Involvement of tumor suppressor protein p53 and p38 MAPK in caffeic acid phenethyl ester-induced apoptosis of C6 glioma cells. Biochem. Pharmacol. 2003, 66, 2281–2289. [Google Scholar] [CrossRef]
- Carrasco-Legleu, C.E.; Sanchez-Perez, Y.; Marquez-Rosado, L.; Fattel-Fazenda, S.; Arce-Popoca, E.; Hernandez-Garcia, S.; Villa-Trevino, S. A single dose of caffeic acid phenethyl ester prevents initiation in a medium-term rat hepatocarcinogenesis model. World J. Gastroenterol. 2006, 12, 6779–6785. [Google Scholar] [CrossRef]
- Wadhwa, R.; Nigam, N.; Bhargava, P.; Dhanjal, J.K.; Goyal, S.; Grover, A.; Sundar, D.; Ishida, Y.; Terao, K.; Kaul, S.C. Molecular Characterization and Enhancement of Anticancer Activity of Caffeic Acid Phenethyl Ester by gamma Cyclodextrin. J. Cancer 2016, 7, 1755–1771. [Google Scholar] [CrossRef]
- Ishida, Y.; Gao, R.; Shah, N.; Bhargava, P.; Furune, T.; Kaul, S.C.; Terao, K.; Wadhwa, R. Anticancer Activity in Honeybee Propolis: Functional Insights to the Role of Caffeic Acid Phenethyl Ester and Its Complex with gamma-Cyclodextrin. Integr. Cancer Ther. 2018, 17, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf. B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef] [PubMed]
- ElBayoumi, T.A.; Torchilin, V.P. Tumor-targeted nanomedicines: Enhanced antitumor efficacy in vivo of doxorubicin-loaded, long-circulating liposomes modified with cancer-specific monoclonal antibody. Clin. Cancer Res. 2009, 15, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
- Wolfram, J.; Zhu, M.; Yang, Y.; Shen, J.; Gentile, E.; Paolino, D.; Fresta, M.; Nie, G.; Chen, C.; Shen, H.; et al. Safety of Nanoparticles in Medicine. Curr. Drug Targets 2015, 16, 1671–1681. [Google Scholar] [CrossRef] [PubMed]
- Barnes, T.A.; Amir, E.; Templeton, A.J.; Gomez-Garcia, S.; Navarro, B.; Seruga, B.; Ocana, A. Efficacy, safety, tolerability and price of newly approved drugs in solid tumors. Cancer Treat. Rev. 2017, 56, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Farokhzad, O.C.; Langer, R. Impact of nanotechnology on drug delivery. ACS Nano 2009, 3, 16–20. [Google Scholar] [CrossRef]
- Navya, P.N.; Daima, H.K. Rational engineering of physicochemical properties of nanomaterials for biomedical applications with nanotoxicological perspectives. Nano Converg. 2016, 3, 1. [Google Scholar] [CrossRef]
- Cosco, D.; Paolino, D.; De Angelis, F.; Cilurzo, F.; Celia, C.; Di Marzio, L.; Russo, D.; Tsapis, N.; Fattal, E.; Fresta, M. Aqueous-core PEG-coated PLA nanocapsules for an efficient entrapment of water soluble anticancer drugs and a smart therapeutic response. Eur. J. Pharm. Biopharm. 2015, 89, 30–39. [Google Scholar] [CrossRef]
- Cosco, D.; Mare, R.; Paolino, D.; Salvatici, M.C.; Cilurzo, F.; Fresta, M. Sclareol-loaded hyaluronan-coated PLGA nanoparticles: Physico-chemical properties and in vitro anticancer features. Int. J. Biol. Macromol. 2019, 132, 550–557. [Google Scholar] [CrossRef]
- Din, F.U.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int. J. Nanomed. 2017, 12, 7291–7309. [Google Scholar] [CrossRef]
- Torchilin, V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Navya, P.N.; Kaphle, A.; Srinivas, S.P.; Bhargava, S.K.; Rotello, V.M.; Daima, H.K. Current trends and challenges in cancer management and therapy using designer nanomaterials. Nano Converg. 2019, 6, 23. [Google Scholar] [CrossRef]
- Wadhwa, R.; Taira, K.; Kaul, S.C. An Hsp70 family chaperone, mortalin/mthsp70/PBP74/Grp75: What, when, and where? Cell Stress Chaperones 2002, 7, 309–316. [Google Scholar] [CrossRef]
- Ran, Q.; Wadhwa, R.; Kawai, R.; Kaul, S.C.; Sifers, R.N.; Bick, R.J.; Smith, J.R.; Pereira-Smith, O.M. Extramitochondrial localization of mortalin/mthsp70/PBP74/GRP75. Biochem. Biophys. Res. Commun. 2000, 275, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, R.; Kaul, S.C.; Mitsui, Y.; Sugimoto, Y. Differential subcellular distribution of mortalin in mortal and immortal mouse and human fibroblasts. Exp. Cell Res. 1993, 207, 442–448. [Google Scholar] [CrossRef]
- Wadhwa, R.; Ryu, J.; Ahn, H.M.; Saxena, N.; Chaudhary, A.; Yun, C.O.; Kaul, S.C. Functional significance of point mutations in stress chaperone mortalin and their relevance to Parkinson disease. J. Biol. Chem. 2015, 290, 8447–8456. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, R.; Takano, S.; Kaur, K.; Deocaris, C.C.; Pereira-Smith, O.M.; Reddel, R.R.; Kaul, S.C. Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int. J. Cancer 2006, 118, 2973–2980. [Google Scholar] [CrossRef]
- Deocaris, C.C.; Lu, W.J.; Kaul, S.C.; Wadhwa, R. Druggability of mortalin for cancer and neuro-degenerative disorders. Curr. Pharm. Des. 2013, 19, 418–429. [Google Scholar] [CrossRef]
- Kaul, Z.; Yaguchi, T.; Harada, J.I.; Ikeda, Y.; Hirano, T.; Chiura, H.X.; Kaul, S.C.; Wadhwa, R. An antibody-conjugated internalizing quantum dot suitable for long-term live imaging of cells. Biochem. Cell Biol. 2007, 85, 133–140. [Google Scholar] [CrossRef]
- Shin, B.K.; Wang, H.; Yim, A.M.; Le Naour, F.; Brichory, F.; Jang, J.H.; Zhao, R.; Puravs, E.; Tra, J.; Michael, C.W.; et al. Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J. Biol. Chem. 2003, 278, 7607–7616. [Google Scholar] [CrossRef]
- Shiota, M.; Ikeda, Y.; Kaul, Z.; Itadani, J.; Kaul, S.C.; Wadhwa, R. Internalizing antibody-based targeted gene delivery for human cancer cells. Hum. Gene Ther. 2007, 18, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Kalra, R.S.; Cheung, C.T.; Chaudhary, A.; Prakash, J.; Kaul, S.C.; Wadhwa, R. CARF (Collaborator of ARF) overexpression in p53-deficient cells promotes carcinogenesis. Mol. Oncol. 2015, 9, 1877–1889. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.K.; Yaguchi, T.; Minoda, Y.; Hirano, T.; Taira, K.; Wadhwa, R.; Kaul, S.C. Alternative reading frame protein (ARF)-independent function of CARF (collaborator of ARF) involves its interactions with p53: Evidence for a novel p53-activation pathway and its negative feedback control. Biochem. J. 2004, 380, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wang, J.; Kaul, S.C.; Wadhwa, R.; Miyako, E. Folic Acid Receptor-Mediated Targeting Enhances the Cytotoxicity, Efficacy, and Selectivity of Withania somnifera Leaf Extract: In vitro and in vivo Evidence. Front. Oncol. 2019, 9, 602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, Y.; Li, S. Polymeric micelles: Nanocarriers for cancer-targeted drug delivery. AAPS Pharm. Sci. Tech. 2014, 15, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, Y.; Li, S. Nanomicellar carriers for targeted delivery of anticancer agents. Ther. Deliv. 2014, 5, 53–68. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, T.; Liu, Q.; He, J. PEG-Derivatized Dual-Functional Nanomicelles for Improved Cancer Therapy. Front. Pharmacol. 2019, 10, 808. [Google Scholar] [CrossRef]
- Trivedi, R.; Kompella, U.B. Nanomicellar formulations for sustained drug delivery: Strategies and underlying principles. Nanomedicine 2010, 5, 485–505. [Google Scholar] [CrossRef]
- Al Bitar, S.; Gali-Muhtasib, H. The Role of the Cyclin Dependent Kinase Inhibitor p21(cip1/waf1) in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers 2019, 11, 1475. [Google Scholar] [CrossRef]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.Z.; Lin, J.; Prewett, M.; Goldstein, N.I.; Fisher, P.B. Apoptosis mediates the selective toxicity of caffeic acid phenethyl ester (CAPE) toward oncogene-transformed rat embryo fibroblast cells. Anticancer Res. 1995, 15, 1841–1848. [Google Scholar] [PubMed]
- Nomura, M.; Kaji, A.; Ma, W.; Miyamoto, K.; Dong, Z. Suppression of cell transformation and induction of apoptosis by caffeic acid phenethyl ester. Mol. Carcinog. 2001, 31, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Chiao, C.; Carothers, A.M.; Grunberger, D.; Solomon, G.; Preston, G.A.; Barrett, J.C. Apoptosis and altered redox state induced by caffeic acid phenethyl ester (CAPE) in transformed rat fibroblast cells. Cancer Res. 1995, 55, 3576–3583. [Google Scholar]
- Na, H.K.; Wilson, M.R.; Kang, K.S.; Chang, C.C.; Grunberger, D.; Trosko, J.E. Restoration of gap junctional intercellular communication by caffeic acid phenethyl ester (CAPE) in a ras-transformed rat liver epithelial cell line. Cancer Lett. 2000, 157, 31–38. [Google Scholar] [CrossRef]
- Izuta, H.; Shimazawa, M.; Tsuruma, K.; Araki, Y.; Mishima, S.; Hara, H. Bee products prevent VEGF-induced angiogenesis in human umbilical vein endothelial cells. BMC Complement Altern. Med. 2009, 9, 45–55. [Google Scholar] [CrossRef]
- Chen, M.J.; Shih, S.C.; Wang, H.Y.; Lin, C.C.; Liu, C.Y.; Wang, T.E.; Chu, C.H.; Chen, Y.J. Caffeic Acid phenethyl ester inhibits epithelial-mesenchymal transition of human pancreatic cancer cells. Evid. Based Complement. Altern. Med. 2013, 2013, 270906–270911. [Google Scholar] [CrossRef]
- Anjaly, K.; Tiku, A.B. Radio-Modulatory Potential of Caffeic Acid Phenethyl Ester: A Therapeutic Perspective. Anticancer Agents Med. Chem. 2018, 18, 468–475. [Google Scholar] [CrossRef]
- Tolba, M.F.; Esmat, A.; Al-Abd, A.M.; Azab, S.S.; Khalifa, A.E.; Mosli, H.A.; Abdel-Rahman, S.Z.; Abdel-Naim, A.B. Caffeic acid phenethyl ester synergistically enhances docetaxel and paclitaxel cytotoxicity in prostate cancer cells. IUBMB Life 2013, 65, 716–729. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, J.X.; Liu, Y.N.; Ge, A.; Gu, H.; Zha, W.J.; Zeng, X.N.; Huang, M. Caffeic acid phenethyl ester alleviates asthma by regulating the airway microenvironment via the ROS-responsive MAPK/Akt pathway. Free Radic. Biol. Med. 2016, 101, 163–175. [Google Scholar] [CrossRef]
- Chung, L.C.; Chiang, K.C.; Feng, T.H.; Chang, K.S.; Chuang, S.T.; Chen, Y.J.; Tsui, K.H.; Lee, J.C.; Juang, H.H. Caffeic acid phenethyl ester upregulates N-myc downstream regulated gene 1 via ERK pathway to inhibit human oral cancer cell growth in vitro and in vivo. Mol. Nutr. Food Res. 2017, 61, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Messerli, S.M.; Ahn, M.R.; Kunimasa, K.; Yanagihara, M.; Tatefuji, T.; Hashimoto, K.; Mautner, V.; Uto, Y.; Hori, H.; Kumazawa, S.; et al. Artepillin C (ARC) in Brazilian green propolis selectively blocks oncogenic PAK1 signaling and suppresses the growth of NF tumors in mice. Phytother. Res. 2009, 23, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Queimada, A.J.; Mota, F.L.; Pinho, S.P.; Macedo, E.A. Solubilities of biologically active phenolic compounds: Measurements and modeling. J. Phys. Chem. B 2009, 113, 3469–3476. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, N.; Andrade, F.; Segovia, N.; Ferrer-Tasies, L.; Sala, S.; Veciana, J.; Ventosa, N. Lipid-based nanovesicles for nanomedicine. Chem. Soc. Rev. 2016, 45, 6520–6545. [Google Scholar] [CrossRef] [PubMed]
- Parveen, S.; Sahoo, S.K. Nanomedicine: Clinical applications of polyethylene glycol conjugated proteins and drugs. Clin. Pharmacokinet. 2006, 45, 965–988. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef]
- Paolino, D.; Accolla, M.L.; Cilurzo, F.; Cristiano, M.C.; Cosco, D.; Castelli, F.; Sarpietro, M.G.; Fresta, M.; Celia, C. Interaction between PEG lipid and DSPE/DSPC phospholipids: An insight of PEGylation degree and kinetics of de-PEGylation. Colloids Surf. B Biointerfaces 2017, 155, 266–275. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CAPE/Polymer (w/w) | Encapsulation Efficiency (%) |
|---|---|
| 1:0.5 | 27.74 ± 0.58 |
| 1:1 | 38.29 ± 2.24 |
| 1:2 | 40.41 ± 1.06 |
| 1:4 | 71.12 ± 6.94 |
| 1: 5 | 75.88 ± 1.99 |
| 1: 10 | 80.91 ± 7.58 |
| 1: 20 | 84.88 ± 8.66 |
| 1: 30 | 77.21. ± 6.78 |
| CAPE/Polymer (w/w) | Loading Efficiency (%) |
|---|---|
| 1:0.5 | 18.50 ± 0.39 |
| 1:1 | 19.65 ± 0.96 |
| 1:2 | 13.47 ± 0.35 |
| 1:4 | 14.22 ± 0.14 |
| 1: 5 | 12.65 ± 0.33 |
| 1: 10 | 7.36 ± 0.69 |
| 1: 20 | 4.04 ± 0.41 |
| 1: 30 | 2.49 ± 0.22 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Bhargava, P.; Yu, Y.; Sari, A.N.; Zhang, H.; Ishii, N.; Yan, K.; Zhang, Z.; Ishida, Y.; Terao, K.; et al. Novel Caffeic Acid Phenethyl Ester-Mortalin Antibody Nanoparticles Offer Enhanced Selective Cytotoxicity to Cancer Cells. Cancers 2020, 12, 2370. https://doi.org/10.3390/cancers12092370
Wang J, Bhargava P, Yu Y, Sari AN, Zhang H, Ishii N, Yan K, Zhang Z, Ishida Y, Terao K, et al. Novel Caffeic Acid Phenethyl Ester-Mortalin Antibody Nanoparticles Offer Enhanced Selective Cytotoxicity to Cancer Cells. Cancers. 2020; 12(9):2370. https://doi.org/10.3390/cancers12092370
Chicago/Turabian StyleWang, Jia, Priyanshu Bhargava, Yue Yu, Anissa Nofita Sari, Huayue Zhang, Noriyuki Ishii, Kangmin Yan, Zhenya Zhang, Yoshiyuki Ishida, Keiji Terao, and et al. 2020. "Novel Caffeic Acid Phenethyl Ester-Mortalin Antibody Nanoparticles Offer Enhanced Selective Cytotoxicity to Cancer Cells" Cancers 12, no. 9: 2370. https://doi.org/10.3390/cancers12092370
APA StyleWang, J., Bhargava, P., Yu, Y., Sari, A. N., Zhang, H., Ishii, N., Yan, K., Zhang, Z., Ishida, Y., Terao, K., Kaul, S. C., Miyako, E., & Wadhwa, R. (2020). Novel Caffeic Acid Phenethyl Ester-Mortalin Antibody Nanoparticles Offer Enhanced Selective Cytotoxicity to Cancer Cells. Cancers, 12(9), 2370. https://doi.org/10.3390/cancers12092370