Targeted Disruption of Bone Marrow Stromal Cell-Derived Gremlin1 Limits Multiple Myeloma Disease Progression In Vivo

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. GREM1 Expression Is Upregulated in MM Bone Marrow Stroma
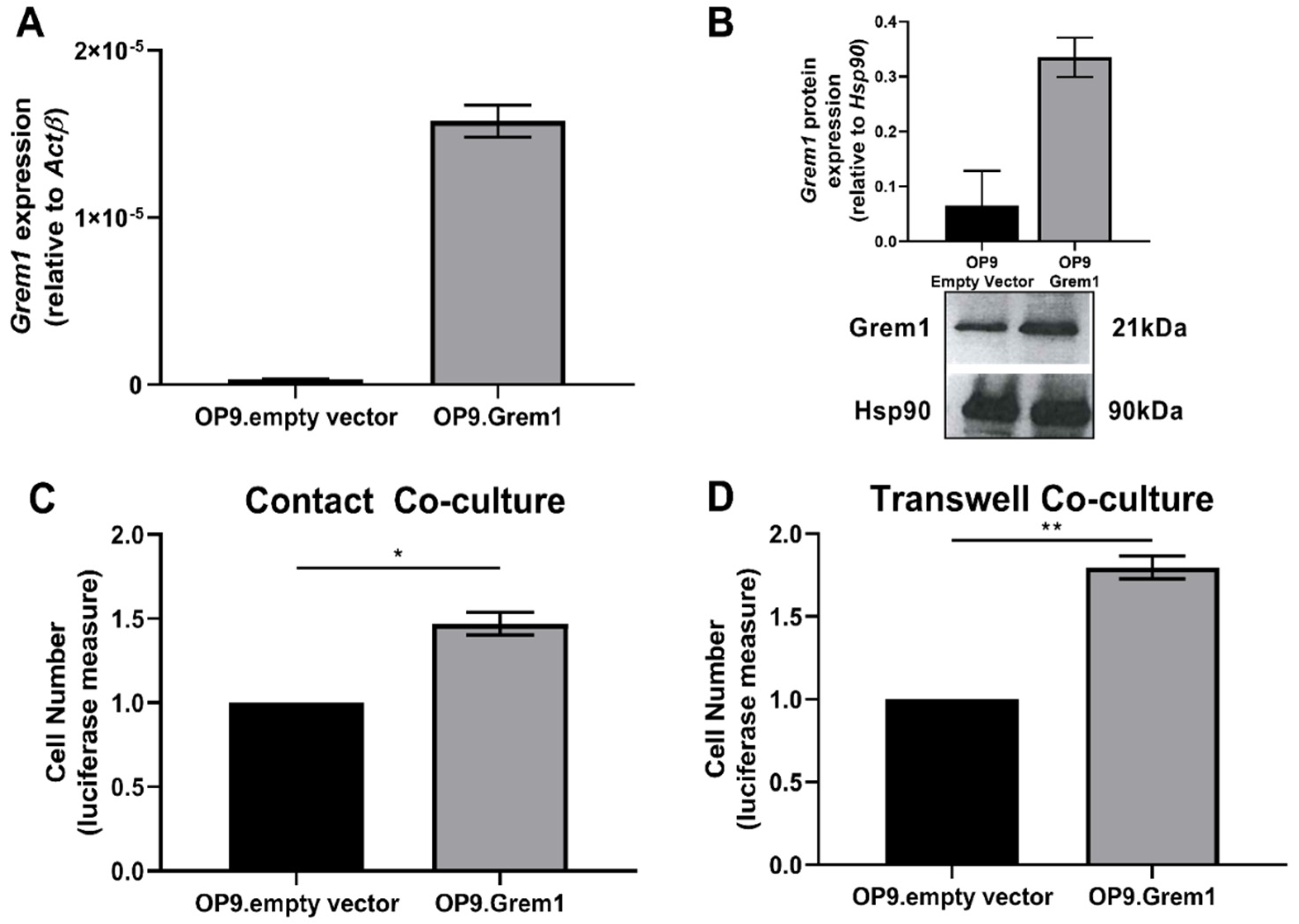
2.2. Increased Grem1 Expression Promotes MM PC Proliferation
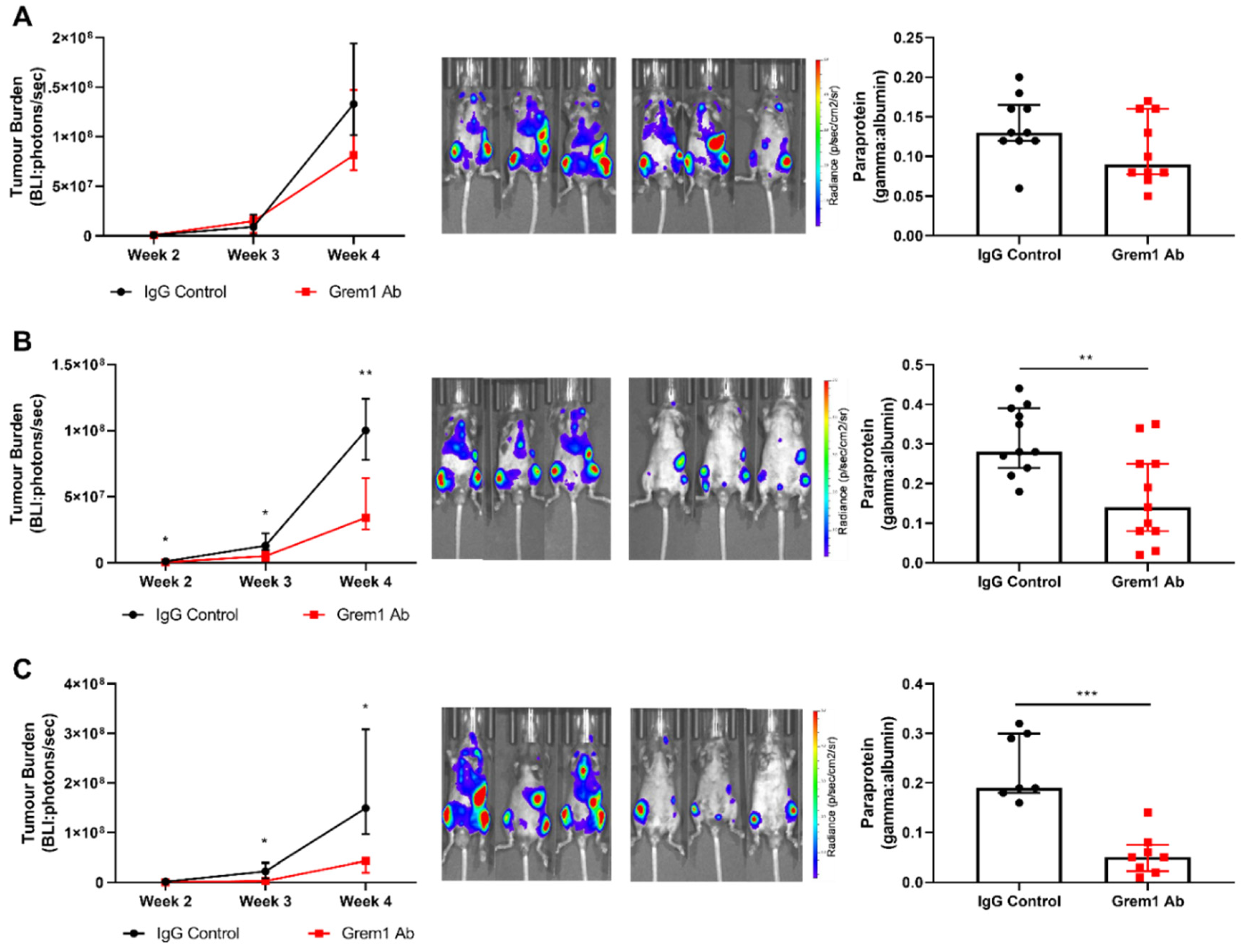
2.3. Targeting Grem1 Reduces MM Tumor Burden In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Human Bone Marrow Stromal Cell Purification and RNA Isolation
4.3. Murine Compact Bone Purification and RNA Isolation
4.4. Quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
4.5. Generation of a Murine Grem1-Overexpressing Stromal Cell Line
4.6. Western Blot Analysis
4.7. Co-Culture of MM PC and BM Stroma
4.8. Luciferase Proliferation Assay
4.9. HEKID1 Reporter Assay
4.10. Confirmation of Anti-Grem1 Antibody Activity
4.11. Targeting Grem1 in an Immunocompetent Murine Model of Systemic MM
4.12. Cell Lineage Flow Cytometric Analysis
4.13. Statistics
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kyle, R.A.; Rajkumar, S.V. Multiple myeloma. N. Engl. J. Med. 2004, 351, 1860–1873. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V. Updated Diagnostic Criteria and Staging System for Multiple Myeloma. Am. Soc. Clin. Oncol. Educ. Book. Am. Soc. Clin. Oncol. Annu. Meet. 2016, 35, e418–e423. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, B.; Binder, M.; Dispenzieri, A.; Kapoor, P.; Buadi, F.; Gertz, M.A.; Lacy, M.; Dingli, D.; Hwa, L.; Leung, N.; et al. Continued improvement in survival in multiple myeloma(MM) including high-risk patients. J. Clin. Oncol. 2019, 37 (Suppl. 15), 8039. [Google Scholar] [CrossRef]
- Cetani, G.; Boccadoro, M.; Oliva, S. A look at treatment strategies for relapsed multiple myeloma. Expert Rev. Anticancer Ther. 2018, 18, 735–750. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef]
- Dutta, A.K.; Fink, J.L.; Grady, J.P.; Morgan, G.J.; Mullighan, C.G.; To, L.B.; Hewett, D.R.; Zannettino, A.C.W. Subclonal evolution in disease progression from MGUS/SMM to multiple myeloma is characterised by clonal stability. Leukemia 2018, 33, 457–468. [Google Scholar] [CrossRef]
- Walker, B.A.; Wardell, C.P.; Melchor, L.; Brioli, A.; Johnson, D.C.; Kaiser, M.F.; Mirabella, F.; Lopez-Corral, L.; Humphray, S.; Murray, L.; et al. Intraclonal heterogeneity is a critical early event in the development of myeloma and precedes the development of clinical symptoms. Leukemia 2014, 28, 384–390. [Google Scholar] [CrossRef]
- Bolli, N.; Maura, F.; Minvielle, S.; Gloznik, D.; Szalat, R.; Fullam, A.; Martincorena, I.; Dawson, K.J.; Samur, M.K.; Zamora, J.; et al. Genomic patterns of progression in smoldering multiple myeloma. Nat. Commun. 2018, 9, 3363. [Google Scholar] [CrossRef]
- Vandyke, K.; Zeissig, M.N.; Hewett, D.R.; Martin, S.K.; Mrozik, K.M.; Cheong, C.M.; Diamond, P.; To, L.B.; Gronthos, S.; Peet, D.J.; et al. HIF-2alpha Promotes Dissemination of Plasma Cells in Multiple Myeloma by Regulating CXCL12/CXCR4 and CCR1. Cancer Res. 2017, 77, 5452–5463. [Google Scholar] [CrossRef]
- Vande Broek, I.; Vanderkerken, K.; Van Camp, B.; Van Riet, I. Extravasation and homing mechanisms in multiple myeloma. Clin. Exp. Metastasis 2008, 25, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Attar-Schneider, O.; Zismanov, V.; Dabbah, M.; Tartakover-Matalon, S.; Drucker, L.; Lishner, M. Multiple myeloma and bone marrow mesenchymal stem cells’ crosstalk: Effect on translation initiation. Mol. Carcinog. 2016, 55, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Noll, J.E.; Williams, S.A.; Purton, L.E.; Zannettino, A.C. Tug of war in the haematopoietic stem cell niche: Do myeloma plasma cells compete for the HSC niche? Blood Cancer J. 2012, 2, e91. [Google Scholar] [CrossRef] [PubMed]
- Gronthos, S. Molecular and cellular characterisation of highly purified stromal stem cells derived from human bone marrow. J. Cell Sci. 2003, 116, 1827–1835. [Google Scholar] [CrossRef] [PubMed]
- Bexell, D.; Gunnarsson, S.; Tormin, A.; Darabi, A.; Gisselsson, D.; Roybon, L.; Scheding, S.; Bengzon, J. Bone marrow multipotent mesenchymal stroma cells act as pericyte-like migratory vehicles in experimental gliomas. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 183–190. [Google Scholar] [CrossRef]
- Sacchetti, B.; Funari, A.; Michienzi, S.; Di Cesare, S.; Piersanti, S.; Saggio, I.; Tagliafico, E.; Ferrari, S.; Robey, P.G.; Riminucci, M.; et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 2007, 131, 324–336. [Google Scholar] [CrossRef]
- Worthley, D.L.; Churchill, M.; Compton, J.T.; Tailor, Y.; Rao, M.; Si, Y.; Levin, D.; Schwartz, M.G.; Uygur, A.; Hayakawa, Y.; et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 2015, 160, 269–284. [Google Scholar] [CrossRef]
- Lawson, M.A.; McDonald, M.M.; Kovacic, N.; Hua Khoo, W.; Terry, R.L.; Down, J.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 2015, 6, 8983. [Google Scholar] [CrossRef]
- Hsu, D.R.; Economides, A.N.; Wang, X.; Eimon, P.M.; Harland, R.M. The Xenopus dorsalizing factor Gremlin identifies a novel family of secreted proteins that antagonize BMP activities. Mol. Cell 1998, 1, 673–683. [Google Scholar] [CrossRef]
- Mitola, S.; Ravelli, C.; Moroni, E.; Salvi, V.; Leali, D.; Ballmer-Hofer, K.; Zammataro, L.; Presta, M. Gremlin is a novel agonist of the major proangiogenic receptor VEGFR2. Blood 2010, 116, 3677–3680. [Google Scholar] [CrossRef]
- Karagiannis, G.S.; Musrap, N.; Saraon, P.; Treacy, A.; Schaeffer, D.F.; Kirsch, R.; Riddell, R.H.; Diamandis, E.P. Bone morphogenetic protein antagonist gremlin-1 regulates colon cancer progression. Biol. Chem. 2015, 396, 163–183. [Google Scholar] [CrossRef] [PubMed]
- Tamminen, J.A.; Parviainen, V.; Ronty, M.; Wohl, A.P.; Murray, L.; Joenvaara, S.; Varjosalo, M.; Lepparanta, O.; Ritvos, O.; Sengle, G.; et al. Gremlin-1 associates with fibrillin microfibrils in vivo and regulates mesothelioma cell survival through transcription factor slug. Oncogenesis 2013, 2, e66. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Cheng, L.; Yan, B.; Zhou, C.; Qian, W.; Xiao, Y.; Qin, T.; Cao, J.; Han, L.; Ma, Q.; et al. Overexpression of Gremlin 1 by sonic hedgehog signaling promotes pancreatic cancer progression. Int. J. Oncol. 2018, 53, 2445–2457. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Smid, M.; Iaria, J.; Salvatori, D.C.; van Dam, H.; Zhu, H.J.; Martens, J.W.; Ten Dijke, P. Cancer-associated fibroblast-derived Gremlin 1 promotes breast cancer progression. Breast Cancer Res. 2019, 21, 1–19. [Google Scholar] [CrossRef]
- Namkoong, H.; Shin, S.M.; Kim, H.K.; Ha, S.A.; Cho, G.W.; Hur, S.Y.; Kim, T.E.; Kim, J.W. The bone morphogenetic protein antagonist gremlin 1 is overexpressed in human cancers and interacts with YWHAH protein. BMC Cancer 2006, 6, 74. [Google Scholar] [CrossRef]
- Sato, M.; Kawana, K.; Fujimoto, A.; Yoshida, M.; Nakamura, H.; Nishida, H.; Inoue, T.; Taguchi, A.; Takahashi, J.; Adachi, K.; et al. Clinical significance of Gremlin 1 in cervical cancer and its effects on cancer stem cell maintenance. Oncol. Rep. 2016, 35, 391–397. [Google Scholar] [CrossRef]
- van Vlodrop, I.J.; Baldewijns, M.M.; Smits, K.M.; Schouten, L.J.; van Neste, L.; van Criekinge, W.; van Poppel, H.; Lerut, E.; Schuebel, K.E.; Ahuja, N.; et al. Prognostic significance of Gremlin1(GREM1) promoter CpG island hypermethylation in clear cell renal cell carcinoma. Am. J. Pathol. 2010, 176, 575–584. [Google Scholar] [CrossRef][Green Version]
- Sneddon, J.B.; Zhen, H.H.; Montgomery, K.; van de Rijn, M.; Tward, A.D.; West, R.; Gladstone, H.; Chang, H.Y.; Morganroth, G.S.; Oro, A.E. Bone morphogenetic protein antagonist gremlin 1 is widely expressed by cancer-associated stromal cells and can promote tumor cell proliferation. Proc. Natl. Acad. Sci. USA 2006, 103, 14842–14847. [Google Scholar] [CrossRef]
- Chen, M.H.; Yeh, Y.C.; Shyr, Y.M.; Jan, Y.H.; Chao, Y.; Li, C.P.; Wang, S.E.; Tzeng, C.H.; Chang, P.M.; Liu, C.Y.; et al. Expression of gremlin 1 correlates with increased angiogenesis and progression-free survival in patients with pancreatic neuroendocrine tumors. J. Gastroenterol. 2013, 48, 101–108. [Google Scholar] [CrossRef]
- Lee, N.; Lee, H.; Moon, S.Y.; Sohn, J.Y.; Hwang, S.M.; Yoon, O.J.; Youn, H.S.; Eom, H.S.; Kong, S.Y. Adverse prognostic impact of bone marrow microvessel density in multiple myeloma. Ann. Lab. Med. 2015, 35, 563–569. [Google Scholar] [CrossRef]
- Chesi, M.; Robbiani, D.F.; Sebag, M.; Chng, W.J.; Affer, M.; Tiedemann, R.; Valdez, R.; Palmer, S.E.; Haas, S.S.; Stewart, A.K.; et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell 2008, 13, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Noll, J.E.; Hewett, D.R.; Williams, S.A.; Vandyke, K.; Kok, C.; To, L.B.; Zannettino, A.C. SAMSN1 is a tumor suppressor gene in multiple myeloma. Neoplasia 2014, 16, 572–585. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, S.; Ciechomska, M.; Cant, R.; van Laar, J.M. Interleukin-6(IL-6) trans signaling drives a STAT3-dependent pathway that leads to hyperactive transforming growth factor-beta(TGF-beta) signaling promoting SMAD3 activation and fibrosis via Gremlin protein. J. Biol. Chem. 2014, 289, 9952–9960. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, H.; Barut, B.A.; Mohrbacher, A.F.; Chauhan, D.; Anderson, K.C. Adhesion of human myeloma-derived cell lines to bone marrow stromal cells stimulates interleukin-6 secretion. Blood 1993, 82, 3712–3720. [Google Scholar] [CrossRef]
- Martinovic, S.; Mazic, S.; Kisic, V.; Basic, N.; Jakic-Razumovic, J.; Borovecki, F.; Batinic, D.; Simic, P.; Grgurevic, L.; Labar, B.; et al. Expression of bone morphogenetic proteins in stromal cells from human bone marrow long-term culture. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2004, 52, 1159–1167. [Google Scholar] [CrossRef]
- Laurila, R.; Parkkila, S.; Isola, J.; Kallioniemi, A.; Alarmo, E.L. The expression patterns of gremlin 1 and noggin in normal adult and tumor tissues. Int. J. Clin. Exp. Pathol. 2013, 6, 1400–1408. [Google Scholar]
- Kumar, S.K.; Dimopoulos, M.A.; Kastritis, E.; Terpos, E.; Nahi, H.; Goldschmidt, H.; Hillengass, J.; Leleu, X.; Beksac, M.; Alsina, M.; et al. Natural history of relapsed myeloma, refractory to immunomodulatory drugs and proteasome inhibitors: A multicenter IMWG study. Leukemia 2017, 31, 2443–2448. [Google Scholar] [CrossRef]
- Kim, H.S.; Shin, M.S.; Cheon, M.S.; Kim, J.W.; Lee, C.; Kim, W.H.; Kim, Y.S.; Jang, B.G. GREM1 is expressed in the cancer-associated myofibroblasts of basal cell carcinomas. PLoS ONE 2017, 12, e0174565. [Google Scholar] [CrossRef]
- Davis, H.; Irshad, S.; Bansal, M.; Rafferty, H.; Boitsova, T.; Bardella, C.; Jaeger, E.; Lewis, A.; Freeman-Mills, L.; Giner, F.C.; et al. Aberrant epithelial GREM1 expression initiates colonic tumorigenesis from cells outside the stem cell niche. Nat. Med. 2015, 21, 62–70. [Google Scholar] [CrossRef]
- Mitsiades, C.S.; Mitsiades, N.S.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. The role of the bone microenvironment in the pathophysiology and therapeutic management of multiple myeloma: Interplay of growth factors, their receptors and stromal interactions. Eur. J. Cancer 2006, 42, 1564–1573. [Google Scholar] [CrossRef]
- Harmer, D.; Falank, C.; Reagan, M.R. Interleukin-6 Interweaves the Bone Marrow Microenvironment, Bone Loss, and Multiple Myeloma. Front. Endocrinol. 2018, 9, 788. [Google Scholar] [CrossRef] [PubMed]
- Berenstein, R.; Nogai, A.; Waechter, M.; Blau, O.; Kuehnel, A.; Schmidt-Hieber, M.; Kunitz, A.; Pezzutto, A.; Dorken, B.; Blau, I.W. Multiple myeloma cells modify VEGF/IL-6 levels and osteogenic potential of bone marrow stromal cells via Notch/miR-223. Mol. Carcinog. 2016, 55, 1927–1939. [Google Scholar] [CrossRef] [PubMed]
- Holien, T.; Vatsveen, T.K.; Hella, H.; Rampa, C.; Brede, G.; Groseth, L.A.; Rekvig, M.; Borset, M.; Standal, T.; Waage, A.; et al. Bone morphogenetic proteins induce apoptosis in multiple myeloma cells by Smad-dependent repression of MYC. Leukemia 2012, 26, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Yoon, S.; Lee, S.; Ha, S.A.; Kim, H.K.; Kim, J.W.; Chung, J. Gremlin-1 induces BMP-independent tumor cell proliferation, migration, and invasion. PLoS ONE 2012, 7, e35100. [Google Scholar] [CrossRef] [PubMed]
- Ciuclan, L.; Sheppard, K.; Dong, L.; Sutton, D.; Duggan, N.; Hussey, M.; Simmons, J.; Morrell, N.W.; Jarai, G.; Edwards, M.; et al. Treatment with anti-gremlin 1 antibody ameliorates chronic hypoxia/SU5416-induced pulmonary arterial hypertension in mice. Am. J. Pathol. 2013, 183, 1461–1473. [Google Scholar] [CrossRef]
- Rowan, S.C.; Jahns, H.; Mthunzi, L.; Piouceau, L.; Cornwell, J.; Doody, R.; Frohlich, S.; Callanan, J.J.; McLoughlin, P. Gremlin 1 depletion in vivo causes severe enteropathy and bone marrow failure. J. Pathol. 2020, 251, 117–122. [Google Scholar] [CrossRef]
- Touzeau, C.; Moreau, P.; Dumontet, C. Monoclonal antibody therapy in multiple myeloma. Leukemia 2017, 31, 1039–1047. [Google Scholar] [CrossRef]
- Durer, C.; Durer, S.; Lee, S.; Chakraborty, R.; Malik, M.N.; Rafae, A.; Zar, M.A.; Kamal, A.; Rosko, N.; Samaras, C.; et al. Treatment of relapsed multiple myeloma: Evidence-based recommendations. Blood Rev. 2019, 39, 100616. [Google Scholar] [CrossRef]
- Laubach, J.; Garderet, L.; Mahindra, A.; Gahrton, G.; Caers, J.; Sezer, O.; Voorhees, P.; Leleu, X.; Johnsen, H.E.; Streetly, M.; et al. Management of relapsed multiple myeloma: Recommendations of the International Myeloma Working Group. Leukemia 2016, 30, 1005–1017. [Google Scholar] [CrossRef]
- Gronthos, S.; Simmons, P.J. The growth factor requirements of STRO-1-positive human bone marrow stromal precursors under serum-deprived conditions in vitro. Blood 1995, 85, 929–940. [Google Scholar] [CrossRef]
- Opperman, K.S.; Vandyke, K.; Clark, K.C.; Coulter, E.A.; Hewett, D.R.; Mrozik, K.M.; Schwarz, N.; Evdokiou, A.; Croucher, P.I.; Psaltis, P.J.; et al. Clodronate-Liposome Mediated Macrophage Depletion Abrogates Multiple Myeloma Tumor Establishment In Vivo. Neoplasia 2019, 21, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Noll, J.E.; Williams, S.A.; Tong, C.M.; Wang, H.; Quach, J.M.; Purton, L.E.; Pilkington, K.; To, L.B.; Evdokiou, A.; Gronthos, S.; et al. Myeloma plasma cells alter the bone marrow microenvironment by stimulating the proliferation of mesenchymal stromal cells. Haematologica 2014, 99, 163–171. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clark, K.C.; Hewett, D.R.; Panagopoulos, V.; Plakhova, N.; Opperman, K.S.; Bradey, A.L.; Mrozik, K.M.; Vandyke, K.; Mukherjee, S.; Davies, G.C.G.; et al. Targeted Disruption of Bone Marrow Stromal Cell-Derived Gremlin1 Limits Multiple Myeloma Disease Progression In Vivo. Cancers 2020, 12, 2149. https://doi.org/10.3390/cancers12082149
Clark KC, Hewett DR, Panagopoulos V, Plakhova N, Opperman KS, Bradey AL, Mrozik KM, Vandyke K, Mukherjee S, Davies GCG, et al. Targeted Disruption of Bone Marrow Stromal Cell-Derived Gremlin1 Limits Multiple Myeloma Disease Progression In Vivo. Cancers. 2020; 12(8):2149. https://doi.org/10.3390/cancers12082149
Chicago/Turabian StyleClark, Kimberley C., Duncan R. Hewett, Vasilios Panagopoulos, Natalya Plakhova, Khatora S. Opperman, Alanah L. Bradey, Krzysztof M. Mrozik, Kate Vandyke, Siddhartha Mukherjee, Gareth C.G. Davies, and et al. 2020. "Targeted Disruption of Bone Marrow Stromal Cell-Derived Gremlin1 Limits Multiple Myeloma Disease Progression In Vivo" Cancers 12, no. 8: 2149. https://doi.org/10.3390/cancers12082149
APA StyleClark, K. C., Hewett, D. R., Panagopoulos, V., Plakhova, N., Opperman, K. S., Bradey, A. L., Mrozik, K. M., Vandyke, K., Mukherjee, S., Davies, G. C. G., Worthley, D. L., & Zannettino, A. C. W. (2020). Targeted Disruption of Bone Marrow Stromal Cell-Derived Gremlin1 Limits Multiple Myeloma Disease Progression In Vivo. Cancers, 12(8), 2149. https://doi.org/10.3390/cancers12082149