Engineering a Humanised Niche to Support Human Haematopoiesis in Mice: Novel Opportunities in Modelling Cancer

 , , , ,
, , , ,  , ,
, ,  , , , and
, , , and 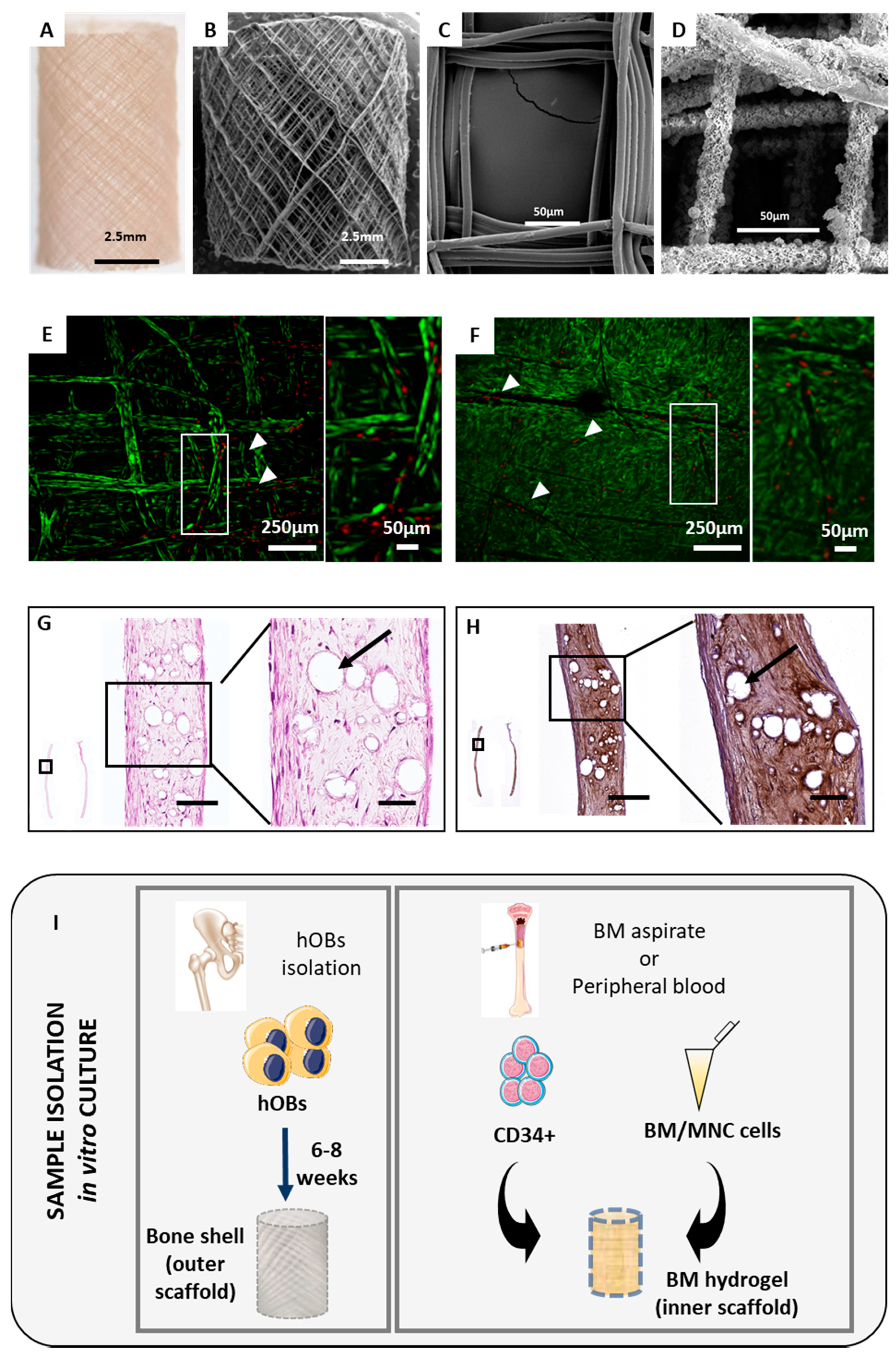{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
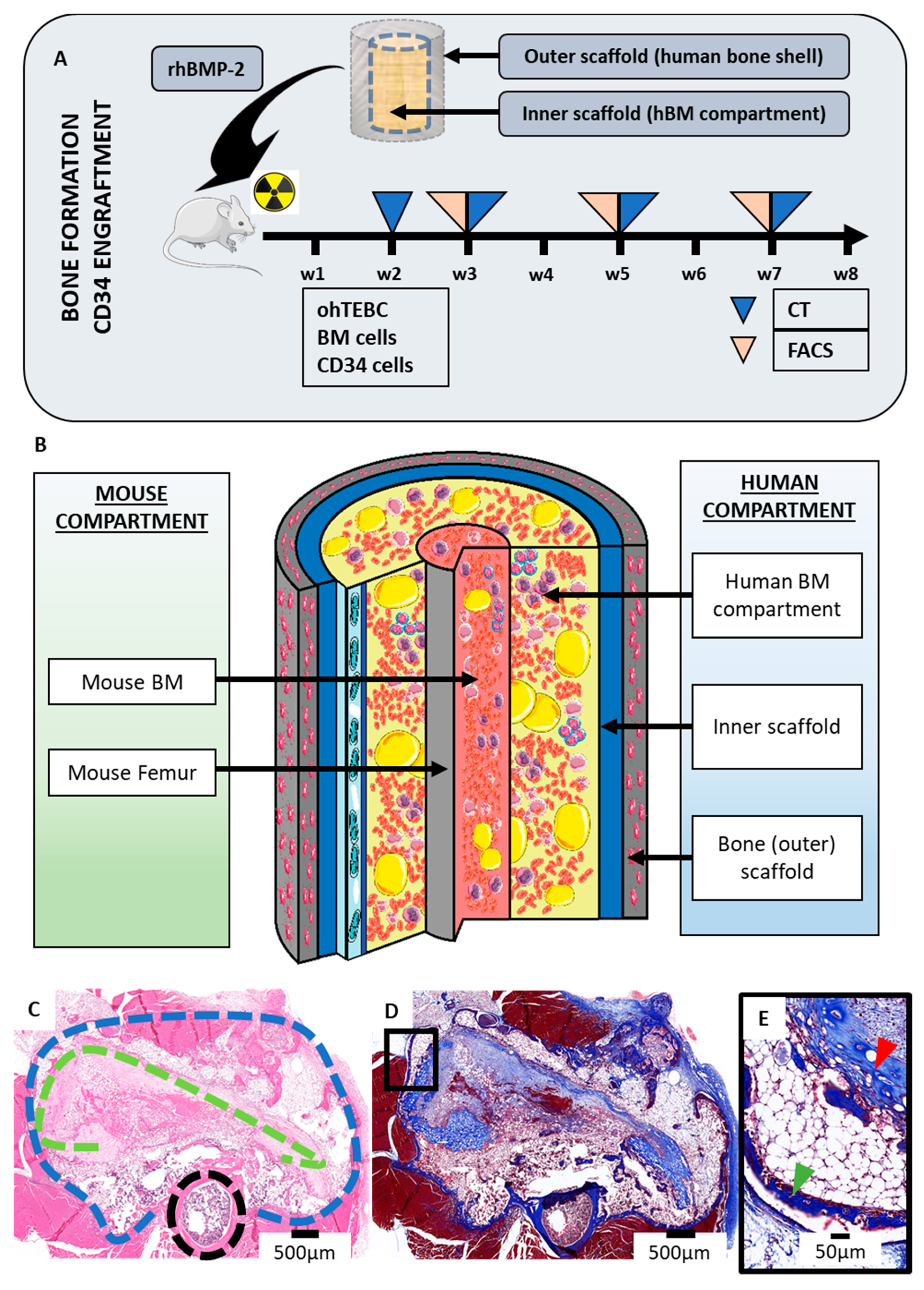
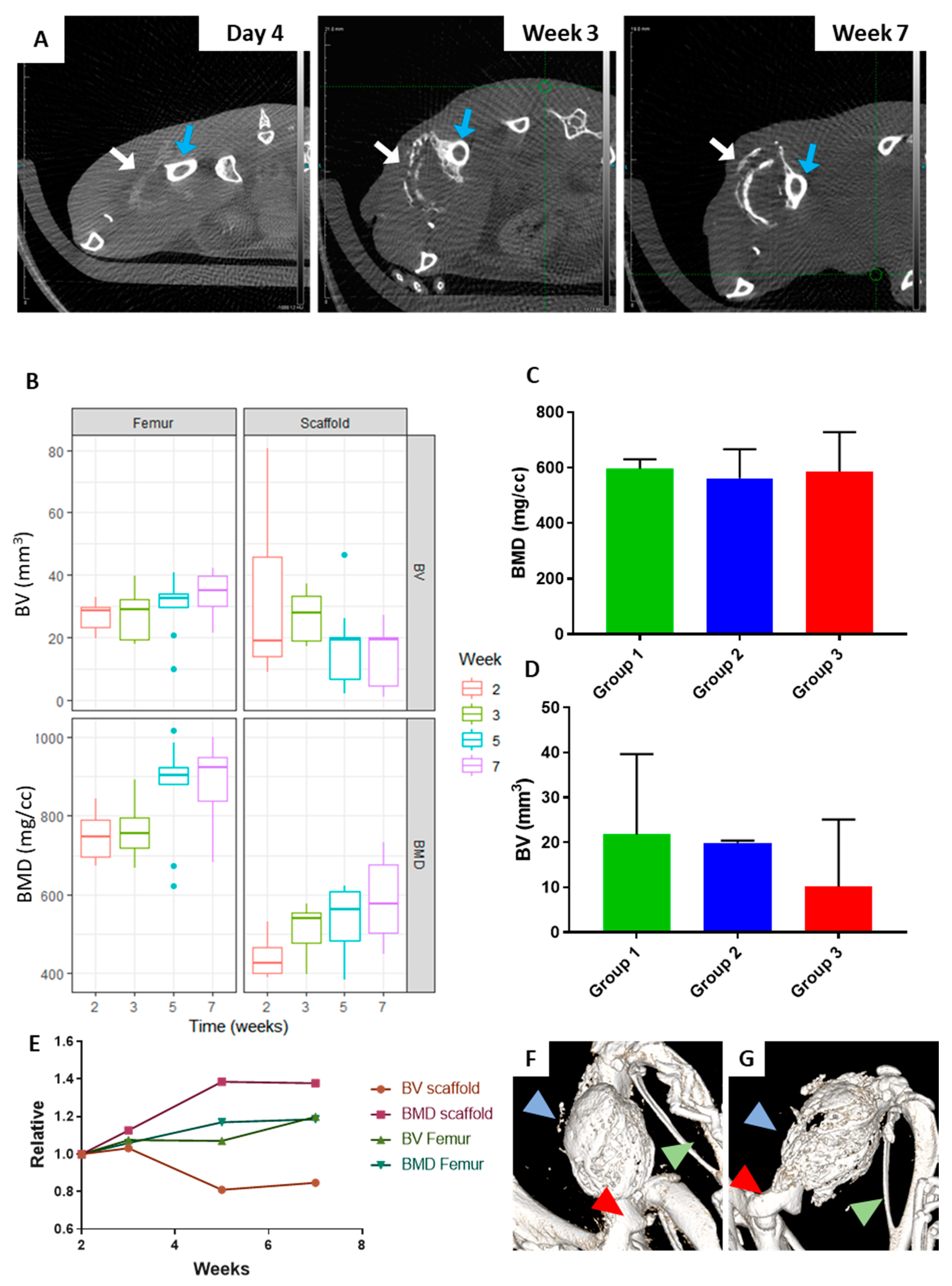
2.1. The Orthotopic Humanised Tissue Engineered Bone Construct (ohTEBC) Forms a Humanised Bone Marrow Niche In Vivo
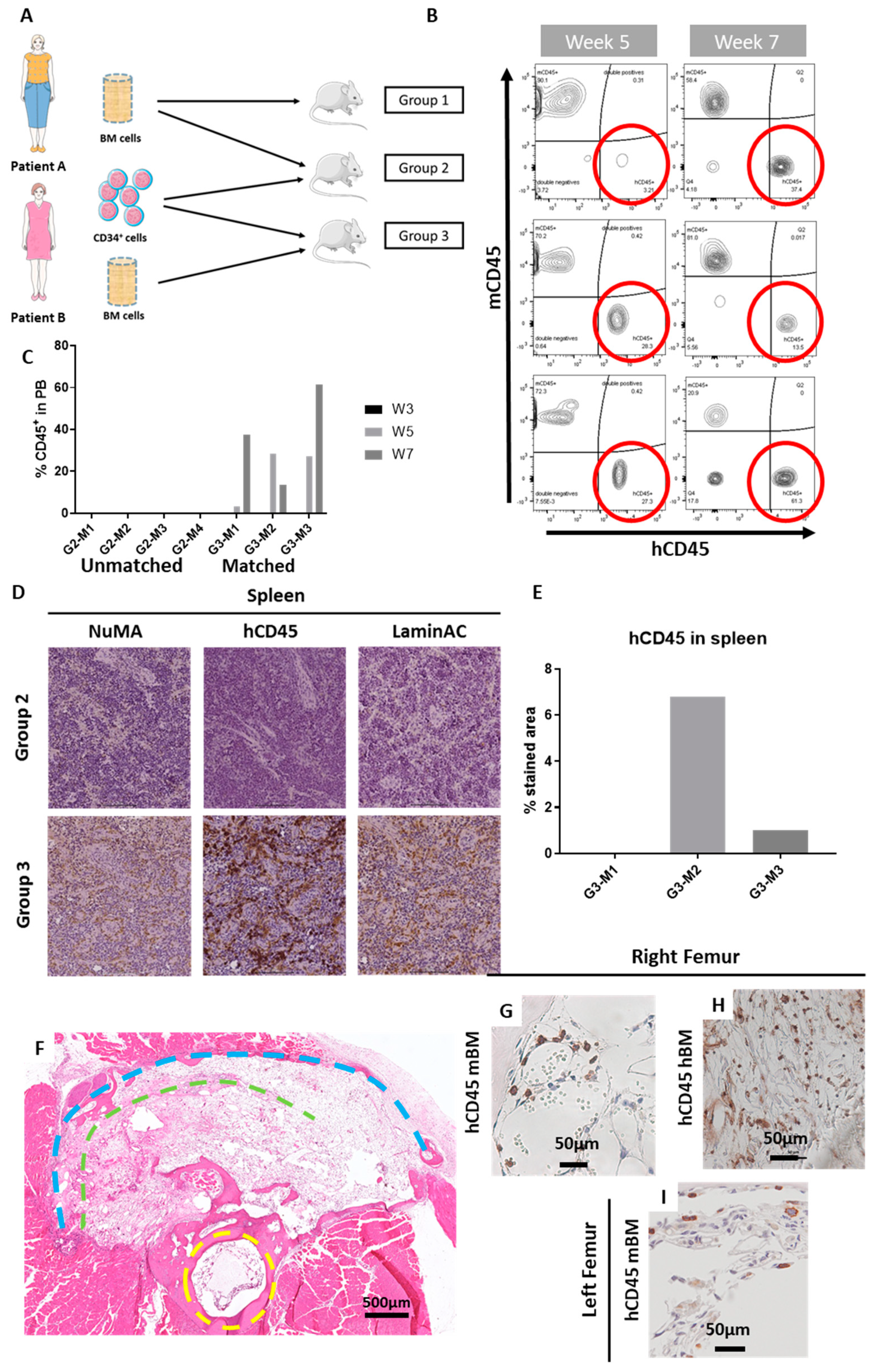
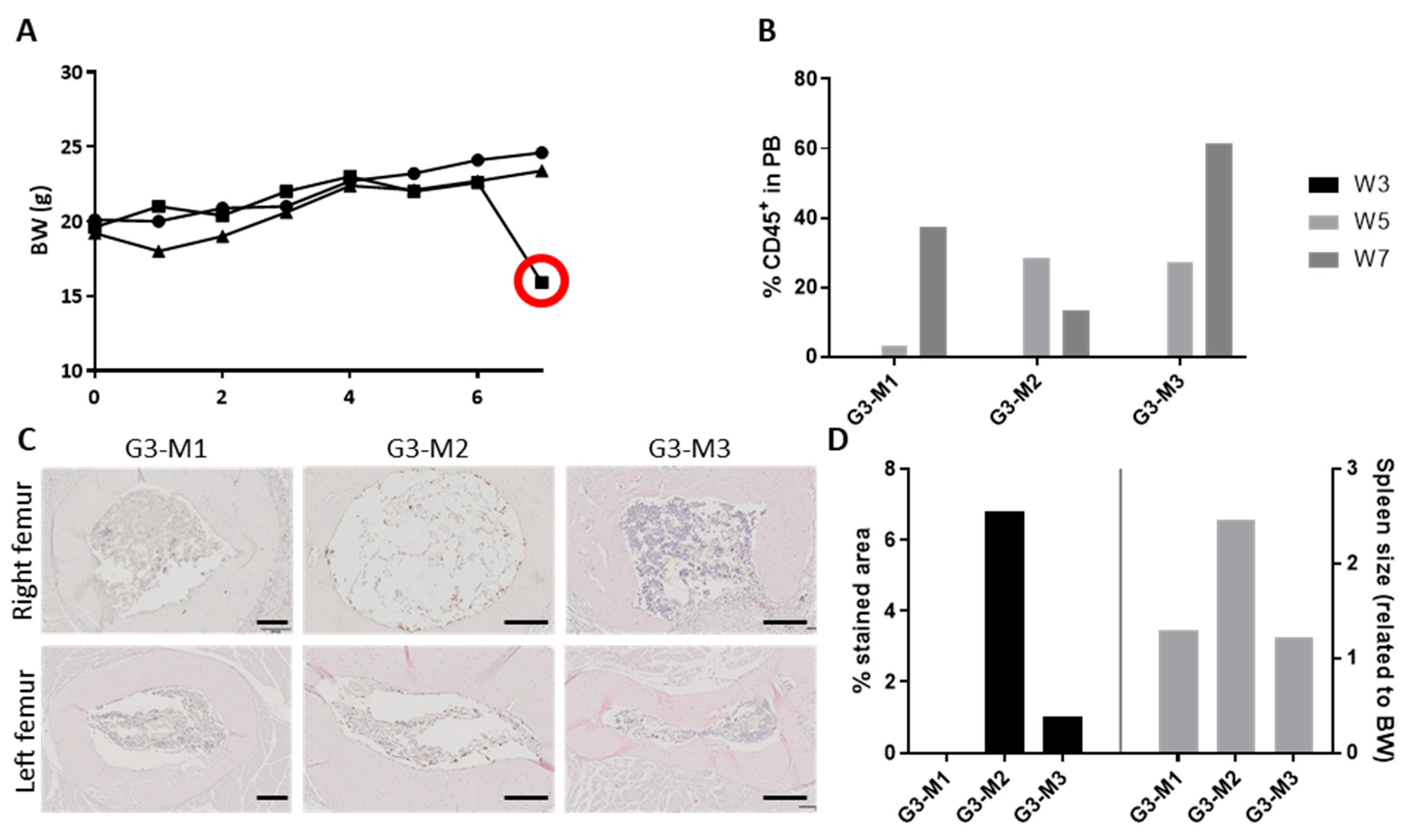
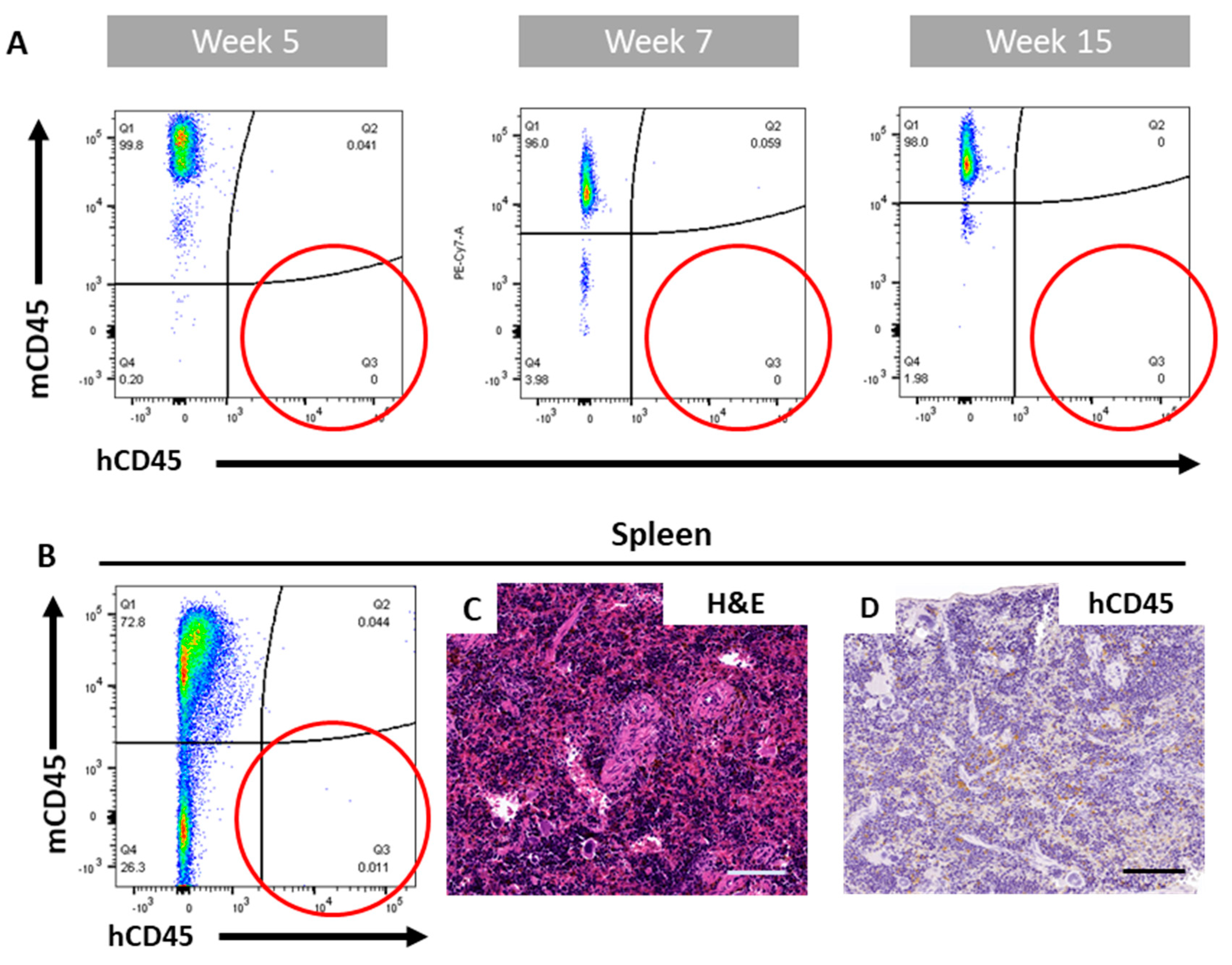
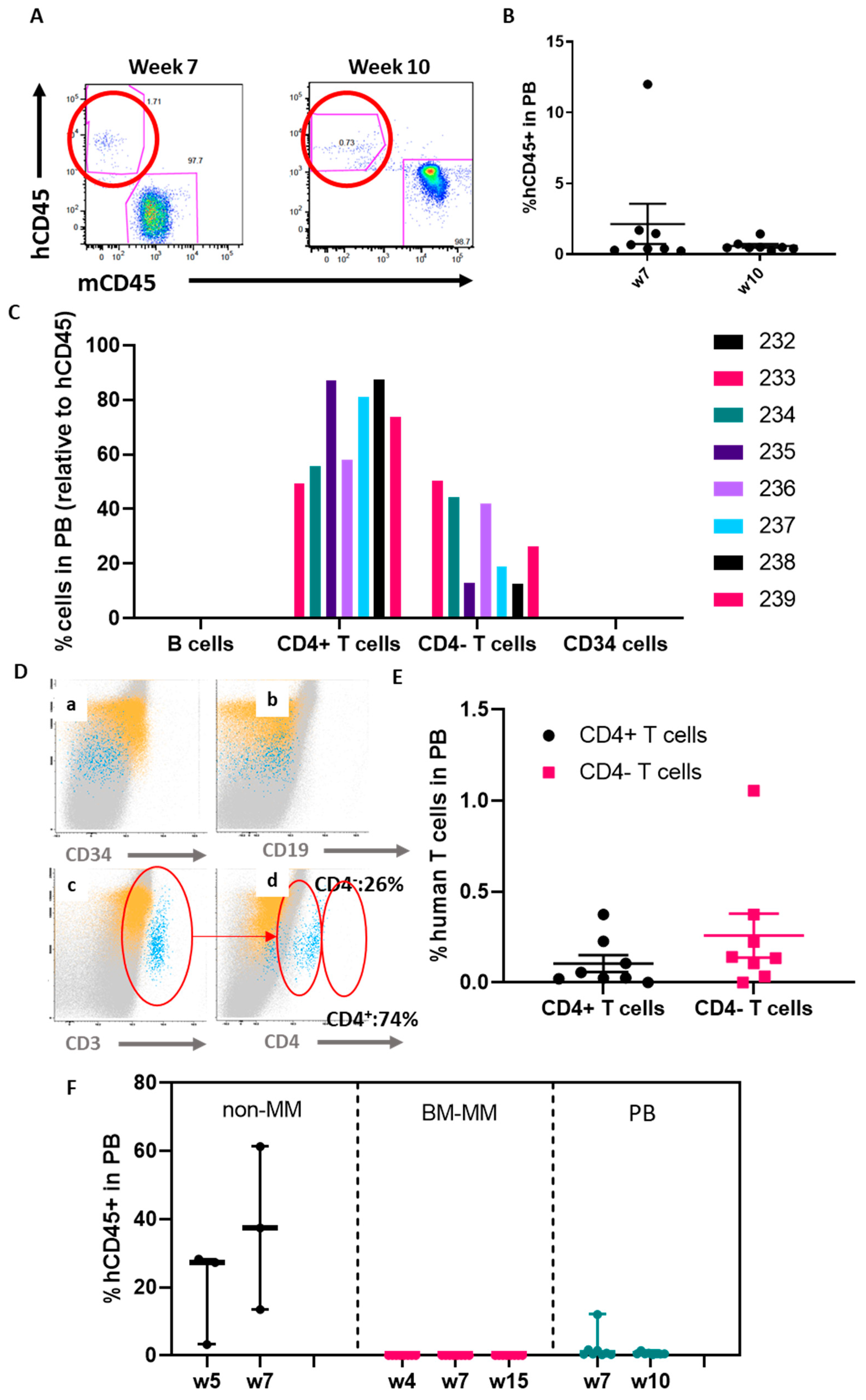
2.2. The ohTEBC Is Able to Support Human Haematopoiesis
2.3. CD34+ Cell Implantation Does Not Significantly Affect Bone Formation
2.4. Diseased Haematopoiesis in the ohTEBC
3. Discussion
4. Materials and Methods
4.1. Patient Sample Processing
4.2. ohTEBC Generation and Characterisation
4.3. In Vivo Implantation
4.4. Bone Formation Monitoring
4.5. Histology and Immunohistochemistry (IHC)
4.6. Statistical Analysis
4.7. Flow Cytometry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Perrin, S. Make mouse studies work. Nature 2014, 507, 423. [Google Scholar] [CrossRef]
- Harrison, R.K. Reason for failure in phase II d Reason for failure in phase III Phase II and phase III failures: 2013–2015. Nat. Rev. Drug Discov. 2016, 15, 817–818. [Google Scholar] [CrossRef]
- Thibaudeau, L.; Quent, V.M.; Holzapfel, B.M.; Taubenberger, A.V.; Straub, M.; Hutmacher, D.W. Mimicking breast cancer-induced bone metastasis in vivo: Current transplantation models and advanced humanized strategies. Cancer Metastasis Rev. 2014, 33, 721–735. [Google Scholar] [CrossRef]
- Holzapfel, B.M.; Thibaudeau, L.; Hesami, P.; Taubenberger, A.; Holzapfel, N.P.; Mayer-Wagner, S.; Power, C.; Clements, J.; Russell, P.; Hutmacher, D.W. Humanised xenograft models of bone metastasis revisited: Novel insights into species-specific mechanisms of cancer cell osteotropism. Cancer Metastasis Rev. 2013, 32, 129–145. [Google Scholar] [CrossRef]
- Zschaler, J.; Schlorke, D.; Arnhold, J. Differences in innate immune response between man and mouse. Crit. Rev. Immunol. 2014, 34, 433–454. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.-E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized Mouse Models of Clinical Disease. Annu. Rev. Pathol. Mech. Dis 2017, 12, 187–215. [Google Scholar] [CrossRef]
- Carrillo, M.A.; Zhen, A.; Kitchen, S.G. The use of the humanized mouse model in gene therapy and immunotherapy for HIV and cancer. Front. Immunol. 2018, 9, 746. [Google Scholar] [CrossRef]
- McCune, J.M.; Namikawa, R.; Kaneshima, H.; Shultz, L.D.; Lieberman, M.; Weissman, I.L. The SCID-hu mouse: Murine model for the analysis of human hematolymphoid differentiation and function. Science (80-.) 1988, 241, 1632–1639. [Google Scholar] [CrossRef]
- Lapidot, T.; Pflumio, F.; Doedens, M.; Murdoch, B.; Williams, D.E.; Dick, J.E. Cytokine stimulation of multilineage hematopoiesis from immature human cells engrafted in SCID Mice. Science (80-.) 1992, 255, 1137–1141. [Google Scholar] [CrossRef]
- Brehm, M.; Shultz, L.; Greiner, D. Humanized Mouse Models to Study Human Diseases. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Holzapfel, B.M.; Wagner, F.; Thibaudeau, L.; Levesque, J.P.; Hutmacher, D.W. Concise review: Humanized models of tumor immunology in the 21st century: Convergence of cancer research and tissue engineering. Stem Cells 2015, 33, 1696–1704. [Google Scholar] [CrossRef] [PubMed]
- Werner-Klein, M.; Proske, J.; Werno, C.; Schneider, K.; Hofmann, H.S.H.-S.; Rack, B.; Buchholz, S.; Ganzer, R.; Blana, A.; Seelbach-Göbel, B.; et al. Immune humanization of immunodeficient mice using diagnostic bone marrow aspirates from carcinoma patients. PLoS ONE 2014, 9, 97860. [Google Scholar] [CrossRef] [PubMed]
- King, M.A.; Covassin, L.; Brehm, M.A.; Racki, W.; Pearson, T.; Leif, J.; Laning, J.; Fodor, W.; Foreman, O.; Burzenski, L.; et al. Human peripheral blood leucocyte non-obese diabetic-severe combined immunodeficiency interleukin-2 receptor gamma chain gene mouse model of xenogeneic graft-versus-host-like disease and the role of host major histocompatibility complex. Clin. Exp. Immunol. 2009, 157, 104–118. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Calimeri, T.; Battista, E.; Conforti, F.; Neri, P.; Di Martino, M.T.; Rossi, M.; Foresta, U.; Piro, E.; Ferrara, F.; Amorosi, A.; et al. A unique three-dimensional SCID-polymeric scaffold (SCID-synth-hu) model for in vivo expansion of human primary multiple myeloma cells. Leukemia 2011, 25, 707–711. [Google Scholar] [CrossRef]
- Wagner, F.; Holzapfel, B.M.; Thibaudeau, L.; Straub, M.; Ling, M.T.; Grifka, J.; Loessner, D.; Levesque, J.P.; Hutmacher, D.W. A Validated Preclinical Animal Model for Primary Bone Tumor Research. J. Bone Jt. Surg. Am. Vol. 2016, 98, 916–925. [Google Scholar] [CrossRef]
- Landgraf, M.; Lahr, C.A.; Sanchez-Herrero, A.; Meinert, C.; Shokoohmand, A.; Pollock, P.M.; Hutmacher, D.W.; Shafiee, A.; McGovern, J.A. Humanized bone facilitates prostate cancer metastasis and recapitulates therapeutic effects of zoledronic acid in vivo. Bone Res. 2019, 7, 31. [Google Scholar] [CrossRef]
- Martine, L.C.; Holzapfel, B.M.; Mcgovern, J.A.; Quent, V.M.; Hesami, P.; Wunner, F.M.; De-juan-pardo, E.M.; Brown, T.D.; Nowlan, B.; Jing, D.; et al. Engineering a humanized bone organ in mice to study bone metastases. Nat. Protoc. 2017, 12. [Google Scholar] [CrossRef]
- Fulciniti, M.; Tassone, P.; Hideshima, T.; Vallet, S.; Nanjappa, P.; Ettenberg, S.A.; Shen, Z.; Patel, N.; Tai, Y.T.; Chauhan, D.; et al. Anti-DKK1 mAb (BHQ880) as a potential therapeutic agent for multiple myeloma. Blood 2009, 114, 371–379. [Google Scholar] [CrossRef]
- Hameed, A.; Brady, J.J.; Dowling, P.; Clynes, M.; O’Gorman, P. Bone disease in multiple myeloma: Pathophysiology and management. Cancer Growth Metastasis 2014, 7, 33–42. [Google Scholar] [CrossRef]
- Hu, J.; Handisides, D.R.; Van Valckenborgh, E.; De Raeve, H.; Menu, E.; Vande Broek, I.; Liu, Q.; Sun, J.D.; Van Camp, B.; Hart, C.P.; et al. Targeting the multiple myeloma hypoxic niche with TH-302, a hypoxia-activated prodrug. Blood 2010, 116, 1524–1527. [Google Scholar] [CrossRef]
- Landgraf, M.; McGovern, J.A.; Friedl, P.; Hutmacher, D.W. Rational Design of Mouse Models for Cancer Research. Trends Biotechnol. 2018, 36, 242–251. [Google Scholar] [CrossRef]
- Holzapfel, B.M.; Hutmacher, D.W.; Nowlan, B.; Barbier, V.; Thibaudeau, L.; Theodoropoulos, C.; Hooper, J.D.; Loessner, D.; Clements, J.A.; Russell, P.J.; et al. Tissue engineered humanized bone supports human hematopoiesis in vivo. Biomaterials 2015, 61, 103–114. [Google Scholar] [CrossRef]
- Thibaudeau, L.; Taubenberger, A.V.; Holzapfel, B.M.; Quent, V.M.; Fuehrmann, T.; Hesami, P.; Brown, T.D.; Dalton, P.D.; Power, C.A.; Hollier, B.G.; et al. A tissue-engineered humanized xenograft model of human breast cancer metastasis to bone. Dis. Model. Mech. 2014, 299–309. [Google Scholar] [CrossRef]
- Baldwin, J.G.; Wagner, F.; Martine, L.C.; Holzapfel, B.M.; Theodoropoulos, C.; Bas, O.; Savi, F.M.; Werner, C.; De-Juan-Pardo, E.M.; Hutmacher, D.W. Periosteum tissue engineering in an orthotopic in vivo platform. Biomaterials 2017, 121, 193–204. [Google Scholar] [CrossRef]
- Brown, T.D.; Slotosch, A.; Thibaudeau, L.; Taubenberger, A.; Loessner, D.; Vaquette, C.; Dalton, P.D.; Hutmacher, D.W. Design and fabrication of tubular scaffolds via direct writing in a melt electrospinning mode. Biointerphases 2012, 7, 1–16. [Google Scholar] [CrossRef]
- Reichert, J.C.; Quent, V.M.C.; Burke, L.J.; Stansfield, S.H.; Clements, J.A.; Hutmacher, D.W. Mineralized human primary osteoblast matrices as a model system to analyse interactions of prostate cancer cells with the bone microenvironment. Biomaterials 2010, 31, 7928–7936. [Google Scholar] [CrossRef]
- Li, Z.; Hardij, J.; Bagchi, D.P.; Scheller, E.L.; MacDougald, O.A. Development, regulation, metabolism and function of bone marrow adipose tissues. Bone 2018, 110, 134–140. [Google Scholar] [CrossRef]
- Liu, H.; He, J.; Koh, S.P.; Zhong, Y.; Liu, Z.; Wang, Z.; Zhang, Y.; Li, Z.; Tam, B.T.; Lin, P.; et al. Reprogrammed marrow adipocytes contribute to myeloma-induced bone disease. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Allegra, A.; Innao, V.; Gerace, D.; Allegra, A.G.; Vaddinelli, D.; Bianco, O.; Musolino, C. The adipose organ and multiple myeloma: Impact of adipokines on tumor growth and potential sites for therapeutic intervention. Eur. J. Intern. Med. 2018, 53, 12–20. [Google Scholar] [CrossRef]
- Zweegman, S.; Engelhardt, M.; Larocca, A. Elderly patients with multiple myeloma: Towards a frailty approach? Curr. Opin. Oncol. 2017, 29, 315–321. [Google Scholar] [CrossRef]
- Winters, S.; Martin, C.; Murphy, D.; Shokar, N.K. Breast Cancer Epidemiology, Prevention, and Screening. In Progress in Molecular Biology and Translational Science; Elsevier B.V.: Amsterdam, The Netherlands, 2017; Volume 151, pp. 1–32. [Google Scholar]
- Hofgaard, P.O.; Jodal, H.C.; Bommert, K.; Huard, B.; Caers, J.; Carlsen, H.; Schwarzer, R.; Schünemann, N.; Jundt, F.; Lindeberg, M.M.; et al. A Novel Mouse Model for Multiple Myeloma (MOPC315.BM) That Allows Noninvasive Spatiotemporal Detection of Osteolytic Disease. PLoS ONE 2012, 7, e51892. [Google Scholar] [CrossRef]
- Ali, N.; Flutter, B.; Rodriguez, R.S.; Sharif-Paghaleh, E.; Barber, L.D.; Lombardi, G.; Nestle, F.O. Xenogeneic Graft-versus-Host-Disease in NOD-scid IL-2Rc null Mice Display a T-Effector Memory Phenotype. PLoS ONE 2012, 7, e44219. [Google Scholar] [CrossRef]
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Gonçalves, F. Bone metastases: An overview. Oncol. Rev. 2017, 11. [Google Scholar] [CrossRef]
- Reagan, M.R.; Rosen, C.J. Navigating the bone marrow niche: Translational insights and cancer-driven dysfunction. Nat. Rev. Rheumatol. 2016, 12, 154. [Google Scholar] [CrossRef]
- Kovacic, N.; Croucher, P.I.; McDonald, M.M. Signaling between tumor cells and the host bone marrow microenvironment. Calcif. Tissue Int. 2014, 94, 125–139. [Google Scholar] [CrossRef]
- Noonan, K.; Borrello, I. The immune microenvironment of myeloma. Cancer Microenviron. 2011, 4, 313–323. [Google Scholar] [CrossRef]
- Balakumaran, A.; Robey, P.G.; Fedarko, N.; Landgren, O. Bone marrow microenvironment in myelomagenesis: Its potential role in early diagnosis. Expert Rev. Mol. Diagn. 2010, 10, 465–480. [Google Scholar] [CrossRef]
- Minoda, Y.; Virshup, I.; Rojas, I.L.; Haigh, O.; Wong, Y.; Miles, J.J.; Wells, C.A.; Radford, K.J. Human CD141+ dendritic cell and CD1c+ dendritic cell undergo concordant early genetic programming after activation in humanized mice in vivo. Front. Immunol. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Haworth, K.G.; Ironside, C.; Norgaard, Z.K.; Obenza, W.M.; Adair, J.E.; Kiem, H.P. In Vivo Murine-Matured Human CD3+Cells as a Preclinical Model for T Cell-Based Immunotherapies. Mol. Ther. Methods Clin. Dev. 2017, 6, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Abarrategi, A.; Foster, K.; Hamilton, A.; Mian, S.A.; Passaro, D.; Gribben, J.; Mufti, G.; Bonnet, D. Versatile humanized niche model enables study of normal and malignant human hematopoiesis. J. Clin. Investig. 2017, 127, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Passaro, D.; Abarrategi, A.; Foster, K.; Ariza-McNaughton, L.; Bonnet, D. Bioengineering of humanized bone marrow microenvironments in mouse and their visualization by live imaging. J. Vis. Exp. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jacamo, R.; Shi, Y.X.; Wang, R.Y.; Battula, V.L.; Konoplev, S.; Strunk, D.; Hofmann, N.A.; Reinisch, A.; Konopleva, M.; et al. Human extramedullary bone marrow in mice: A novel in vivo model of genetically controlled hematopoietic microenvironment. Blood 2012, 119, 4971–4980. [Google Scholar] [CrossRef]
- Groen, R.W.J.; Noort, W.A.; Raymakers, R.A.; Prins, H.J.; Aalders, L.; Hofhuis, F.M.; Moerer, P.; Van Velzen, J.F.; Bloem, A.C.; Van Kessel, B.; et al. Reconstructing the human hematopoietic niche in immunodeficient mice: Opportunities for studying primary multiple myeloma. Blood 2012, 120. [Google Scholar] [CrossRef]
- Ishikawa, F.; Yasukawa, M.; Lyons, B.; Yoshida, S.; Miyamoto, T.; Yoshimoto, G.; Watanabe, T.; Akashi, K.; Shultz, L.D.; Harada, M. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor γ chain null mice. Blood 2005, 106, 1565–1573. [Google Scholar] [CrossRef]
- Gao, Z.; Fackler, M.J.; Leung, W.; Lumkul, R.; Ramirez, M.; Theobald, N.; Malech, H.L.; Civin, C.I. Human CD34+ cell preparations contain over 100-fold greater NOD/SCID mouse engrafting capacity than do CD34- cell preparations. Proc. Exp. Hematol. 2001, 29, 910–921. [Google Scholar] [CrossRef]
- Noll, J.E.; Williams, S.A.; Purton, L.E.; Zannettino, A.C.W. Tug of war in the haematopoietic stem cell niche: Do myeloma plasma cells compete for the HSC niche? Blood Cancer J. 2012, 2. [Google Scholar] [CrossRef]
- Bruns, I.; Cadeddu, R.P.; Brueckmann, I.; Fröbel, J.; Geyh, S.; Büst, S.; Fischer, J.C.; Roels, F.; Wilk, C.M.; Schildberg, F.A.; et al. Multiple myeloma-related deregulation of bone marrow-derived CD34 + hematopoietic stem and progenitor cells. Blood 2012, 120, 2620–2630. [Google Scholar] [CrossRef]
- Calcinotto, A.; Ponzoni, M.; Ria, R.; Grioni, M.; Cattaneo, E.; Villa, I.; Teresa, M.; Bertilaccio, S.; Chesi, M.; Rubinacci, A.; et al. Modifications of the mouse bone marrow microenvironment favor angiogenesis and correlate with disease progression from asymptomatic to symptomatic multiple myeloma. Oncoimmunology 2015, 4, e1008850. [Google Scholar] [CrossRef]
- Sze, D.M.Y.; Giesajtis, G.; Brown, R.D.; Raitakari, M.; Gibson, J.; Ho, J.; Baxter, A.G.; De Groth, B.F.S.; Basten, A.; Joshua, D.E. Clonal cytotoxic T cells are expanded in myeloma and reside in the CD8+CD57+CD28- compartment. Blood 2001, 98, 2817–2827. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Ferrari, S.; Graham, R.; Russell, G. Denosumab and bisphosphonates: Different mechanisms of action and effects. Bone 2011, 48, 677–692. [Google Scholar] [CrossRef]
- Dimopoulos, K.; Gimsing, P.; Grønbæk, K. The role of epigenetics in the biology of multiple myeloma. Blood Cancer J. 2014, 4, e207. [Google Scholar] [CrossRef] [PubMed]
- Canella, A.; Nieves, H.C.; Sborov, D.W.; Cascione, L.; Radomska, H.S.; Smith, E.; Stiff, A.; Consiglio, J.; Caserta, E.; Rizzotto, L.; et al. HDAC inhibitor AR-42 decreases CD44 expression and sensitizes myeloma cells to lenalidomide. Oncotarget 2015, 6, 31134–31150. [Google Scholar] [CrossRef] [PubMed]
- Sondergeld, P.; van de Donk, N.W.C.J.; Richardson, P.G.; Plesner, T. Monoclonal antibodies in myeloma. Clin. Adv. Hematol. Oncol. 2015, 13, 599–609. [Google Scholar] [PubMed]
- Berz, D.; McCormack, E.M.; Winer, E.S.; Colvin, G.A.; Quesenberry, P.J. Cryopreservation of hematopoietic stem cells. Am. J. Hematol. 2007, 82, 463–472. [Google Scholar] [CrossRef]
- Vaquette, C.; Ivanovski, S.; Hamlet, S.M.; Hutmacher, D.W. Effect of culture conditions and calcium phosphate coating on ectopic bone formation. Biomaterials 2013, 34, 5538–5551. [Google Scholar] [CrossRef]
- Muerza-Cascante, M.L.; Shokoohmand, A.; Khosrotehrani, K.; Haylock, D.; Dalton, P.D.; Hutmacher, D.W.; Loessner, D. Endosteal-like extracellular matrix expression on melt electrospun written scaffolds. Acta Biomater. 2016, 52, 145–158. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchez-Herrero, A.; Calvo, I.A.; Flandes-Iparraguirre, M.; Landgraf, M.; Lahr, C.A.; Shafiee, A.; Granero-Molto, F.; Saez, B.; Mazo, M.M.; Paiva, B.; et al. Engineering a Humanised Niche to Support Human Haematopoiesis in Mice: Novel Opportunities in Modelling Cancer. Cancers 2020, 12, 2205. https://doi.org/10.3390/cancers12082205
Sanchez-Herrero A, Calvo IA, Flandes-Iparraguirre M, Landgraf M, Lahr CA, Shafiee A, Granero-Molto F, Saez B, Mazo MM, Paiva B, et al. Engineering a Humanised Niche to Support Human Haematopoiesis in Mice: Novel Opportunities in Modelling Cancer. Cancers. 2020; 12(8):2205. https://doi.org/10.3390/cancers12082205
Chicago/Turabian StyleSanchez-Herrero, Alvaro, Isabel A. Calvo, Maria Flandes-Iparraguirre, Marietta Landgraf, Christoph A. Lahr, Abbas Shafiee, Froilán Granero-Molto, Borja Saez, Manuel M. Mazo, Bruno Paiva, and et al. 2020. "Engineering a Humanised Niche to Support Human Haematopoiesis in Mice: Novel Opportunities in Modelling Cancer" Cancers 12, no. 8: 2205. https://doi.org/10.3390/cancers12082205
APA StyleSanchez-Herrero, A., Calvo, I. A., Flandes-Iparraguirre, M., Landgraf, M., Lahr, C. A., Shafiee, A., Granero-Molto, F., Saez, B., Mazo, M. M., Paiva, B., de Juan Pardo, E., Nicol, A., Prosper, F., Bray, L. J., & McGovern, J. A. (2020). Engineering a Humanised Niche to Support Human Haematopoiesis in Mice: Novel Opportunities in Modelling Cancer. Cancers, 12(8), 2205. https://doi.org/10.3390/cancers12082205