Distinct Mutation Patterns Reveal Melanoma Subtypes and Influence Immunotherapy Response in Advanced Melanoma Patients

 , , , , , , ,
, , , , , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Tumor Tissue
2.2. Sequencing
2.3. Tumor Mutational Burden
2.4. Gene Set Enrichment Analysis
2.5. Statistics
3. Results
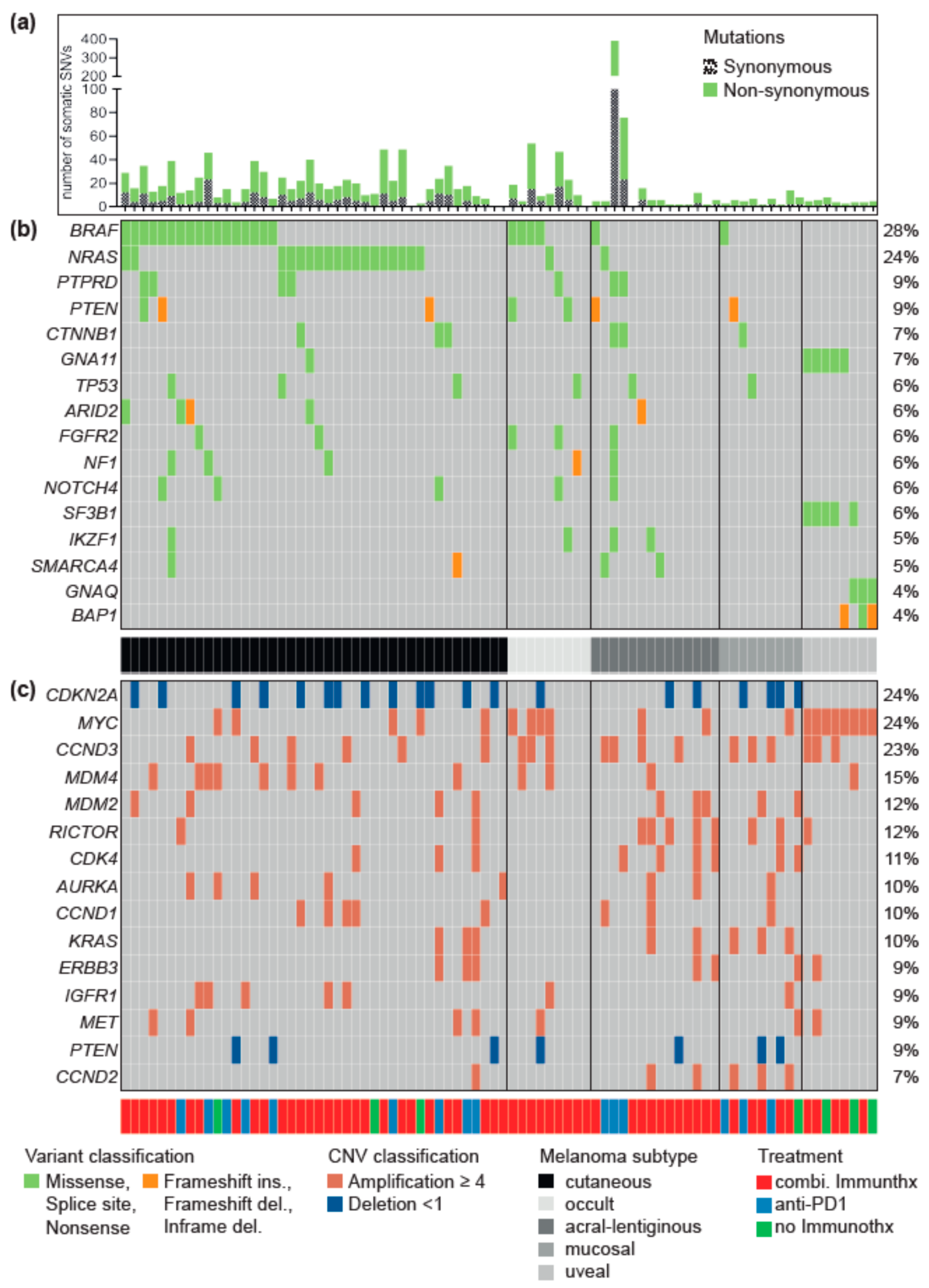
3.1. Driver Alterations and Signaling Cascade
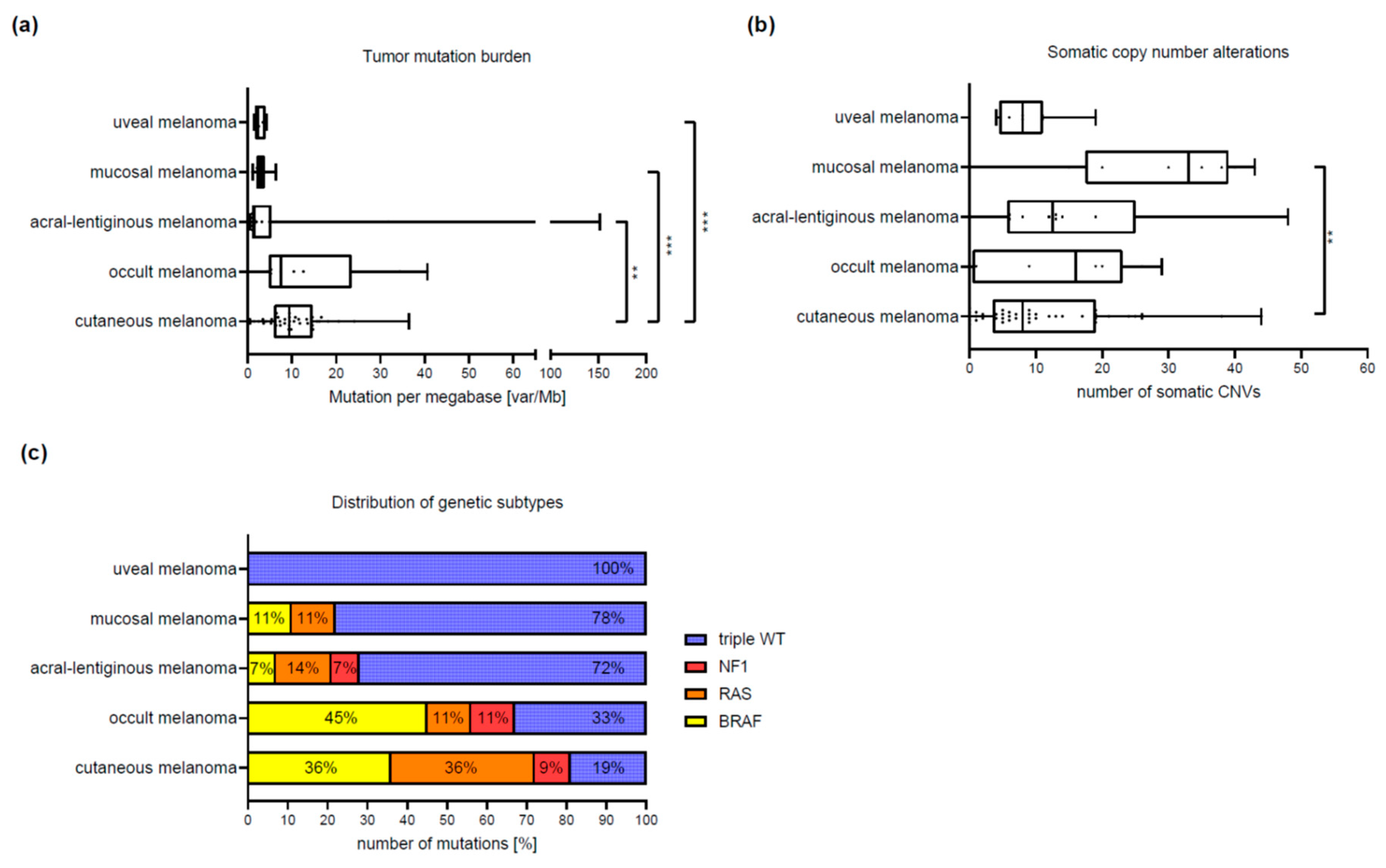
3.2. Differences between Melanoma Subtypes
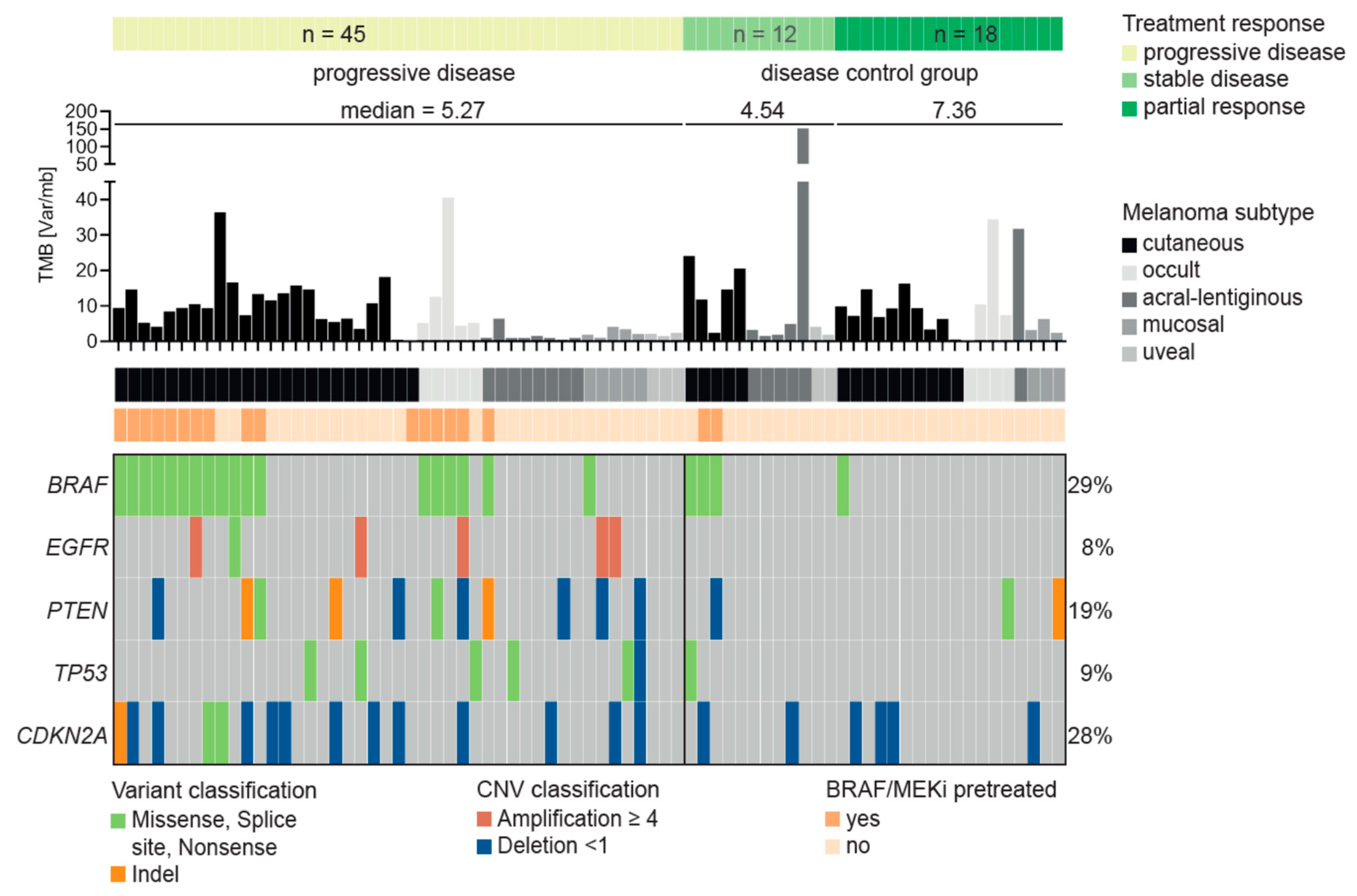
3.3. Characteristics of Patients Treated with Immune Checkpoint Inhibitors
3.4. Resistance Predictor for Immune Checkpoint Inhibitor Therapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data availability
References
- Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Krauthammer, M.; Kong, Y.; Bacchiocchi, A.; Evans, P.; Pornputtapong, N.; Wu, C.; McCusker, J.P.; Ma, S.; Cheng, E.; Straub, R.; et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat. Genet. 2015, 47, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Liang, W.S.; Hendricks, W.; Kiefer, J.; Schmidt, J.; Sekar, S.; Carpten, J.; Craig, D.W.; Adkins, J.; Cuyugan, L.; Manojlovic, Z.; et al. Integrated genomic analyses reveal frequent TERT aberrations in acral melanoma. Genome Res. 2017, 27, 524–532. [Google Scholar] [CrossRef]
- Newell, F.; Kong, Y.; Wilmott, J.S.; Johansson, P.A.; Ferguson, P.M.; Cui, C.; Li, Z.; Kazakoff, S.H.; Burke, H.; Dodds, T.J.; et al. Whole-genome landscape of mucosal melanoma reveals diverse drivers and therapeutic targets. Nat. Commun. 2019, 10, 3163. [Google Scholar] [CrossRef]
- Garman, B.; Anastopoulos, I.N.; Krepler, C.; Brafford, P.; Sproesser, K.; Jiang, Y.; Wubbenhorst, B.; Amaravadi, R.; Bennett, J.; Beqiri, M.; et al. Genetic and Genomic Characterization of 462 Melanoma Patient-Derived Xenografts, Tumor Biopsies, and Cell Lines. Cell Rep. 2017, 21, 1936–1952. [Google Scholar] [CrossRef]
- Krepler, C.; Sproesser, K.; Brafford, P.; Beqiri, M.; Garman, B.; Xiao, M.; Shannan, B.; Watters, A.; Perego, M.; Zhang, G.; et al. A Comprehensive Patient-Derived Xenograft Collection Representing the Heterogeneity of Melanoma. Cell Rep. 2017, 21, 1953–1967. [Google Scholar] [CrossRef]
- Kato, S.; Goodman, A.; Walavalkar, V.; Barkauskas, D.A.; Sharabi, A.; Kurzrock, R. Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin. Cancer Res. 2017, 23, 4242–4250. [Google Scholar] [CrossRef]
- Forschner, A.; Niessner, H.; Moller, Y.; Horak, P.; Frohlich, M.; Warsow, G.; Stenzinger, A.; Frohling, S.; Glimm, H.; Klumpp, B.; et al. Genomics of Immunotherapy-Associated Hyperprogressors-Letter. Clin. Cancer Res. 2017, 23, 6374–6375. [Google Scholar] [CrossRef]
- Forschner, A.; Hilke, F.J.; Bonzheim, I.; Gschwind, A.; Demidov, G.; Amaral, T.; Ossowski, S.; Riess, O.; Schroeder, C.; Martus, P.; et al. MDM2, MDM4 and EGFR Amplifications and Hyperprogression in Metastatic Acral and Mucosal Melanoma. Cancers 2020, 12, 540. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.A.; Luke, J.J.; Zha, Y.; Segal, J.P.; Ritterhouse, L.L.; Spranger, S.; Matijevich, K.; Gajewski, T.F. Secondary resistance to immunotherapy associated with beta-catenin pathway activation or PTEN loss in metastatic melanoma. J. Immunother. Cancer 2019, 7, 295. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Leonardelli, S.; Sucker, A.; Schadendorf, D.; Griewank, K.G.; Paschen, A. Tumor CDKN2A-Associated JAK2 Loss and Susceptibility to Immunotherapy Resistance. J. Natl. Cancer Inst. 2018, 110, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404. [Google Scholar] [CrossRef]
- Boshuizen, J.; Koopman, L.A.; Krijgsman, O.; Shahrabi, A.; van den Heuvel, E.G.; Ligtenberg, M.A.; Vredevoogd, D.W.; Kemper, K.; Kuilman, T.; Song, J.Y.; et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. Nat. Med. 2018, 24, 203–212. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Kallberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef]
- Demidov, G.; Ossowski, S. ClinCNV: Novel method for allele-specific somatic copy-number alterations detection. bioRxiv 2019. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Cingolani, P.; Patel, V.M.; Coon, M.; Nguyen, T.; Land, S.J.; Ruden, D.M.; Lu, X. Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front. Genet. 2012, 3, 35. [Google Scholar] [CrossRef] [PubMed]
- Tamborero, D.; Rubio-Perez, C.; Deu-Pons, J.; Schroeder, M.P.; Vivancos, A.; Rovira, A.; Tusquets, I.; Albanell, J.; Rodon, J.; Tabernero, J.; et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Assoun, S.; Theou-Anton, N.; Nguenang, M.; Cazes, A.; Danel, C.; Abbar, B.; Pluvy, J.; Gounant, V.; Khalil, A.; Namour, C.; et al. Association of TP53 mutations with response and longer survival under immune checkpoint inhibitors in advanced non-small-cell lung cancer. Lung Cancer 2019, 132, 65–71. [Google Scholar] [CrossRef]
- Brenner, E.; Schorg, B.F.; Ahmetlic, F.; Wieder, T.; Hilke, F.J.; Simon, N.; Schroeder, C.; Demidov, G.; Riedel, T.; Fehrenbacher, B.; et al. Cancer immune control needs senescence induction by interferon-dependent cell cycle regulator pathways in tumours. Nat. Commun. 2020, 11, 1335. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Parrella, P.; Caballero, O.L.; Sidransky, D.; Merbs, S.L. Detection of c-myc amplification in uveal melanoma by fluorescent in situ hybridization. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1679–1684. [Google Scholar]
- Lin, X.; Sun, R.; Zhao, X.; Zhu, D.; Zhao, X.; Gu, Q.; Dong, X.; Zhang, D.; Zhang, Y.; Li, Y.; et al. C-myc overexpression drives melanoma metastasis by promoting vasculogenic mimicry via c-myc/snail/Bax signaling. J. Mol. Med. 2017, 95, 53–67. [Google Scholar] [CrossRef]
- Wolfer, A.; Ramaswamy, S. MYC and metastasis. Cancer Res. 2011, 71, 2034–2037. [Google Scholar] [CrossRef]
- McCarthy, C.; Kalirai, H.; Lake, S.L.; Dodson, A.; Damato, B.E.; Coupland, S.E. Insights into genetic alterations of liver metastases from uveal melanoma. Pigment Cell Melanoma Res. 2016, 29, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Park, J.J.; Diefenbach, R.J.; Joshua, A.M.; Kefford, R.F.; Carlino, M.S.; Rizos, H. Oncogenic signaling in uveal melanoma. Pigment Cell Melanoma Res. 2018, 31, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Egberts, F.; Bergner, I.; Kruger, S.; Haag, J.; Behrens, H.M.; Hauschild, A.; Rocken, C. Metastatic melanoma of unknown primary resembles the genotype of cutaneous melanomas. Ann. Oncol. 2014, 25, 246–250. [Google Scholar] [CrossRef]
- Heppt, M.V.; Tietze, J.K.; Reinholz, M.; Rahimi, F.; Jung, A.; Kirchner, T.; Ruzicka, T.; Flaig, M.J.; Berking, C. Disease kinetics but not disease burden is relevant for survival in melanoma of unknown primary tumor. Discov. Med. 2015, 20, 231–237. [Google Scholar]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.H.; Aiba, S.; Brocker, E.B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef]
- Furney, S.J.; Pedersen, M.; Gentien, D.; Dumont, A.G.; Rapinat, A.; Desjardins, L.; Turajlic, S.; Piperno-Neumann, S.; de la Grange, P.; Roman-Roman, S.; et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013, 3, 1122–1129. [Google Scholar] [CrossRef]
- Fruh, M.; Peters, S. EGFR mutation subtype impacts efficacy of immune checkpoint inhibitors in non-small-cell lung cancer. Ann. Oncol. 2019, 30, 1190–1192. [Google Scholar] [CrossRef]
- Lee, C.K.; Man, J.; Lord, S.; Links, M.; Gebski, V.; Mok, T.; Yang, J.C. Checkpoint Inhibitors in Metastatic EGFR-Mutated Non-Small Cell Lung Cancer-A Meta-Analysis. J. Thorac. Oncol. 2017, 12, 403–407. [Google Scholar] [CrossRef]
- Ozaki, Y.; Muto, S.; Takagi, H.; Watanabe, M.; Inoue, T.; Fukuhara, M.; Yamaura, T.; Okabe, N.; Matsumura, Y.; Hasegawa, T.; et al. Tumor mutation burden and immunological, genomic, and clinicopathological factors as biomarkers for checkpoint inhibitor treatment of patients with non-small-cell lung cancer. Cancer Immunol. Immunother. 2020, 69, 127–134. [Google Scholar] [CrossRef]
- Johnson, D.B.; Pectasides, E.; Feld, E.; Ye, F.; Zhao, S.; Johnpulle, R.; Merritt, R.; McDermott, D.F.; Puzanov, I.; Lawrence, D.; et al. Sequencing Treatment in BRAFV600 Mutant Melanoma: Anti-PD-1 Before and After BRAF Inhibition. J. Immunother. 2017, 40, 31–35. [Google Scholar] [CrossRef]
- Moser, J.C.; Chen, D.; Hu-Lieskovan, S.; Grossmann, K.F.; Patel, S.; Colonna, S.V.; Ying, J.; Hyngstrom, J.R. Real-world survival of patients with advanced BRAF V600 mutated melanoma treated with front-line BRAF/MEK inhibitors, anti-PD-1 antibodies, or nivolumab/ipilimumab. Cancer Med. 2019, 8, 7637–7643. [Google Scholar] [CrossRef] [PubMed]
- Schilling, B.; Martens, A.; Geukes Foppen, M.H.; Gebhardt, C.; Hassel, J.C.; Rozeman, E.A.; Gesierich, A.; Gutzmer, R.; Kahler, K.C.; Livingstone, E.; et al. First-line therapy-stratified survival in BRAF-mutant melanoma: A retrospective multicenter analysis. Cancer. Immunol. Immunother. 2019, 68, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book/ASCO Am. Soc. Clin. Oncol. Educ. Meet. 2019, 39, 147–164. [Google Scholar] [CrossRef]
- Kong, Y.; Sheng, X.; Wu, X.; Yan, J.; Ma, M.; Yu, J.; Si, L.; Chi, Z.; Cui, C.; Dai, J.; et al. Frequent Genetic Aberrations in the CDK4 Pathway in Acral Melanoma indicate the potential for CDK4/6 Inhibitors in Targeted Therapy. Clin. Cancer Res. 2017, 23, 6946–6957. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Sheng, X.; Kong, Y.; Chi, Z.; Si, L.; Cui, C.; Yan, X.; Mao, L.; Lian, B.; Li, S.; et al. Palbociclib for treatment of metastatic melanoma with copy number variations of CDK4 pathway: Case report. Chin. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Young, R.J.; Waldeck, K.; Martin, C.; Foo, J.H.; Cameron, D.P.; Kirby, L.; Do, H.; Mitchell, C.; Cullinane, C.; Liu, W.; et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res. 2014, 27, 590–600. [Google Scholar] [CrossRef]
- Posch, C.; Sanlorenzo, M.; Ma, J.; Kim, S.T.; Zekhtser, M.; Ortiz-Urda, S. MEK/CDK4,6 co-targeting is effective in a subset of NRAS, BRAF and ‘wild type’ melanomas. Oncotarget 2018, 9, 34990–34995. [Google Scholar] [CrossRef]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmana, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef]
- Johnson, D.B.; Frampton, G.M.; Rioth, M.J.; Yusko, E.; Xu, Y.; Guo, X.; Ennis, R.C.; Fabrizio, D.; Chalmers, Z.R.; Greenbowe, J.; et al. Targeted Next Generation Sequencing Identifies Markers of Response to PD-1 Blockade. Cancer Immunol. Res. 2016, 4, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Forschner, A.; Battke, F.; Hadaschik, D.; Schulze, M.; Weissgraeber, S.; Han, C.T.; Kopp, M.; Frick, M.; Klumpp, B.; Tietze, N.; et al. Tumor mutation burden and circulating tumor DNA in combined CTLA-4 and PD-1 antibody therapy in metastatic melanoma—results of a prospective biomarker study. J. Immunother. Cancer 2019, 7, 180. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Characteristics | |
|---|---|
| Age at the first diagnosis of melanoma | years |
| Median | 57 (46–67) |
| Range | 17–85 |
| Sex | no of patients (%) |
| Female | 37(45) |
| Male | 45 (55) |
| Melanoma type | no. of patients (%) |
| Cutaneous | 42 (51) |
| Acral | 14 (17) |
| Uveal | 8 (10) |
| Mucosal | 9 (11) |
| Occult | 9 (11) |
| Tumor thickness of primary melanoma | mm |
| Median (IQR) | 3.3 (1.8–5.1) |
| Range | 0.38–5.1 |
| The tumor stage at the time of tumor sequencing | no. of patients (%) |
| Stage II | 5 (6) |
| Stage IV | 77 (94) |
| Immunotherapy | 76/82 (93) |
| Targeted therapy | 21 (26) |
| Chemotherapy | 4 (5) |
| No systemic treatment | 5 (6) |
| Origin of the tissue sequenced | no of patients (%) |
| Lymph node | 19 (23) |
| Other metastasis | 57 (70) |
| Primary melanoma | 6 (7) |
| Pretreatment of tissue sequenced | no. of patients (%) |
| Tissue therapy naïve | 52 (63) |
| Tissue progressive under ICI | 18 (22) |
| Tissue progressive under targeted therapy | 8 (10) |
| Tissue progressive under chemotherapy | 4 (5) |
| Genetic Characteristics of the 82 Patients | Cutaneous | Acral | Mucosal | Uveal | Occult |
|---|---|---|---|---|---|
| (n = 42) | (n = 14) | (n = 9) | (n = 8) | (n = 9) | |
| Tumor mutation burden (TMB) | |||||
| Median (IQR) | 9.4 (6–14.6) | 1.5 (1–5.3) | 3.2 (2–3.8) | 2.8 (1.6–4) | 7.5 (4.8–23.5) |
| Range | 0–36.444 | 0.51–151.8 | 1.1–6.a | 1.5–4.2 | 0–40.6 |
| Comparison to cutaneous subtype 1 | p = 0.0027 * | p = 0.0003 * | p = 0.0003 * | p = 0.837 | |
| Single nucleotide variants (SNVs) | |||||
| Median (IQR) | 10 (6–15) | 3 (1–7.3) | 3 (2–4.5) | 3.5 (2–4) | 11 (4–25) |
| Range | 0–33 | 0–156 | 1–6 | 2–4 | 1–44 |
| Comparison to cutaneous subtype 1 | p = 0.0134 | p = 0.0005 * | p = 0.0008 * | p = 0.995 | |
| Copy number variants (CNVs) | |||||
| Median (IQR) | 8 (3.5–19) | 12.5 (5.8–25) | 33 (17.5–39) | 8 (4.5–11) | 16 (0.5–23) |
| Range | 0–44 | 0–48 | 0–43 | 4–19 | 0–29 |
| Comparison to cutaneous subtype 1 | p = 0.321 | p = 0.002 | p = 0.995 | p = 0.587 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hilke, F.J.; Sinnberg, T.; Gschwind, A.; Niessner, H.; Demidov, G.; Amaral, T.; Ossowski, S.; Bonzheim, I.; Röcken, M.; Riess, O.; et al. Distinct Mutation Patterns Reveal Melanoma Subtypes and Influence Immunotherapy Response in Advanced Melanoma Patients. Cancers 2020, 12, 2359. https://doi.org/10.3390/cancers12092359
Hilke FJ, Sinnberg T, Gschwind A, Niessner H, Demidov G, Amaral T, Ossowski S, Bonzheim I, Röcken M, Riess O, et al. Distinct Mutation Patterns Reveal Melanoma Subtypes and Influence Immunotherapy Response in Advanced Melanoma Patients. Cancers. 2020; 12(9):2359. https://doi.org/10.3390/cancers12092359
Chicago/Turabian StyleHilke, Franz J., Tobias Sinnberg, Axel Gschwind, Heike Niessner, German Demidov, Teresa Amaral, Stephan Ossowski, Irina Bonzheim, Martin Röcken, Olaf Riess, and et al. 2020. "Distinct Mutation Patterns Reveal Melanoma Subtypes and Influence Immunotherapy Response in Advanced Melanoma Patients" Cancers 12, no. 9: 2359. https://doi.org/10.3390/cancers12092359
APA StyleHilke, F. J., Sinnberg, T., Gschwind, A., Niessner, H., Demidov, G., Amaral, T., Ossowski, S., Bonzheim, I., Röcken, M., Riess, O., Garbe, C., Schroeder, C., & Forschner, A. (2020). Distinct Mutation Patterns Reveal Melanoma Subtypes and Influence Immunotherapy Response in Advanced Melanoma Patients. Cancers, 12(9), 2359. https://doi.org/10.3390/cancers12092359