Overhauling CAR T Cells to Improve Efficacy, Safety and Cost

 , , ,
, , ,
Abstract
1. Introduction
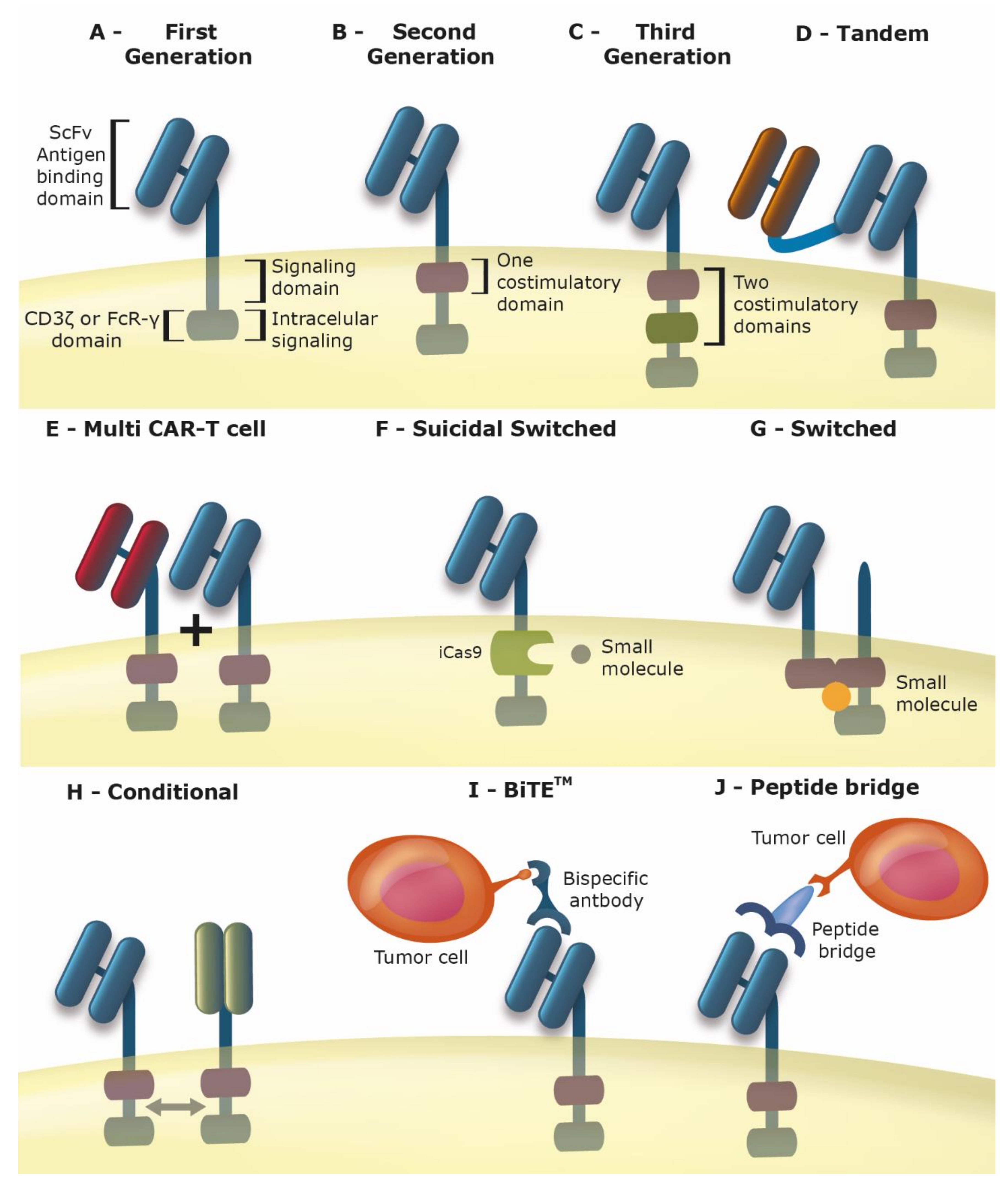
2. Chimeric Antigen Receptor T Cells (Car-T Cells)
3. Improving the Car for Increased Safety and Efficacy
4. Alternatives to Primary T Cells
4.1. Induced Pluripotent Stem Cells (iPSC)
4.2. Natural Killer (NK) Cells
4.3. Other Cell Types as Carriers of CAR
5. New Routes for Gene Transfer: Bringing the Car to the Cell
5.1. Transposons for CAR Gene Transfer
5.2. Site-Specific Insertion of CAR
5.3. Introduction of CAR Using mRNA
5.4. Systemic Administration of CAR Using Nanoparticles
6. Improved Pre-Clinical Models
7. Combining Car with Other Immunotherapies
8. Reducing Cost
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Facts & Figures 2020 Reports Largest One-Year Drop in Cancer Mortality. Available online: https://www.cancer.org/latest-news/facts-and-figures-2020.html (accessed on 11 August 2020).
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR-T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Boyiadzis, M.M.; Dhodapkar, M.V.; Brentjens, R.J.; Kochenderfer, J.N.; Neelapu, S.S.; Maus, M.V.; Porter, D.L.; Maloney, D.G.; Grupp, S.A.; Mackall, C.L.; et al. Chimeric antigen receptor (CAR) T therapies for the treatment of hematologic malignancies: Clinical perspective and significance. J. Immunother. Cancer 2018, 6, 137. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, O.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Porter, D.; Frey, N.; Wood, P.A.; Weng, Y.; Grupp, S.A. Grading of cytokine release syndrome associated with the CAR-T cell therapy tisagenlecleucel. J. Hematol. Oncol. 2018, 11, 35. [Google Scholar] [CrossRef]
- Wang, Z.; Han, W. Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy. Biomark. Res. 2018, 6, 4. [Google Scholar] [CrossRef]
- Hartmann, J.; Schussler-Lenz, M.; Bondanza, A.; Buchholz, C.J. Clinical development of CAR-T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 2017, 9, 1183–1197. [Google Scholar] [CrossRef]
- Perales, M.A.; Kebriaei, P.; Kean, L.S.; Sadelain, M. Building a Safer and Faster CAR: Seatbelts, Airbags, and CRISPR. Biol. Blood Marrow Transplant. 2018, 24, 27–31. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Antigen-Specific T-Cell Activation Independently of the MHC: Chimeric Antigen Receptor-Redirected T Cells. Front. Immunol. 2013, 4, 371. [Google Scholar] [CrossRef]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol. Res. 2015, 3, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Norelli, M.; Casucci, M.; Bonini, C.; Bondanza, A. Clinical pharmacology of CAR-T cells: Linking cellular pharmacodynamics to pharmacokinetics and antitumor effects. Biochim. Biophys. Acta 2016, 1865, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Bajgain, P.; Sukumaran, S.; Ansari, S.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology 2016, 5, e1253656. [Google Scholar] [CrossRef] [PubMed]
- Duong, M.T.; Collinson-Pautz, M.R.; Morschl, E.; Lu, A.; Szymanski, S.P.; Zhang, M.; Brandt, M.E.; Chang, W.C.; Sharp, K.L.; Toler, S.M.; et al. Two-Dimensional Regulation of CAR-T Cell Therapy with Orthogonal Switches. Mol. Ther. Oncolytics 2019, 12, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR-T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef]
- Aldoss, I.; Bargou, R.C.; Nagorsen, D.; Friberg, G.R.; Baeuerle, P.A.; Forman, S.J. Redirecting T cells to eradicate B-cell acute lymphoblastic leukemia: Bispecific T-cell engagers and chimeric antigen receptors. Leukemia 2017, 31, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Slaney, C.Y.; Wang, P.; Darcy, P.K.; Kershaw, M.H. CARs versus BiTEs: A Comparison between T Cell-Redirection Strategies for Cancer Treatment. Cancer Discov. 2018, 8, 924–934. [Google Scholar] [CrossRef]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR-T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef]
- Wu, C.Y.; Roybal, K.T.; Puchner, E.M.; Onuffer, J.; Lim, W.A. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 2015, 350, aab4077. [Google Scholar] [CrossRef]
- Straathof, K.C.; Pule, M.A.; Yotnda, P.; Dotti, G.; Vanin, E.F.; Brenner, M.K.; Heslop, H.E.; Spencer, D.M.; Rooney, C.M. An inducible caspase 9 safety switch for T-cell therapy. Blood 2005, 105, 4247–4254. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, A.; Santoro, S.; Tse, T.; Candelario-Chopra, T.; Gomes, T.; Campos, J.; Spelman, M.; Seacrest, S.; Coyle, S.; Rupp, L.; et al. Evaluating the reversible control of an engineered CAR-T cell ON-switch. J. Clin. Oncol. 2017, 35, e14550. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, Y.; Allen, M.E.; Pan, Y.; Kyriakakis, P.; Lu, S.; Chang, Y.J.; Wang, X.; Chien, S.; Wang, Y. Engineering light-controllable CAR-T cells for cancer immunotherapy. Sci. Adv. 2020, 6, eaay9209. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wang, Y.; Tan, X.; Zheng, X.; Wang, F.; Ke, K.; Zhang, C.; Liao, N.; Dang, Y.; Shi, Y.; et al. An Optogenetic Controllable T Cell System for Hepatocellular Carcinoma Immunotherapy. Theranostics 2019, 9, 1837–1850. [Google Scholar] [CrossRef]
- Ma, J.S.; Kim, J.Y.; Kazane, S.A.; Choi, S.H.; Yun, H.Y.; Kim, M.S.; Rodgers, D.T.; Pugh, H.M.; Singer, O.; Sun, S.B.; et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E450–E458. [Google Scholar] [CrossRef]
- Kudo, K.; Imai, C.; Lorenzini, P.; Kamiya, T.; Kono, K.; Davidoff, A.M.; Chng, W.J.; Campana, D. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. 2014, 74, 93–103. [Google Scholar] [CrossRef]
- Tamada, K.; Geng, D.; Sakoda, Y.; Bansal, N.; Srivastava, R.; Li, Z.; Davila, E. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin. Cancer Res. 2012, 18, 6436–6445. [Google Scholar] [CrossRef]
- Urbanska, K.; Lanitis, E.; Poussin, M.; Lynn, R.C.; Gavin, B.P.; Kelderman, S.; Yu, J.; Scholler, N.; Powell, D.J., Jr. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 2012, 72, 1844–1852. [Google Scholar] [CrossRef]
- Rodgers, D.T.; Mazagova, M.; Hampton, E.N.; Cao, Y.; Ramadoss, N.S.; Hardy, I.R.; Schulman, A.; Du, J.; Wang, F.; Singer, O.; et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc. Natl. Acad. Sci. USA 2016, 113, E459–E468. [Google Scholar] [CrossRef]
- Raj, D.; Yang, M.H.; Rodgers, D.; Hampton, E.N.; Begum, J.; Mustafa, A.; Lorizio, D.; Garces, I.; Propper, D.; Kench, J.G.; et al. Switchable CAR-T cells mediate remission in metastatic pancreatic ductal adenocarcinoma. Gut 2018. [Google Scholar] [CrossRef]
- Kim, M.S.; Ma, J.S.; Yun, H.; Cao, Y.; Kim, J.Y.; Chi, V.; Wang, D.; Woods, A.; Sherwood, L.; Caballero, D.; et al. Redirection of genetically engineered CAR-T cells using bifunctional small molecules. J. Am. Chem. Soc. 2015, 137, 2832–2835. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Rodgers, D.T.; Du, J.; Ahmad, I.; Hampton, E.N.; Ma, J.S.; Mazagova, M.; Choi, S.H.; Yun, H.Y.; Xiao, H.; et al. Design of Switchable Chimeric Antigen Receptor T Cells Targeting Breast Cancer. Angew. Chem. 2016, 55, 7520–7524. [Google Scholar] [CrossRef] [PubMed]
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef]
- Hay, K.A.; Hanafi, L.A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; Lopez, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR-T Cell Therapy. Mol. Ther. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef]
- Ding, L.; Hu, Y.; Zhao, K.; Wei, G.; Wu, W.; Wu, Z.; Xiao, L.; Huang, H. Pleural cavity cytokine release syndrome in CD19-directed chimeric antigen receptor-modified T cell therapy: A case report. Medicine 2018, 97, e9992. [Google Scholar] [CrossRef]
- Van Loenen, M.M.; de Boer, R.; Hagedoorn, R.S.; Jankipersadsing, V.; Amir, A.L.; Falkenburg, J.H.; Heemskerk, M.H. Multi-cistronic vector encoding optimized safety switch for adoptive therapy with T-cell receptor-modified T cells. Gene Ther. 2013, 20, 861–867. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Patel, A.; Hossain, S.; Kaufman, H.L. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am. J. Clin. Dermatol. 2017, 18, 1–15. [Google Scholar] [CrossRef]
- Juillerat, A.; Marechal, A.; Filhol, J.M.; Valton, J.; Duclert, A.; Poirot, L.; Duchateau, P. Design of chimeric antigen receptors with integrated controllable transient functions. Sci. Rep. 2016, 6, 18950. [Google Scholar] [CrossRef]
- Ramos, C.A.; Asgari, Z.; Liu, E.; Yvon, E.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. An inducible caspase 9 suicide gene to improve the safety of mesenchymal stromal cell therapies. Stem Cells 2010, 28, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Dotti, G.; Krance, R.A.; Martinez, C.A.; Naik, S.; Kamble, R.T.; Durett, A.G.; Dakhova, O.; Savoldo, B.; Di Stasi, A.; et al. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood 2015, 125, 4103–4113. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.; Rotem-Yehudar, R.; Slama, G.; Landes, S.; Kneller, A.; Leiba, M.; Koren-Michowitz, M.; Shimoni, A.; Nagler, A. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin. Cancer Res. 2008, 14, 3044–3051. [Google Scholar] [CrossRef]
- John, L.B.; Kershaw, M.H.; Darcy, P.K. Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology 2013, 2, e26286. [Google Scholar] [CrossRef]
- John, L.B.; Devaud, C.; Duong, C.P.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef]
- Hill, B.T.; Roberts, Z.J.; Xue, A.; Rossi, J.M.; Smith, M.R. Rapid tumor regression from PD-1 inhibition after anti-CD19 chimeric antigen receptor T-cell therapy in refractory diffuse large B-cell lymphoma. Bone Marrow Transpl. 2020, 55, 1184–1187. [Google Scholar] [CrossRef]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR-T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef]
- Prosser, M.E.; Brown, C.E.; Shami, A.F.; Forman, S.J.; Jensen, M.C. Tumor PD-L1 co-stimulates primary human CD8(+) cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol. Immunol. 2012, 51, 263–272. [Google Scholar] [CrossRef]
- Schlenker, R.; Olguin-Contreras, L.F.; Leisegang, M.; Schnappinger, J.; Disovic, A.; Ruhland, S.; Nelson, P.J.; Leonhardt, H.; Harz, H.; Wilde, S.; et al. Chimeric PD-1:28 Receptor Upgrades Low-Avidity T cells and Restores Effector Function of Tumor-Infiltrating Lymphocytes for Adoptive Cell Therapy. Cancer Res. 2017, 77, 3577–3590. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Rousseau, F.; Guilhot, F.; Malinge, P.; Magistrelli, G.; Herren, S.; Jones, S.A.; Jones, G.W.; Scheller, J.; Lissilaa, R.; et al. Novel Insights into Interleukin 6 (IL-6) Cis- and Trans-signaling Pathways by Differentially Manipulating the Assembly of the IL-6 Signaling Complex. J. Biol. Chem. 2015, 290, 26943–26953. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro-Vornhagen, A.; Godel, P.; Subklewe, M.; Stemmler, H.J.; Schlosser, H.A.; Schlaak, M.; Kochanek, M.; Boll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef]
- Uciechowski, P.; Dempke, W.C.M. Interleukin-6: A Masterplayer in the Cytokine Network. Oncology 2020, 98, 131–137. [Google Scholar] [CrossRef]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schonfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther. Nucleic Acids 2013, 2, e105. [Google Scholar] [CrossRef]
- Hombach, A.A.; Rappl, G.; Abken, H. Blocking CD30 on T Cells by a Dual Specific CAR for CD30 and Colon Cancer Antigens Improves the CAR-T Cell Response against CD30(-) Tumors. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 1825–1835. [Google Scholar] [CrossRef]
- Anurathapan, U.; Chan, R.C.; Hindi, H.F.; Mucharla, R.; Bajgain, P.; Hayes, B.C.; Fisher, W.E.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; et al. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 623–633. [Google Scholar] [CrossRef]
- Casucci, M.; Falcone, L.; Camisa, B.; Norelli, M.; Porcellini, S.; Stornaiuolo, A.; Ciceri, F.; Traversari, C.; Bordignon, C.; Bonini, C.; et al. Extracellular NGFR Spacers Allow Efficient Tracking and Enrichment of Fully Functional CAR-T Cells Co-Expressing a Suicide Gene. Front. Immunol. 2018, 9, 507. [Google Scholar] [CrossRef]
- Blake, Z.; Marks, D.K.; Gartrell, R.D.; Hart, T.; Horton, P.; Cheng, S.K.; Taback, B.; Horst, B.A.; Saenger, Y.M. Complete intracranial response to talimogene laherparepvec (T-Vec), pembrolizumab and whole brain radiotherapy in a patient with melanoma brain metastases refractory to dual checkpoint-inhibition. J. Immunother. Cancer 2018, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Veillette, A.; Davidson, D. Developing combination immunotherapies against cancer that make sense. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.; Heuser, C.; Gerken, M.; Fischer, B.; Lewalter, K.; Diehl, V.; Pohl, C.; Abken, H. T cell activation by recombinant FcepsilonRI gamma-chain immune receptors: An extracellular spacer domain impairs antigen-dependent T cell activation but not antigen recognition. Gene Ther. 2000, 7, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.E. Cellular Therapy of Cancer: Development of Gene Therapy Based Approaches; World Scientific: Hackensack, NJ, USA, 2014. [Google Scholar]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Timmermans, F.; Velghe, I.; Vanwalleghem, L.; De Smedt, M.; Van Coppernolle, S.; Taghon, T.; Moore, H.D.; Leclercq, G.; Langerak, A.W.; Kerre, T.; et al. Generation of T cells from human embryonic stem cell-derived hematopoietic zones. J. Immunol. 2009, 182, 6879–6888. [Google Scholar] [CrossRef]
- Kennedy, M.; Awong, G.; Sturgeon, C.M.; Ditadi, A.; LaMotte-Mohs, R.; Zuniga-Pflucker, J.C.; Keller, G. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012, 2, 1722–1735. [Google Scholar] [CrossRef]
- Nishimura, T.; Kaneko, S.; Kawana-Tachikawa, A.; Tajima, Y.; Goto, H.; Zhu, D.; Nakayama-Hosoya, K.; Iriguchi, S.; Uemura, Y.; Shimizu, T.; et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013, 12, 114–126. [Google Scholar] [CrossRef]
- Vizcardo, R.; Masuda, K.; Yamada, D.; Ikawa, T.; Shimizu, K.; Fujii, S.; Koseki, H.; Kawamoto, H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell 2013, 12, 31–36. [Google Scholar] [CrossRef]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192. [Google Scholar] [CrossRef]
- Garreta, E.; Sanchez, S.; Lajara, J.; Montserrat, N.; Belmonte, J.C.I. Roadblocks in the Path of iPSC to the Clinic. Curr. Transpl. Rep. 2018, 5, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Doubrovina, E.; Oflaz-Sozmen, B.; Prockop, S.E.; Kernan, N.A.; Abramson, S.; Teruya-Feldstein, J.; Hedvat, C.; Chou, J.F.; Heller, G.; Barker, J.N.; et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood 2012, 119, 2644–2656. [Google Scholar] [CrossRef] [PubMed]
- Provasi, E.; Genovese, P.; Lombardo, A.; Magnani, Z.; Liu, P.Q.; Reik, A.; Chu, V.; Paschon, D.E.; Zhang, L.; Kuball, J.; et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 2012, 18, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Guillerey, C.; Huntington, N.D.; Smyth, M.J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 2016, 17, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.M., Jr.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Marten, A.; von Lilienfeld-Toal, M.; Buchler, M.W.; Schmidt, J. Soluble MIC is elevated in the serum of patients with pancreatic carcinoma diminishing gammadelta T cell cytotoxicity. Int. J. Cancer 2006, 119, 2359–2365. [Google Scholar] [CrossRef]
- Salih, H.R.; Rammensee, H.G.; Steinle, A. Cutting edge: Down-regulation of MICA on human tumors by proteolytic shedding. J. Immunol. 2002, 169, 4098–4102. [Google Scholar] [CrossRef]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef]
- Ashiru, O.; Boutet, P.; Fernandez-Messina, L.; Aguera-Gonzalez, S.; Skepper, J.N.; Vales-Gomez, M.; Reyburn, H.T. Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res. 2010, 70, 481–489. [Google Scholar] [CrossRef]
- Klingemann, H. Challenges of cancer therapy with natural killer cells. Cytotherapy 2015, 17, 245–249. [Google Scholar] [CrossRef]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef]
- Muller, T.; Uherek, C.; Maki, G.; Chow, K.U.; Schimpf, A.; Klingemann, H.G.; Tonn, T.; Wels, W.S. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol. Immunother. 2008, 57, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Deng, Y.; Benson, D.M.; He, S.; Hughes, T.; Zhang, J.; Peng, Y.; Mao, H.; Yi, L.; Ghoshal, K.; et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014, 28, 917–927. [Google Scholar] [CrossRef]
- Seidel, D.; Shibina, A.; Siebert, N.; Wels, W.S.; Reynolds, C.P.; Huebener, N.; Lode, H.N. Disialoganglioside-specific human natural killer cells are effective against drug-resistant neuroblastoma. Cancer Immunol. Immunother. 2015, 64, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Wada, M.; Firor, A.E.; Pinz, K.G.; Jares, A.; Liu, H.; Salman, H.; Golightly, M.; Lan, F.; Jiang, X.; et al. Novel anti-CD3 chimeric antigen receptor targeting of aggressive T cell malignancies. Oncotarget 2016, 7, 56219–56232. [Google Scholar] [CrossRef] [PubMed]
- Raikar, S.S.; Fleischer, L.C.; Moot, R.; Fedanov, A.; Paik, N.Y.; Knight, K.A.; Doering, C.B.; Spencer, H.T. Development of chimeric antigen receptors targeting T-cell malignancies using two structurally different anti-CD5 antigen binding domains in NK and CRISPR-edited T cell lines. Oncoimmunology 2018, 7, e1407898. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bonig, H.; Kohl, U.; Kloess, S.; et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef]
- Zhang, Q.; Xu, J.; Ding, J.; Liu, H.; Li, H.; Lu, M.; Miao, Y.; Wang, Z.; Fu, Q.; Zheng, J. Bortezomib improves adoptive carbonic anhydrase IX specific chimeric antigen receptormodified NK92 cell therapy in mouse models of human renal cell carcinoma. Oncol. Rep. 2018, 40, 3714–3724. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, H.; Ding, J.; Liu, H.; Li, H.; Lu, M.; Miao, Y.; Li, L.; Zheng, J. Combination Therapy with EpCAM-CAR-NK-92 Cells and Regorafenib against Human Colorectal Cancer Models. J. Immunol. Res. 2018, 2018, 4263520. [Google Scholar] [CrossRef]
- Chen, X.; Han, J.; Chu, J.; Zhang, L.; Zhang, J.; Chen, C.; Chen, L.; Wang, Y.; Wang, H.; Yi, L.; et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget 2016, 7, 27764–27777. [Google Scholar] [CrossRef]
- Imai, C.; Iwamoto, S.; Campana, D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 2005, 106, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.H.; Connolly, J.; Shimasaki, N.; Mimura, K.; Kono, K.; Campana, D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013, 73, 1777–1786. [Google Scholar] [CrossRef] [PubMed]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef] [PubMed]
- Fujisaki, H.; Kakuda, H.; Shimasaki, N.; Imai, C.; Ma, J.; Lockey, T.; Eldridge, P.; Leung, W.H.; Campana, D. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res. 2009, 69, 4010–4017. [Google Scholar] [CrossRef] [PubMed]
- Denman, C.J.; Senyukov, V.V.; Somanchi, S.S.; Phatarpekar, P.V.; Kopp, L.M.; Johnson, J.L.; Singh, H.; Hurton, L.; Maiti, S.N.; Huls, M.H.; et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE 2012, 7, e30264. [Google Scholar] [CrossRef]
- Li, L.; Liu, L.N.; Feller, S.; Allen, C.; Shivakumar, R.; Fratantoni, J.; Wolfraim, L.A.; Fujisaki, H.; Campana, D.; Chopas, N.; et al. Expression of chimeric antigen receptors in natural killer cells with a regulatory-compliant non-viral method. Cancer Gene Ther. 2010, 17, 147–154. [Google Scholar] [CrossRef]
- Shimasaki, N.; Fujisaki, H.; Cho, D.; Masselli, M.; Lockey, T.; Eldridge, P.; Leung, W.; Campana, D. A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy 2012, 14, 830–840. [Google Scholar] [CrossRef]
- Spanholtz, J.; Preijers, F.; Tordoir, M.; Trilsbeek, C.; Paardekooper, J.; de Witte, T.; Schaap, N.; Dolstra, H. Clinical-grade generation of active NK cells from cord blood hematopoietic progenitor cells for immunotherapy using a closed-system culture process. PLoS ONE 2011, 6, e20740. [Google Scholar] [CrossRef]
- Woll, P.S.; Grzywacz, B.; Tian, X.; Marcus, R.K.; Knorr, D.A.; Verneris, M.R.; Kaufman, D.S. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood 2009, 113, 6094–6101. [Google Scholar] [CrossRef]
- Kruschinski, A.; Moosmann, A.; Poschke, I.; Norell, H.; Chmielewski, M.; Seliger, B.; Kiessling, R.; Blankenstein, T.; Abken, H.; Charo, J. Engineering antigen-specific primary human NK cells against HER-2 positive carcinomas. Proc. Natl. Acad. Sci. USA 2008, 105, 17481–17486. [Google Scholar] [CrossRef]
- Altvater, B.; Landmeier, S.; Pscherer, S.; Temme, J.; Schweer, K.; Kailayangiri, S.; Campana, D.; Juergens, H.; Pule, M.; Rossig, C. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin. Cancer Res. 2009, 15, 4857–4866. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Kailayangiri, S.; Altvater, B.; Spurny, C.; Jamitzky, S.; Schelhaas, S.; Jacobs, A.H.; Wiek, C.; Roellecke, K.; Hanenberg, H.; Hartmann, W.; et al. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immune-inhibitory HLA-G. Oncoimmunology 2017, 6, e1250050. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Nikzad, R.; Angelo, L.S.; Aviles-Padilla, K.; Le, D.T.; Singh, V.K.; Bimler, L.; Vukmanovic-Stejic, M.; Vendrame, E.; Ranganath, T.; Simpson, L.; et al. Human natural killer cells mediate adaptive immunity to viral antigens. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, T.E.; Sun, J.C.; Lanier, L.L. Natural Killer Cell Memory. Immunity 2015, 43, 634–645. [Google Scholar] [CrossRef]
- Harrer, D.C.; Dorrie, J.; Schaft, N. Chimeric Antigen Receptors in Different Cell Types: New Vehicles Join the Race. Hum. Gene Ther. 2018, 29, 547–558. [Google Scholar] [CrossRef]
- De Oliveira, S.N.; Ryan, C.; Giannoni, F.; Hardee, C.L.; Tremcinska, I.; Katebian, B.; Wherley, J.; Sahaghian, A.; Tu, A.; Grogan, T.; et al. Modification of hematopoietic stem/progenitor cells with CD19-specific chimeric antigen receptors as a novel approach for cancer immunotherapy. Hum. Gene Ther. 2013, 24, 824–839. [Google Scholar] [CrossRef]
- Yong, C.S.; John, L.B.; Devaud, C.; Prince, M.H.; Johnstone, R.W.; Trapani, J.A.; Darcy, P.K.; Kershaw, M.H. A role for multiple chimeric antigen receptor-expressing leukocytes in antigen-specific responses to cancer. Oncotarget 2016, 7, 34582–34598. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020. [Google Scholar] [CrossRef]
- Mirzaei, H.R.; Mirzaei, H.; Lee, S.Y.; Hadjati, J.; Till, B.G. Prospects for chimeric antigen receptor (CAR) gammadelta T cells: A potential game changer for adoptive T cell cancer immunotherapy. Cancer Lett. 2016, 380, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Burga, R.A.; Powell, A.B.; Chorvinsky, E.A.; Hoq, N.; McCormack, S.E.; Van Pelt, S.N.; Hanley, P.J.; Cruz, C.R.Y. Beyond CAR-T Cells: Other Cell-Based Immunotherapeutic Strategies against Cancer. Front. Oncol. 2019, 9, 196. [Google Scholar] [CrossRef] [PubMed]
- Fransson, M.; Piras, E.; Burman, J.; Nilsson, B.; Essand, M.; Lu, B.; Harris, R.A.; Magnusson, P.U.; Brittebo, E.; Loskog, A.S. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J. Neuroinflamm. 2012, 9, 112. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, K.G.; Hoeppli, R.E.; Huang, Q.; Gillies, J.; Luciani, D.S.; Orban, P.C.; Broady, R.; Levings, M.K. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J. Clin. Investig. 2016, 126, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Zanatta, D.B.; Tsujita, M.; Borelli, P.; Aguiar, R.B.; Ferrari, D.G.; Strauss, B.E. Genetic barcode sequencing for screening altered population dynamics of hematopoietic stem cells transduced with lentivirus. Mol. Ther. Methods Clin. Dev. 2014, 1, 14052. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Riviere, I. Clinical manufacturing of CAR-T cells: Foundation of a promising therapy. Mol. Ther. Oncolytics 2016, 3, 16015. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef]
- Shah, N.N.; Qin, H.; Yates, B.; Su, L.; Shalabi, H.; Raffeld, M.; Ahlman, M.A.; Stetler-Stevenson, M.; Yuan, C.; Guo, S.; et al. Clonal expansion of CAR-T cells harboring lentivector integration in the CBL gene following anti-CD22 CAR T-cell therapy. Blood Adv. 2019, 3, 2317–2322. [Google Scholar] [CrossRef]
- Kolata, G. Gene Therapy Hits a Peculiar Roadblock: A Virus Shortage. The New York Times, 27 November.
- Hackett, P.B.; Largaespada, D.A.; Cooper, L.J. A transposon and transposase system for human application. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 674–683. [Google Scholar] [CrossRef]
- Chicaybam, L.; Sodre, A.L.; Curzio, B.A.; Bonamino, M.H. An efficient low cost method for gene transfer to T lymphocytes. PLoS ONE 2013, 8, e60298. [Google Scholar] [CrossRef]
- Ramanayake, S.; Bilmon, I.; Bishop, D.; Dubosq, M.C.; Blyth, E.; Clancy, L.; Gottlieb, D.; Micklethwaite, K. Low-cost generation of Good Manufacturing Practice-grade CD19-specific chimeric antigen receptor-expressing T cells using piggyBac gene transfer and patient-derived materials. Cytotherapy 2015, 17, 1251–1267. [Google Scholar] [CrossRef] [PubMed]
- Hamada, M.; Nishio, N.; Okuno, Y.; Suzuki, S.; Kawashima, N.; Muramatsu, H.; Tsubota, S.; Wilson, M.H.; Morita, D.; Kataoka, S.; et al. Integration Mapping of piggyBac-Mediated CD19 Chimeric Antigen Receptor T Cells Analyzed by Novel Tagmentation-Assisted PCR. EBioMedicine 2018, 34, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Monjezi, R.; Miskey, C.; Gogishvili, T.; Schleef, M.; Schmeer, M.; Einsele, H.; Ivics, Z.; Hudecek, M. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia 2017, 31, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, T.; Iwase, N.; Kawakami, K.; Iwasaki, M.; Yamamoto, C.; Ohmine, K.; Uchibori, R.; Teruya, T.; Ido, H.; Saga, Y.; et al. The Tol2 transposon system mediates the genetic engineering of T-cells with CD19-specific chimeric antigen receptors for B-cell malignancies. Gene Ther. 2015, 22, 209–215. [Google Scholar] [CrossRef]
- Kebriaei, P.; Singh, H.; Huls, M.H.; Figliola, M.J.; Bassett, R.; Olivares, S.; Jena, B.; Dawson, M.J.; Kumaresan, P.R.; Su, S.; et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR-T cells. J. Clin. Investig. 2016, 126, 3363–3376. [Google Scholar] [CrossRef]
- He, J.; Zhang, Z.; Lv, S.; Liu, X.; Cui, L.; Jiang, D.; Zhang, Q.; Li, L.; Qin, W.; Jin, H.; et al. Engineered CAR-T cells targeting mesothelin by piggyBac transposon system for the treatment of pancreatic cancer. Cell. Immunol. 2018, 329, 31–40. [Google Scholar] [CrossRef]
- Crossland, D.L.; Denning, W.L.; Ang, S.; Olivares, S.; Mi, T.; Switzer, K.; Singh, H.; Huls, H.; Gold, K.S.; Glisson, B.S.; et al. Antitumor activity of CD56-chimeric antigen receptor T cells in neuroblastoma and SCLC models. Oncogene 2018, 37, 3686–3697. [Google Scholar] [CrossRef]
- Li, H.; Huang, Y.; Jiang, D.Q.; Cui, L.Z.; He, Z.; Wang, C.; Zhang, Z.W.; Zhu, H.L.; Ding, Y.M.; Li, L.F.; et al. Antitumor activity of EGFR-specific CAR-T cells against non-small-cell lung cancer cells in vitro and in mice. Cell Death Dis. 2018, 9, 177. [Google Scholar] [CrossRef]
- Nakazawa, Y.; Matsuda, K.; Kurata, T.; Sueki, A.; Tanaka, M.; Sakashita, K.; Imai, C.; Wilson, M.H.; Koike, K. Anti-proliferative effects of T cells expressing a ligand-based chimeric antigen receptor against CD116 on CD34(+) cells of juvenile myelomonocytic leukemia. J. Hematol. Oncol. 2016, 9, 27. [Google Scholar] [CrossRef]
- Huang, X.; Park, H.; Greene, J.; Pao, J.; Mulvey, E.; Zhou, S.X.; Albert, C.M.; Moy, F.; Sachdev, D.; Yee, D.; et al. IGF1R- and ROR1-Specific CAR-T Cells as a Potential Therapy for High Risk Sarcomas. PLoS ONE 2015, 10, e0133152. [Google Scholar] [CrossRef]
- Krishnamurthy, J.; Rabinovich, B.A.; Mi, T.; Switzer, K.C.; Olivares, S.; Maiti, S.N.; Plummer, J.B.; Singh, H.; Kumaresan, P.R.; Huls, H.M.; et al. Genetic Engineering of T Cells to Target HERV-K, an Ancient Retrovirus on Melanoma. Clin. Cancer Res. 2015, 21, 3241–3251. [Google Scholar] [CrossRef] [PubMed]
- Chicaybam, L.; Abdo, L.; Bonamino, M.H. Generation of CAR+ T Lymphocytes Using the Sleeping Beauty Transposon System. Methods Mol. Biol. 2020, 2086, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfo, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR-T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef] [PubMed]
- Stadtmauer, E.A.; Cohen, A.D.; Weber, K.; Lacey, S.F.; Gonzalez, V.E.; Melenhorst, J.J.; Fraietta, J.A.; Plesa, G.; Shea, J.; Matlawski, T.; et al. First-in-Human Assessment of Feasibility and Safety of Multiplexed Genetic Engineering of Autologous T Cells Expressing NY-ESO -1 TCR and CRISPR/Cas9 Gene Edited to Eliminate Endogenous TCR and PD-1 (NYCE T cells) in Advanced Multiple Myeloma (MM) and Sarcoma. Blood 2019, 134, 49. [Google Scholar] [CrossRef]
- Chicaybam, L.; Abdo, L.; Viegas, M.; Marques, L.V.C.; de Sousa, P.; Batista-Silva, L.R.; Alves-Monteiro, V.; Bonecker, S.; Monte-Mor, B.; Bonamino, M.H. Transposon-mediated generation of CAR-T cells shows efficient anti B-cell leukemia response after ex vivo expansion. Gene Ther. 2020, 27, 85–95. [Google Scholar] [CrossRef]
- de Macedo Abdo, L.; Barros, L.R.C.; Saldanha Viegas, M.; Vieira Codeco Marques, L.; de Sousa Ferreira, P.; Chicaybam, L.; Bonamino, M.H. Development of CAR-T cell therapy for B-ALL using a point-of-care approach. Oncoimmunology 2020, 9, 1752592. [Google Scholar] [CrossRef]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gonen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef]
- Qin, D.Y.; Huang, Y.; Li, D.; Wang, Y.S.; Wang, W.; Wei, Y.Q. Paralleled comparison of vectors for the generation of CAR-T cells. Anticancer Drugs 2016, 27, 711–722. [Google Scholar] [CrossRef]
- Foster, J.B.; Choudhari, N.; Perazzelli, J.; Storm, J.; Hofmann, T.J.; Jain, P.; Storm, P.B.; Pardi, N.; Weissman, D.; Waanders, A.J.; et al. Purification of mRNA Encoding Chimeric Antigen Receptor Is Critical for Generation of a Robust T-Cell Response. Hum. Gene Ther. 2018. [Google Scholar] [CrossRef]
- Olden, B.R.; Cheng, Y.; Yu, J.L.; Pun, S.H. Cationic polymers for non-viral gene delivery to human T cells. J. Control Release 2018, 282, 140–147. [Google Scholar] [CrossRef]
- Singh, N.; Liu, X.; Hulitt, J.; Jiang, S.; June, C.H.; Grupp, S.A.; Barrett, D.M.; Zhao, Y. Nature of tumor control by permanently and transiently modified GD2 chimeric antigen receptor T cells in xenograft models of neuroblastoma. Cancer Immunol. Res. 2014, 2, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Moon, E.; Carpenito, C.; Paulos, C.M.; Liu, X.; Brennan, A.L.; Chew, A.; Carroll, R.G.; Scholler, J.; Levine, B.L.; et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010, 70, 9053–9061. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef]
- Panjwani, M.K.; Smith, J.B.; Schutsky, K.; Gnanandarajah, J.; O’Connor, C.M.; Powell, D.J., Jr.; Mason, N.J. Feasibility and Safety of RNA-transfected CD20-specific Chimeric Antigen Receptor T Cells in Dogs with Spontaneous B Cell Lymphoma. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 1602–1614. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.T.; Stephan, S.B.; Moffett, H.F.; McKnight, L.E.; Ji, W.; Reiman, D.; Bonagofski, E.; Wohlfahrt, M.E.; Pillai, S.P.S.; Stephan, M.T. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol. 2017, 12, 813–820. [Google Scholar] [CrossRef]
- Frank, A.M.; Buchholz, C.J. Surface-Engineered Lentiviral Vectors for Selective Gene Transfer into Subtypes of Lymphocytes. Mol. Ther. Methods Clin. Dev. 2019, 12, 19–31. [Google Scholar] [CrossRef]
- Zhang, X.; Claerhout, S.; Prat, A.; Dobrolecki, L.E.; Petrovic, I.; Lai, Q.; Landis, M.D.; Wiechmann, L.; Schiff, R.; Giuliano, M.; et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013, 73, 4885–4897. [Google Scholar] [CrossRef]
- Rongvaux, A.; Willinger, T.; Martinek, J.; Strowig, T.; Gearty, S.V.; Teichmann, L.L.; Saito, Y.; Marches, F.; Halene, S.; Palucka, A.K.; et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 2014, 32, 364–372. [Google Scholar] [CrossRef]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR-T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef]
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR-T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Kersten, K.; de Visser, K.E.; van Miltenburg, M.H.; Jonkers, J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol. Med. 2017, 9, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Dichwalkar, T.; Chang, J.Y.H.; Cossette, B.; Garafola, D.; Zhang, A.Q.; Fichter, M.; Wang, C.; Liang, S.; Silva, M.; et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science 2019, 365, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR-T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef]
- Ajina, A.; Maher, J. Synergistic combination of oncolytic virotherapy with CAR T-cell therapy. Prog. Mol. Biol. Transl. Sci. 2019, 164, 217–292. [Google Scholar] [CrossRef]
- Lanitis, E.; Coukos, G.; Irving, M. All systems go: Converging synthetic biology and combinatorial treatment for CAR-T cell therapy. Curr. Opin. Biotechnol. 2020, 65, 75–87. [Google Scholar] [CrossRef]
- Guedan, S.; Alemany, R. CAR-T Cells and Oncolytic Viruses: Joining Forces to Overcome the Solid Tumor Challenge. Front. Immunol. 2018, 9, 2460. [Google Scholar] [CrossRef]
- Tanoue, K.; Rosewell Shaw, A.; Watanabe, N.; Porter, C.; Rana, B.; Gottschalk, S.; Brenner, M.; Suzuki, M. Armed Oncolytic Adenovirus-Expressing PD-L1 Mini-Body Enhances Antitumor Effects of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Res. 2017, 77, 2040–2051. [Google Scholar] [CrossRef]
- Wing, A.; Fajardo, C.A.; Posey, A.D., Jr.; Shaw, C.; Da, T.; Young, R.M.; Alemany, R.; June, C.H.; Guedan, S. Improving CART-Cell Therapy of Solid Tumors with Oncolytic Virus-Driven Production of a Bispecific T-cell Engager. Cancer Immunol. Res. 2018, 6, 605–616. [Google Scholar] [CrossRef]
- Li, Y.; Xiao, F.; Zhang, A.; Zhang, D.; Nie, W.; Xu, T.; Han, B.; Seth, P.; Wang, H.; Yang, Y.; et al. Oncolytic adenovirus targeting TGF-beta enhances anti-tumor responses of mesothelin-targeted chimeric antigen receptor T cell therapy against breast cancer. Cell. Immunol. 2020, 348, 104041. [Google Scholar] [CrossRef] [PubMed]
- Mock, U.; Nickolay, L.; Philip, B.; Cheung, G.W.; Zhan, H.; Johnston, I.C.D.; Kaiser, A.D.; Peggs, K.; Pule, M.; Thrasher, A.J.; et al. Automated manufacturing of chimeric antigen receptor T cells for adoptive immunotherapy using CliniMACS prodigy. Cytotherapy 2016, 18, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Granzin, M.; Soltenborn, S.; Muller, S.; Kollet, J.; Berg, M.; Cerwenka, A.; Childs, R.W.; Huppert, V. Fully automated expansion and activation of clinical-grade natural killer cells for adoptive immunotherapy. Cytotherapy 2015, 17, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.E.; Chicaybam, L.; Stein, R.T.; Tanuri, A.; Delgado-Canedo, A.; Bonamino, M.H. Retroviral vectors and transposons for stable gene therapy: Advances, current challenges and perspectives. J. Transl. Med. 2016, 14, 288. [Google Scholar] [CrossRef]
- Ghassemi, S.; Nunez-Cruz, S.; O’Connor, R.S.; Fraietta, J.A.; Patel, P.R.; Scholler, J.; Barrett, D.M.; Lundh, S.M.; Davis, M.M.; Bedoya, F.; et al. Reducing Ex Vivo Culture Improves the Antileukemic Activity of Chimeric Antigen Receptor (CAR) T Cells. Cancer Immunol. Res. 2018, 6, 1100–1109. [Google Scholar] [CrossRef]
- Haque, T.; Wilkie, G.M.; Jones, M.M.; Higgins, C.D.; Urquhart, G.; Wingate, P.; Burns, D.; McAulay, K.; Turner, M.; Bellamy, C.; et al. Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: Results of a phase 2 multicenter clinical trial. Blood 2007, 110, 1123–1131. [Google Scholar] [CrossRef]
- Melenhorst, J.J.; Leen, A.M.; Bollard, C.M.; Quigley, M.F.; Price, D.A.; Rooney, C.M.; Brenner, M.K.; Barrett, A.J.; Heslop, H.E. Allogeneic virus-specific T cells with HLA alloreactivity do not produce GVHD in human subjects. Blood 2010, 116, 4700–4702. [Google Scholar] [CrossRef]
- Cruz, C.R.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood 2013, 122, 2965–2973. [Google Scholar] [CrossRef]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR-T cells. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR-T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef]
- Torikai, H.; Reik, A.; Soldner, F.; Warren, E.H.; Yuen, C.; Zhou, Y.; Crossland, D.L.; Huls, H.; Littman, N.; Zhang, Z.; et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 2013, 122, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Kasakovski, D.; Xu, L.; Li, Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J. Hematol. Oncol. 2018, 11, 91. [Google Scholar] [CrossRef] [PubMed]
- Ajina, A.; Maher, J. Strategies to Address Chimeric Antigen Receptor Tonic Signaling. Mol. Cancer Ther. 2018, 17, 1795–1815. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, A.S.; Peterson, C.W.; Kiem, H.P. Chimeric antigen receptor T-cell approaches to HIV cure. Curr. Opin. HIV AIDS 2018, 13, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, H.; Kimura, T.; Rurik, J.G.; Hancock, A.S.; Leibowitz, M.S.; Li, L.; Scholler, J.; Monslow, J.; Lo, A.; Han, W.; et al. Targeting cardiac fibrosis with engineered T cells. Nature 2019, 573, 430–433. [Google Scholar] [CrossRef]
- Zhang, Q.; Lu, W.; Liang, C.L.; Chen, Y.; Liu, H.; Qiu, F.; Dai, Z. Chimeric Antigen Receptor (CAR) Treg: A Promising Approach to Inducing Immunological Tolerance. Front. Immunol. 2018, 9, 2359. [Google Scholar] [CrossRef]
- Maldini, C.R.; Ellis, G.I.; Riley, J.L. CAR-T cells for infection, autoimmunity and allotransplantation. Nat. Rev. Immunol. 2018, 18, 605–616. [Google Scholar] [CrossRef]


{kind=link}
| Location (Year/Agency) | Company | Product | Description |
|---|---|---|---|
| China (2003/SFDA) | Shenzhen Gentech SiBiono, Shenzhen, China | Gendicine (Ad-p53) | Non-replicating adenoviral vector expressing wild-type p53 for the treatment of head and neck cancer. |
| China (2005/SFDA) | Shanghai Sunway Biotech, Pudong, Shanghai, China | Oncorine/H101 (Onyx-015) | Conditionally replicating adenovirus containing a mutant E1b protein which confers tumor-specific oncolysis. Approved for treatment of head and neck cancers. |
| European Union (2012/EMA) | Amsterdam Molecular Therapeutics, Amsterdam, The Netherlands | Glybera *(alipogene tiparvovec) | Adeno-associated virus encoding the Ser(447)X variant of human lipoprotein lipase (LPL) gene for the treatment of familial LPL deficiency. |
| USA (2015/FDA) European Union (2015/EMA) | BioVex Inc(a subsidiary ofAmgen, Inc), Woburn, MA, USA | Imlygic (talimogene laherparepvec, T-Vec, oncovex-GMCSF) | Oncolytic herpes virus encoding GM-CSF for the treatment of melanoma. |
| European Union (2016/EMA) | GlaxoSmithKline (GSK), Uxbridge, Middlesex, UK | Strimvelis (GSK2696273) | Retroviral vector encoding adenosine deaminase (ADA) for the treatment of ADA-SCID. |
| USA (2017/FDA) European Union (2018/EMA) | Novartis, Basel, Switzerland | Kymriah (tisagenlecleucel, CTL019) | CAR-T cell targeting CD19 for the treatment of B cell acute lymphocytic leukemia. |
| USA (2017/FDA) European Union (2018/EMA) | Kite Pharma (acquired by Gilead Sciences), Santa Monica, CA, USA | Yescarta (axicabtagene ciloleucel, Axi-cel) | CAR-T cell targeting CD19 for the treatment of diffuse large B cell lymphoma. |
| USA (2017/FDA) European Union (2018/EMA) | Spark Therapeutics, Philadelphia, PA, USA | Luxturna (Voretigene neparvovec) | AAV encoding RPE65 for the treatment of Leber’s Congenital Amaurosis. |
| USA (2019/FDA) | Avexis (a subsidiary of Novartis), Bannockburn, IL, USA | Zolgensma (onasemnogene abeparvovec-xioi) | AAV encoding SMN1 for the treatment of spinal muscular atrophy (SMA). |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chicaybam, L.; Bonamino, M.H.; Luckow Invitti, A.; Bortman Rozenchan, P.; de Luna Vieira, I.; Strauss, B.E. Overhauling CAR T Cells to Improve Efficacy, Safety and Cost. Cancers 2020, 12, 2360. https://doi.org/10.3390/cancers12092360
Chicaybam L, Bonamino MH, Luckow Invitti A, Bortman Rozenchan P, de Luna Vieira I, Strauss BE. Overhauling CAR T Cells to Improve Efficacy, Safety and Cost. Cancers. 2020; 12(9):2360. https://doi.org/10.3390/cancers12092360
Chicago/Turabian StyleChicaybam, Leonardo, Martín H. Bonamino, Adriana Luckow Invitti, Patricia Bortman Rozenchan, Igor de Luna Vieira, and Bryan E. Strauss. 2020. "Overhauling CAR T Cells to Improve Efficacy, Safety and Cost" Cancers 12, no. 9: 2360. https://doi.org/10.3390/cancers12092360
APA StyleChicaybam, L., Bonamino, M. H., Luckow Invitti, A., Bortman Rozenchan, P., de Luna Vieira, I., & Strauss, B. E. (2020). Overhauling CAR T Cells to Improve Efficacy, Safety and Cost. Cancers, 12(9), 2360. https://doi.org/10.3390/cancers12092360