Aberrant BUB1 Overexpression Promotes Mitotic Segregation Errors and Chromosomal Instability in Multiple Myeloma

 and
and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. BUB1 Overexpression in Patient-Derived Primary Myeloma Cells and Human Myeloma-Derived Cell Lines (HMCLs)
2.2. Generation of BUB1-Reduced HMCLs
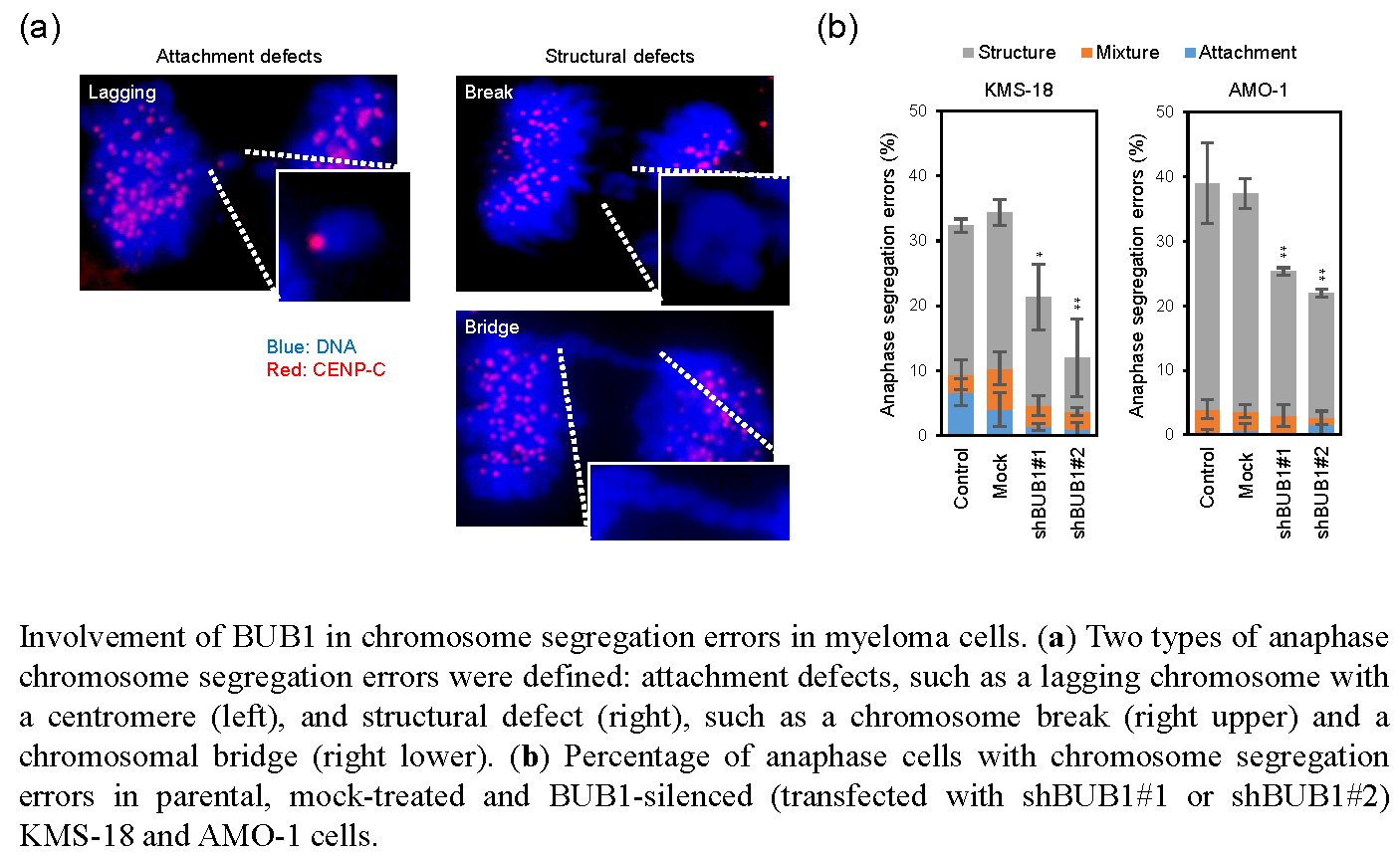
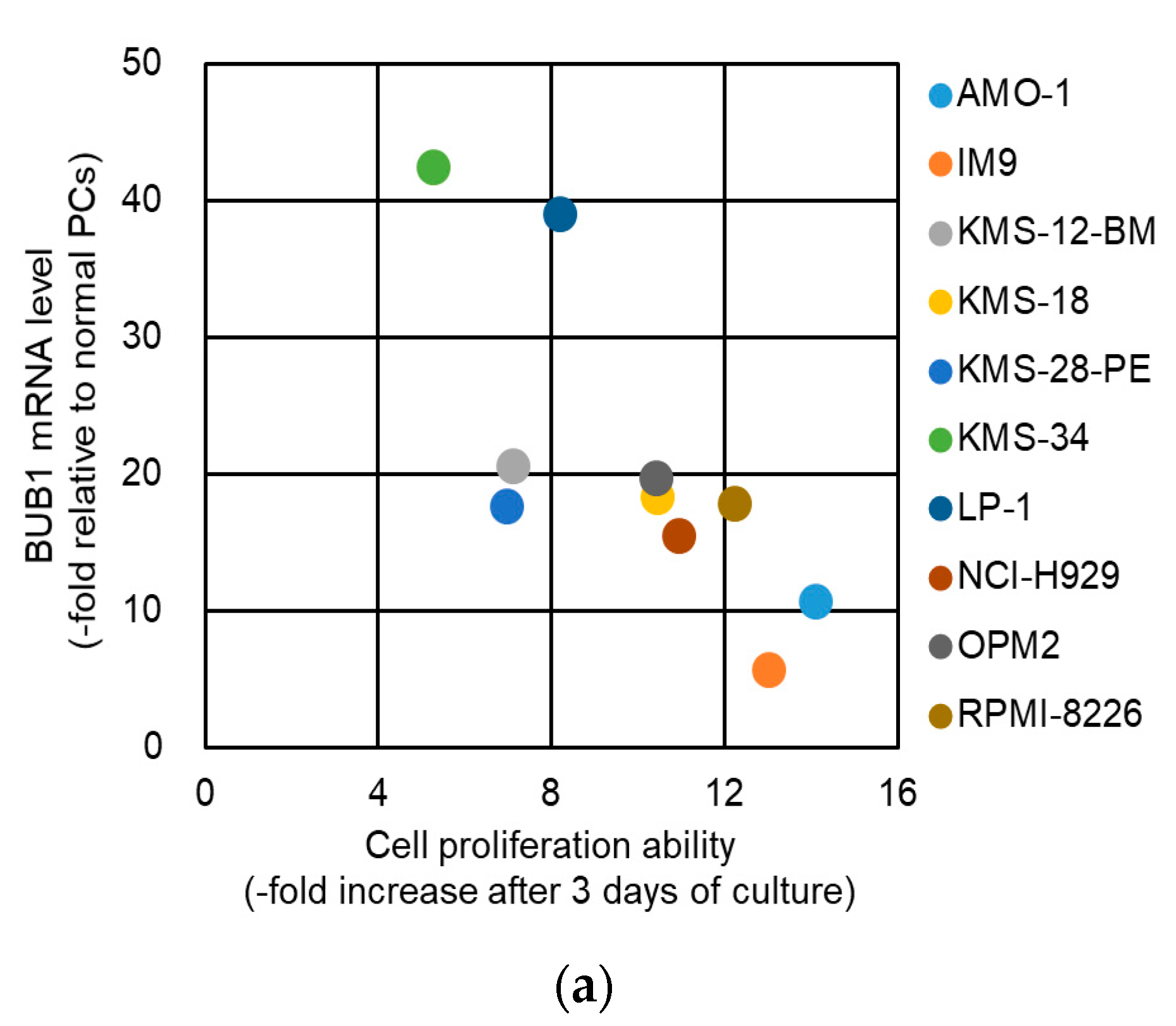
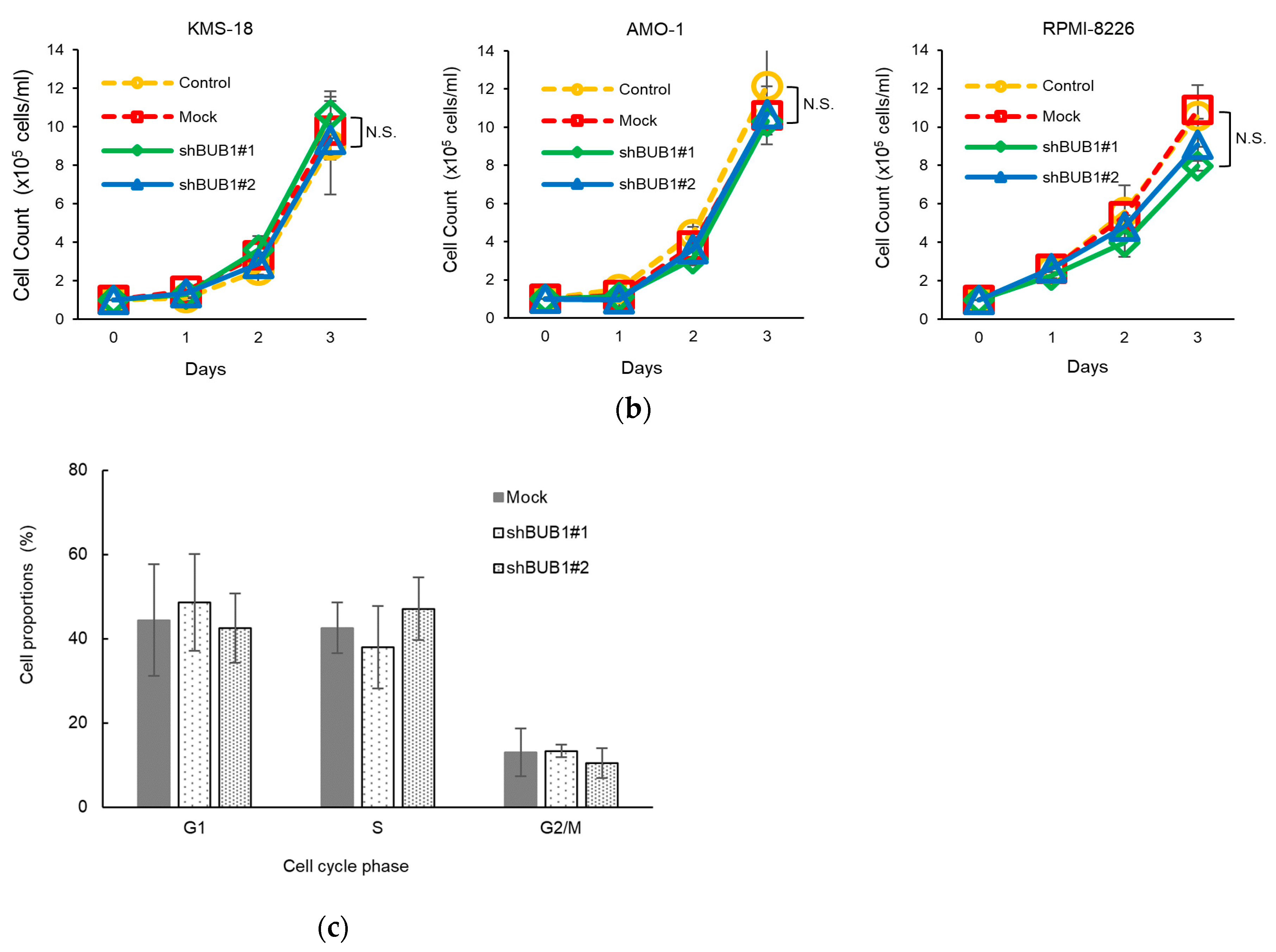
2.3. No Association of BUB1 Expression with Short-Term Proliferative Potency in HMCLs
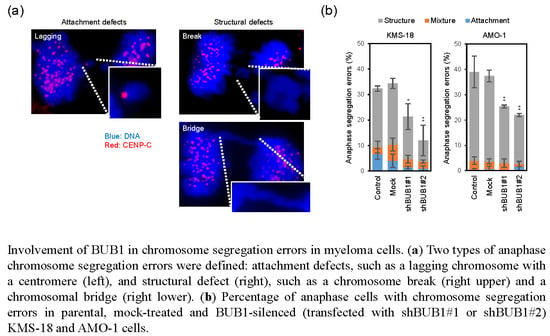
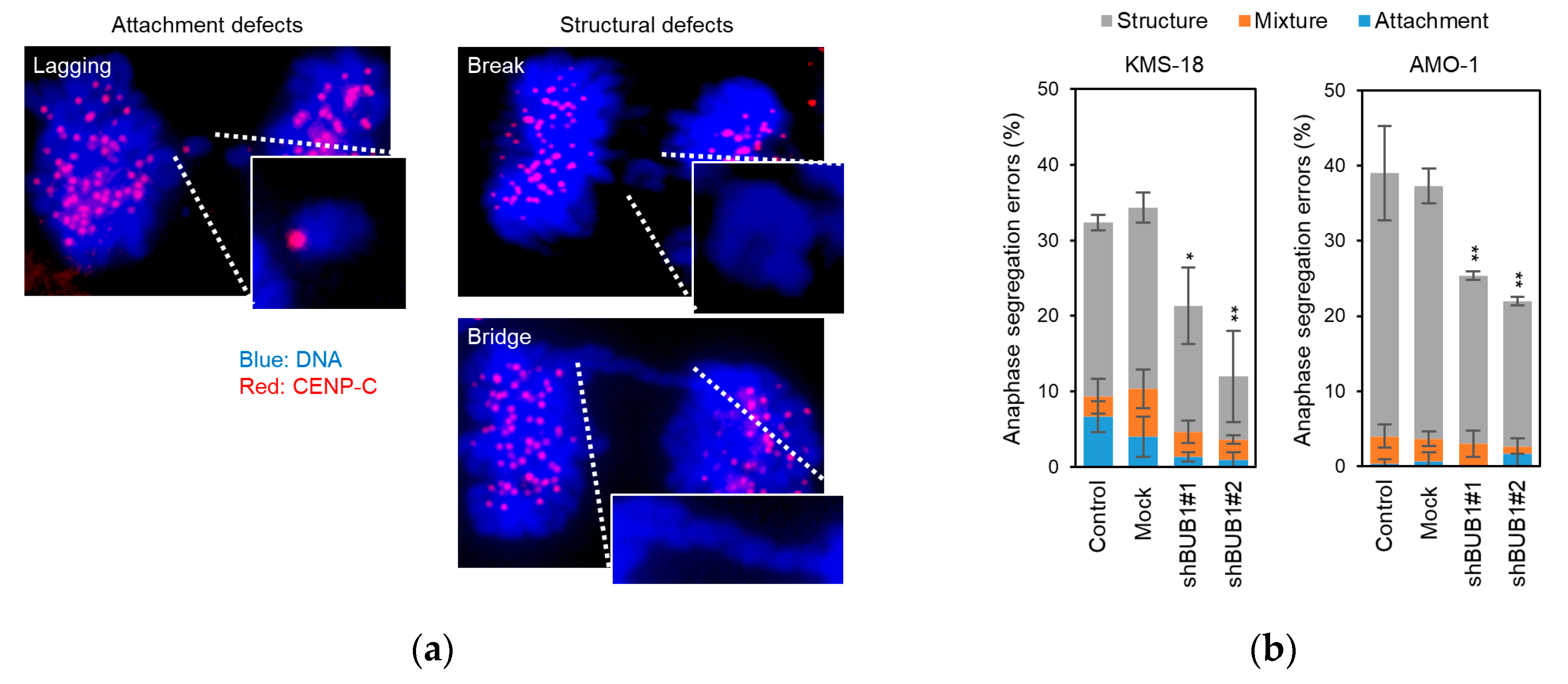
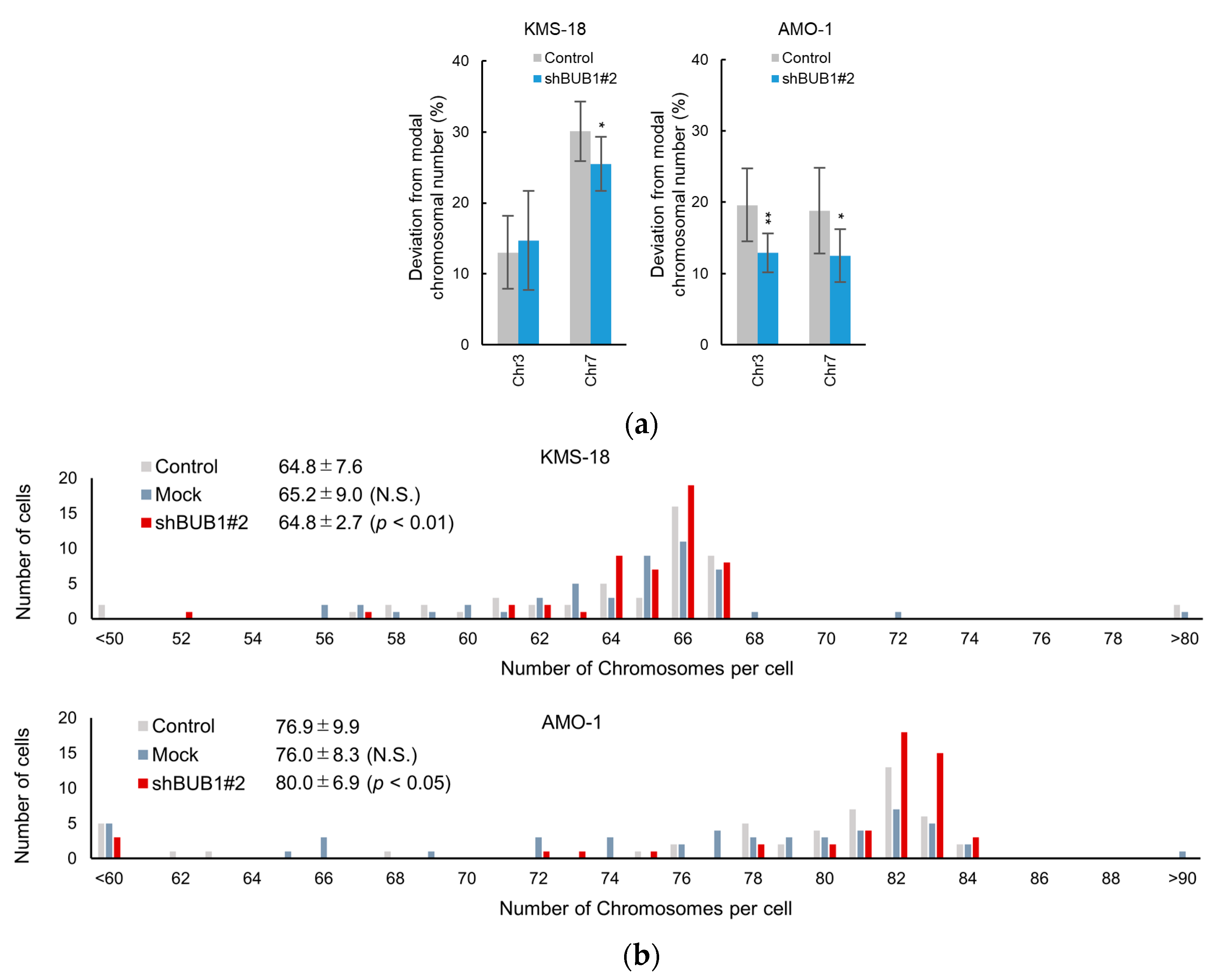
2.4. BUB1 Overexpression Promotes Chromosome Segregation Defects and Chromosome Instability (CIN) in Myeloma Cells
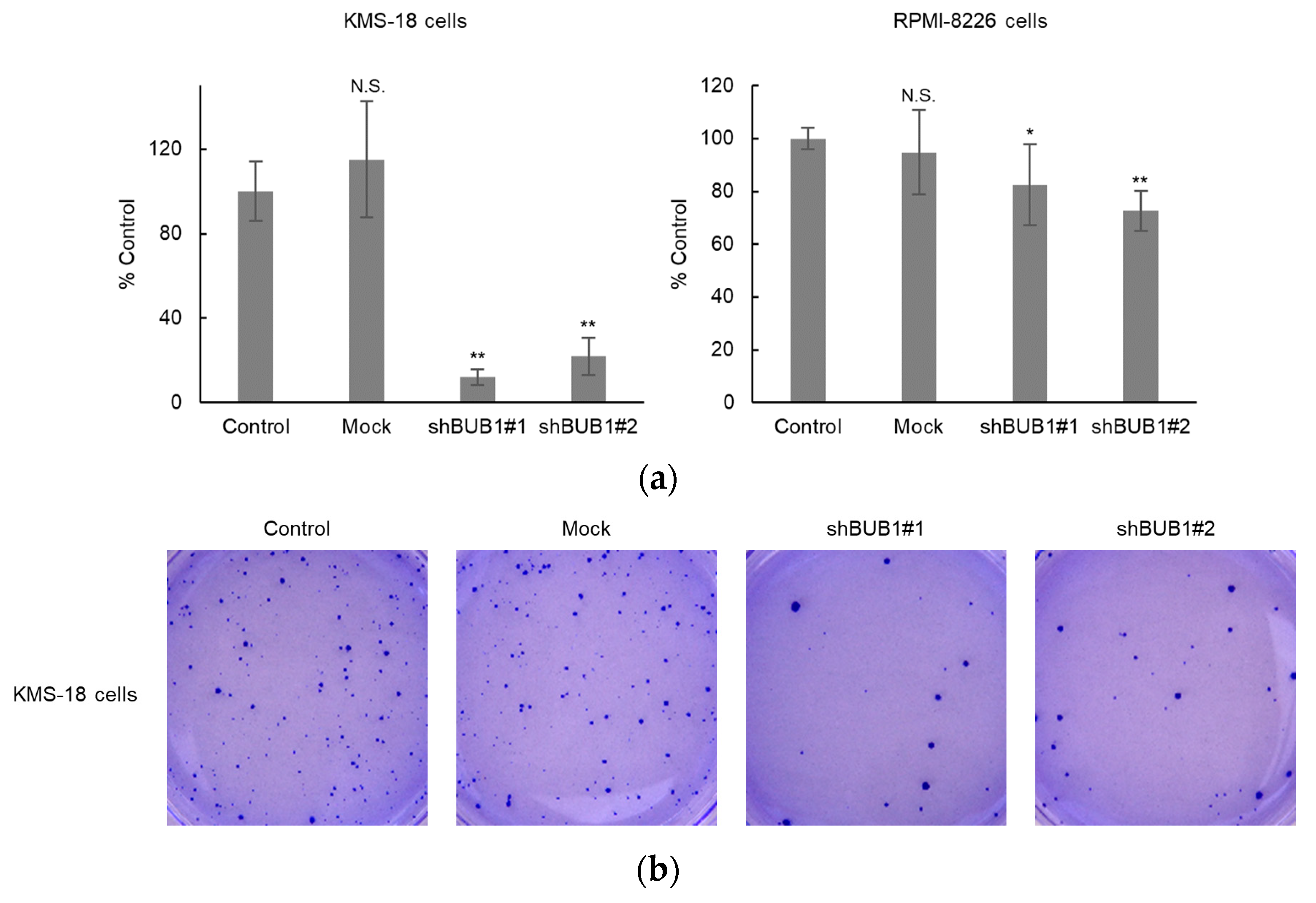
2.5. BUB1 Knockdown Significantly Reduces Colony Formation of Myeloma Cells
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. Cell Lines and Reagents
4.3. Quantitative RT-PCR
4.4. Western Blotting
4.5. Lentiviral Preparation and Infection
4.6. Immunofluorescence Microscopy
4.7. Chromosomal Analysis
4.8. Assays for Cell Proliferation and Cell Cycle
4.9. Colony-Formation Assay
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Walker, B.A.; Wardell, C.P.; Melchor, L.; Hulkki, S.; Potter, N.E.; Johnson, D.C.; Fenwick, K.; Kozarewa, I.; Gonzalez, D.; Lord, C.J.; et al. Intraclonal heterogeneity and distinct molecular mechanisms characterize the development of t(4;14) and t(11;14) myeloma. Blood 2012, 120, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Wardell, C.P.; Melchor, L.; Brioli, A.; Johnson, D.C.; Kaiser, M.F.; Mirabella, F.; Lopez-Corral, L.; Humphray, S.; Murray, L.; et al. Intraclonal heterogeneity is a critical early event in the development of myeloma and precedes the development of clinical symptoms. Leukemia 2014, 28, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Magrangeas, F.; Avet-Loiseau, H.; Gouraud, W.; Lode, L.; Decaux, O.; Godmer, P.; Garderet, L.; Voillat, L.; Facon, T.; Stoppa, A.M.; et al. Minor clone provides a reservoir for relapse in multiple myeloma. Leukemia 2013, 27, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Chesi, M.; Egan, J.B.; Garbitt, V.M.; Palmer, S.E.; Braggio, E.; Van Wier, S.; Blackburn, P.R.; Baker, A.S.; Dispenzieri, A.; et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012, 120, 1067–1076. [Google Scholar] [CrossRef]
- Egan, J.B.; Shi, C.X.; Tembe, W.; Christoforides, A.; Kurdoglu, A.; Sinari, S.; Middha, S.; Asmann, Y.; Schmidt, J.; Braggio, E.; et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood 2012, 120, 1060–1066. [Google Scholar] [CrossRef]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol 2017, 14, 100–113. [Google Scholar] [CrossRef]
- Yamazaki, H.; Shirakawa, K.; Matsumoto, T.; Hirabayashi, S.; Murakawa, Y.; Kobayashi, M.; Sarca, A.D.; Kazuma, Y.; Matsui, H.; Maruyama, W.; et al. Endogenous APOBEC3B Overexpression Constitutively Generates DNA Substitutions and Deletions in Myeloma Cells. Sci. Rep. 2019, 9, 7122. [Google Scholar] [CrossRef]
- Walker, B.A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Melchor, L.; Pawlyn, C.; Kaiser, M.F.; et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat. Commun. 2015, 6, 6997. [Google Scholar] [CrossRef]
- Ohguchi, H.; Hideshima, T.; Anderson, K.C. The biological significance of histone modifiers in multiple myeloma: Clinical applications. Blood Cancer J. 2018, 8, 83. [Google Scholar] [CrossRef]
- Schvartzman, J.M.; Sotillo, R.; Benezra, R. Mitotic chromosomal instability and cancer: Mouse modelling of the human disease. Nat. Rev. Cancer 2010, 10, 102–115. [Google Scholar] [CrossRef]
- Lara-Gonzalez, P.; Westhorpe, F.G.; Taylor, S.S. The spindle assembly checkpoint. Curr. Biol. 2012, 22, R966–R980. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, S.; Gorbsky, G.J. Spatiotemporal regulation of the anaphase-promoting complex in mitosis. Nat. Rev. Mol. Cell Biol. 2015, 16, 82–94. [Google Scholar] [CrossRef]
- Williams, G.L.; Roberts, T.M.; Gjoerup, O.V. Bub1: Escapades in a cellular world. Cell Cycle 2007, 6, 1699–1704. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Sun, Y.; Harley, S.E.; Zou, H.; Yu, H. Human Bub1 protects centromeric sister-chromatid cohesion through Shugoshin during mitosis. Proc. Natl. Acad. Sci. USA 2004, 101, 18012–18017. [Google Scholar] [CrossRef] [PubMed]
- Klebig, C.; Korinth, D.; Meraldi, P. Bub1 regulates chromosome segregation in a kinetochore-independent manner. J. Cell Biol. 2009, 185, 841–858. [Google Scholar] [CrossRef]
- Cahill, D.P.; Lengauer, C.; Yu, J.; Riggins, G.J.; Willson, J.K.; Markowitz, S.D.; Kinzler, K.W.; Vogelstein, B. Mutations of mitotic checkpoint genes in human cancers. Nature 1998, 392, 300–303. [Google Scholar] [CrossRef]
- Ohshima, K.; Haraoka, S.; Yoshioka, S.; Hamasaki, M.; Fujiki, T.; Suzumiya, J.; Kawasaki, C.; Kanda, M.; Kikuchi, M. Mutation analysis of mitotic checkpoint genes (hBUB1 and hBUBR1) and microsatellite instability in adult T-cell leukemia/lymphoma. Cancer Lett. 2000, 158, 141–150. [Google Scholar] [CrossRef]
- Hempen, P.M.; Kurpad, H.; Calhoun, E.S.; Abraham, S.; Kern, S.E. A double missense variation of the BUB1 gene and a defective mitotic spindle checkpoint in the pancreatic cancer cell line Hs766T. Hum. Mutat. 2003, 21, 445. [Google Scholar] [CrossRef]
- Grabsch, H.I.; Askham, J.M.; Morrison, E.E.; Pomjanski, N.; Lickvers, K.; Parsons, W.J.; Boecking, A.; Gabbert, H.E.; Mueller, W. Expression of BUB1 protein in gastric cancer correlates with the histological subtype, but not with DNA ploidy or microsatellite instability. J. Pathol. 2004, 202, 208–214. [Google Scholar] [CrossRef]
- Grabsch, H.; Takeno, S.; Parsons, W.J.; Pomjanski, N.; Boecking, A.; Gabbert, H.E.; Mueller, W. Overexpression of the mitotic checkpoint genes BUB1, BUBR1, and BUB3 in gastric cancer--association with tumour cell proliferation. J. Pathol. 2003, 200, 16–22. [Google Scholar] [CrossRef]
- Shigeishi, H.; Oue, N.; Kuniyasu, H.; Wakikawa, A.; Yokozaki, H.; Ishikawa, T.; Yasui, W. Expression of Bub1 gene correlates with tumor proliferating activity in human gastric carcinomas. Pathobiology 2001, 69, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Stahl, D.; Braun, M.; Gentles, A.J.; Lingohr, P.; Walter, A.; Kristiansen, G.; Gutgemann, I. Low BUB1 expression is an adverse prognostic marker in gastric adenocarcinoma. Oncotarget 2017, 8, 76329–76339. [Google Scholar] [CrossRef] [PubMed]
- Shichiri, M.; Yoshinaga, K.; Hisatomi, H.; Sugihara, K.; Hirata, Y. Genetic and epigenetic inactivation of mitotic checkpoint genes hBUB1 and hBUBR1 and their relationship to survival. Cancer Res. 2002, 62, 13–17. [Google Scholar] [PubMed]
- Piao, J.; Zhu, L.; Sun, J.; Li, N.; Dong, B.; Yang, Y.; Chen, L. High expression of CDK1 and BUB1 predicts poor prognosis of pancreatic ductal adenocarcinoma. Gene 2019, 701, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Gu, Z.Y.; Li, Q.; Liu, Q.H.; Yang, X.Y.; Zhang, J.J. Identification of significant genes with poor prognosis in ovarian cancer via bioinformatical analysis. J. Ovarian Res. 2019, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhang, S.; Ibrahim, A.N.; Deng, Z.; Wang, M. Serine/threonine kinase BUB1 promotes proliferation and radio-resistance in glioblastoma. Pathol. Res. Pr. 2019, 215, 152508. [Google Scholar] [CrossRef]
- Tatekawa, S.; Chinen, Y.; Ri, M.; Narita, T.; Shimura, Y.; Matsumura-Kimoto, Y.; Tsukamoto, T.; Kobayashi, T.; Kawata, E.; Uoshima, N.; et al. Epigenetic repression of miR-375 is the dominant mechanism for constitutive activation of the PDPK1/RPS6KA3 signalling axis in multiple myeloma. Br. J. Haematol. 2017, 178, 534–546. [Google Scholar] [CrossRef]
- Morales, A.G.; Pezuk, J.A.; Brassesco, M.S.; de Oliveira, J.C.; de Paula Queiroz, R.G.; Machado, H.R.; Carlotti, C.G., Jr.; Neder, L.; de Oliveira, H.F.; Scrideli, C.A.; et al. BUB1 and BUBR1 inhibition decreases proliferation and colony formation, and enhances radiation sensitivity in pediatric glioblastoma cells. Childs Nerv. Syst. 2013, 29, 2241–2248. [Google Scholar] [CrossRef]
- Perera, D.; Tilston, V.; Hopwood, J.A.; Barchi, M.; Boot-Handford, R.P.; Taylor, S.S. Bub1 maintains centromeric cohesion by activation of the spindle checkpoint. Dev. Cell 2007, 13, 566–579. [Google Scholar] [CrossRef]
- Wang, Z.; Katsaros, D.; Shen, Y.; Fu, Y.; Canuto, E.M.; Benedetto, C.; Lu, L.; Chu, W.M.; Risch, H.A.; Yu, H. Biological and Clinical Significance of MAD2L1 and BUB1, Genes Frequently Appearing in Expression Signatures for Breast Cancer Prognosis. PLoS ONE 2015, 10, e0136246. [Google Scholar] [CrossRef]
- Han, J.Y.; Han, Y.K.; Park, G.Y.; Kim, S.D.; Lee, C.G. Bub1 is required for maintaining cancer stem cells in breast cancer cell lines. Sci. Rep. 2015, 5, 15993. [Google Scholar] [CrossRef] [PubMed]
- Chinen, Y.; Kuroda, J.; Shimura, Y.; Nagoshi, H.; Kiyota, M.; Yamamoto-Sugitani, M.; Mizutani, S.; Sakamoto, N.; Ri, M.; Kawata, E.; et al. Phosphoinositide protein kinase PDPK1 is a crucial cell signaling mediator in multiple myeloma. Cancer Res. 2014, 74, 7418–7429. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instability in colorectal cancers. Nature 1997, 386, 623–627. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Silkworth, W.T.; Nardi, I.K.; Nicholson, J.M.; Compton, D.A.; Cimini, D. The mitotic origin of chromosomal instability. Curr. Biol. 2014, 24, R148–R149. [Google Scholar] [CrossRef] [PubMed]
- Schürch, C.M.; Rasche, L.; Frauenfeld, L.; Weinhold, N.; Fend, F. A review on tumor heterogeneity and evolution in multiple myeloma: Pathological, radiological, molecular genetics, and clinical integration. Virchows Arch. 2020, 476, 337–351. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 548–567. [Google Scholar] [CrossRef]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689.e3. [Google Scholar] [CrossRef]
- Pinto, A.E.; Pires, A.; Silva, G.; Bicho, C.; Andre, S.; Soares, J. Ploidy and S-phase fraction as predictive markers of response to radiotherapy in cervical cancer. Pathol. Res. Pr. 2011, 207, 623–627. [Google Scholar] [CrossRef]
- Duijf, P.H.; Schultz, N.; Benezra, R. Cancer cells preferentially lose small chromosomes. Int. J. Cancer 2013, 132, 2316–2326. [Google Scholar] [CrossRef]
- Mizuno, Y.; Tsukamoto, T.; Kawata, E.; Uoshima, N.; Uchiyama, H.; Yokota, I.; Maegawa, S.; Takimoto, T.; Tanba, K.; Matsumura-Kimoto, Y.; et al. Chromosomal abnormality variation detected by G-banding is associated with prognosis of diffuse large B-cell lymphoma treated by R-CHOP-based therapy. Cancer Med. 2018, 7, 655–664. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2019, 33, 159–170. [Google Scholar] [CrossRef]
- Baysal, M.; Demirci, U.; Umit, E.; Kirkizlar, H.O.; Atli, E.I.; Gurkan, H.; Gulsaran, S.K.; Bas, V.; Mail, C.; Demir, A.M. Concepts of Double Hit and Triple Hit Disease in Multiple Myeloma, Entity and Prognostic Significance. Sci. Rep. 2020, 10, 5991. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Zhang, P. Aneuploidy and tumorigenesis. Semin. Cell Dev. Biol. 2011, 22, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Ricke, R.M.; Jeganathan, K.B.; van Deursen, J.M. Bub1 overexpression induces aneuploidy and tumor formation through Aurora B kinase hyperactivation. J. Cell Biol. 2011, 193, 1049–1064. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.F.; Lin, P.M.; Yang, M.C.; Liu, T.C.; Chang, J.G.; Sue, Y.C.; Chen, T.P. Expression of hBUB1 in acute myeloid leukemia. Leuk. Lymphoma 2002, 43, 385–391. [Google Scholar] [CrossRef]
- Pagotto, S.; Veronese, A.; Soranno, A.; Balatti, V.; Ramassone, A.; Guanciali-Franchi, P.E.; Palka, G.; Innocenti, I.; Autore, F.; Rassenti, L.Z.; et al. HNRNPL Restrains miR-155 Targeting of BUB1 to Stabilize Aberrant Karyotypes of Transformed Cells in Chronic Lymphocytic Leukemia. Cancers 2019, 11, 575. [Google Scholar] [CrossRef]
- Roberto, G.M.; Engel, E.E.; Scrideli, C.A.; Tone, L.G.; Brassesco, M.S. Downregulation of miR-10B* is correlated with altered expression of mitotic kinases in osteosarcoma. Pathol. Res. Pr. 2018, 214, 213–216. [Google Scholar] [CrossRef]
- Jessulat, M.; Malty, R.H.; Nguyen-Tran, D.H.; Deineko, V.; Aoki, H.; Vlasblom, J.; Omidi, K.; Jin, K.; Minic, Z.; Hooshyar, M.; et al. Spindle Checkpoint Factors Bub1 and Bub2 Promote DNA Double-Strand Break Repair by Nonhomologous End Joining. Mol. Cell Biol. 2015, 35, 2448–2463. [Google Scholar] [CrossRef]
- Sheltzer, J.M.; Amon, A. The aneuploidy paradox: Costs and benefits of an incorrect karyotype. Trends Genet. 2011, 27, 446–453. [Google Scholar] [CrossRef]
- Pfau, S.J.; Silberman, R.E.; Knouse, K.A.; Amon, A. Aneuploidy impairs hematopoietic stem cell fitness and is selected against in regenerating tissues in vivo. Genes Dev. 2016, 30, 1395–1408. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Takimoto-Shimomura, T.; Tsukamoto, T.; Maegawa, S.; Fujibayashi, Y.; Matsumura-Kimoto, Y.; Mizuno, Y.; Chinen, Y.; Shimura, Y.; Mizutani, S.; Horiike, S.; et al. Dual targeting of bromodomain-containing 4 by AZD5153 and BCL2 by AZD4320 against B-cell lymphomas concomitantly overexpressing c-MYC and BCL2. Investig. New Drugs 2019, 37, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, T.; Nakahata, S.; Sato, R.; Kanai, A.; Nakano, M.; Chinen, Y.; Maegawa-Matsui, S.; Matsumura-Kimoto, Y.; Takimoto-Shimomura, T.; Mizuno, Y.; et al. BRD4-Regulated Molecular Targets in Mantle Cell Lymphoma: Insights into Targeted Therapeutic Approach. Cancer Genom. Proteom. 2020, 17, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Chinen, Y.; Tsukamoto, T.; Takimoto-Shimomura, T.; Matsumura-Kimoto, Y.; Fujibayashi, Y.; Kuwahara-Ota, S.; Fujino, T.; Nishiyama, D.; Shimura, Y.; et al. A novel method of amplified fluorescent in situ hybridization for detection of chromosomal microdeletions in B cell lymphoma. Int. J. Hematol. 2019, 109, 593–602. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujibayashi, Y.; Isa, R.; Nishiyama, D.; Sakamoto-Inada, N.; Kawasumi, N.; Yamaguchi, J.; Kuwahara-Ota, S.; Matsumura-Kimoto, Y.; Tsukamoto, T.; Chinen, Y.; et al. Aberrant BUB1 Overexpression Promotes Mitotic Segregation Errors and Chromosomal Instability in Multiple Myeloma. Cancers 2020, 12, 2206. https://doi.org/10.3390/cancers12082206
Fujibayashi Y, Isa R, Nishiyama D, Sakamoto-Inada N, Kawasumi N, Yamaguchi J, Kuwahara-Ota S, Matsumura-Kimoto Y, Tsukamoto T, Chinen Y, et al. Aberrant BUB1 Overexpression Promotes Mitotic Segregation Errors and Chromosomal Instability in Multiple Myeloma. Cancers. 2020; 12(8):2206. https://doi.org/10.3390/cancers12082206
Chicago/Turabian StyleFujibayashi, Yuto, Reiko Isa, Daichi Nishiyama, Natsumi Sakamoto-Inada, Norichika Kawasumi, Junko Yamaguchi, Saeko Kuwahara-Ota, Yayoi Matsumura-Kimoto, Taku Tsukamoto, Yoshiaki Chinen, and et al. 2020. "Aberrant BUB1 Overexpression Promotes Mitotic Segregation Errors and Chromosomal Instability in Multiple Myeloma" Cancers 12, no. 8: 2206. https://doi.org/10.3390/cancers12082206
APA StyleFujibayashi, Y., Isa, R., Nishiyama, D., Sakamoto-Inada, N., Kawasumi, N., Yamaguchi, J., Kuwahara-Ota, S., Matsumura-Kimoto, Y., Tsukamoto, T., Chinen, Y., Shimura, Y., Kobayashi, T., Horiike, S., Taniwaki, M., Handa, H., & Kuroda, J. (2020). Aberrant BUB1 Overexpression Promotes Mitotic Segregation Errors and Chromosomal Instability in Multiple Myeloma. Cancers, 12(8), 2206. https://doi.org/10.3390/cancers12082206