MR1-Restricted T Cells in Cancer Immunotherapy




Abstract
1. Introduction
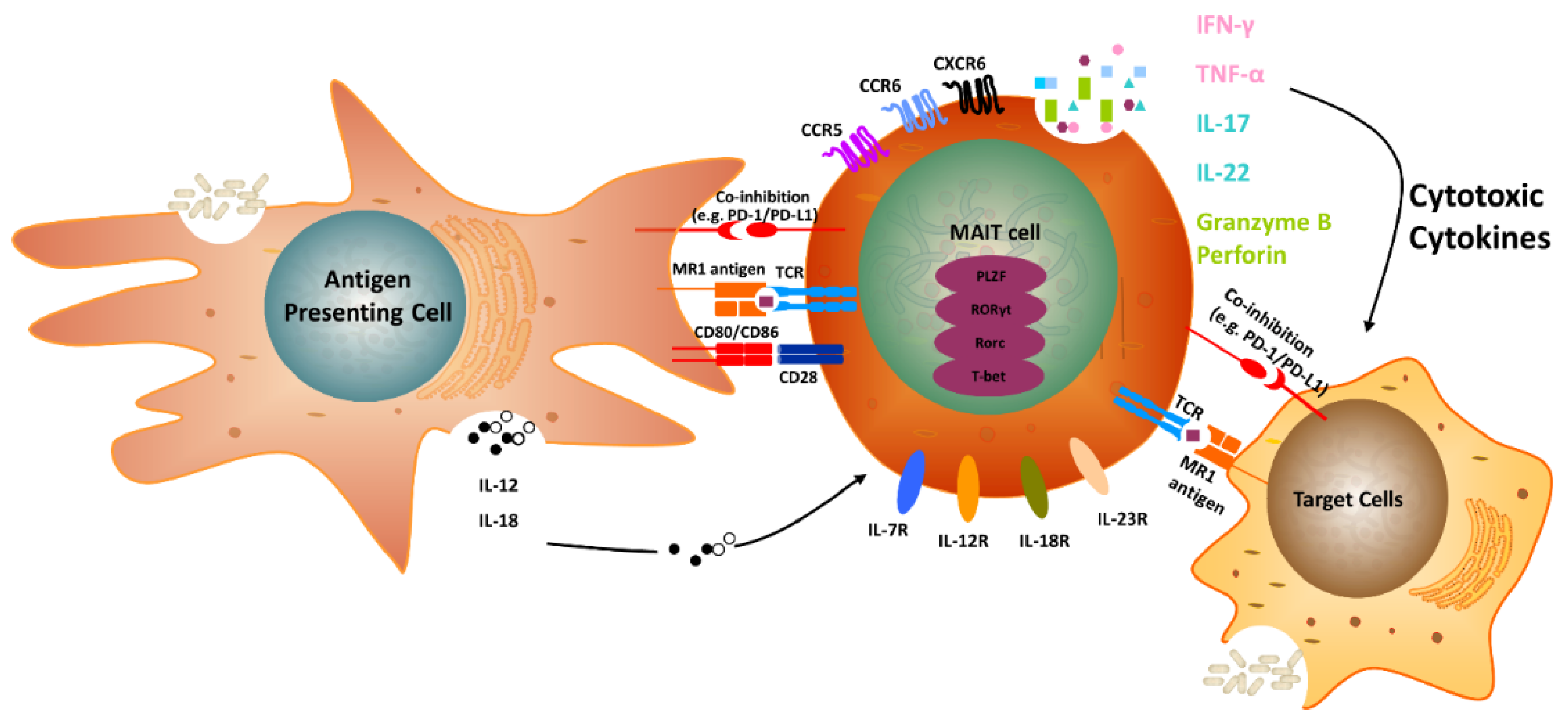
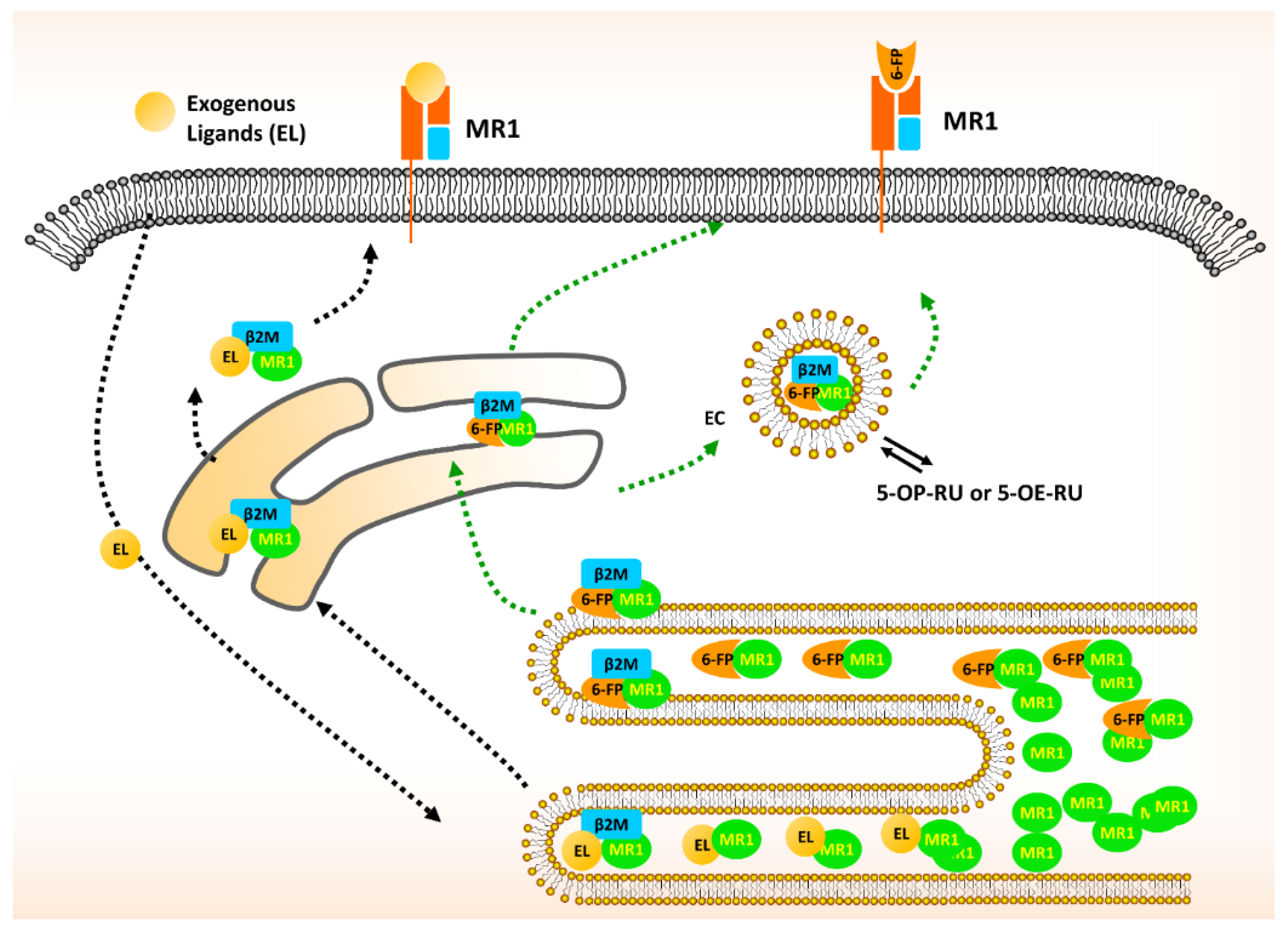
2. The Biology of MR1 in MAIT Cells
3. Classical MAIT Cells with TRAV1-2
4. Non-classical MAIT Cells without TRAV1-2
5. MAIT Cells and Cancer
6. MAIT Cells and IL-17 Family Members
7. Atypical MR1-Restricted T cells
8. TCR Antigens of MAIT Versus MR1T Cells
9. MR1-Restricted γδ T Cells
10. MR1T Cells in Cancer Immunotherapy
11. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Vojinovic, D.; Radjabzadeh, D.; Kurilshikov, A.; Amin, N.; Wijmenga, C.; Franke, L.; Ikram, M.A.; Uitterlinden, A.G.; Zhernakova, A.; Fu, J.; et al. Relationship between gut microbiota and circulating metabolites in population-based cohorts. Nat. Commun. 2019, 10, 5813. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Felix, K.M.; Tahsin, S.; Wu, H.J. Host-microbiota interplay in mediating immune disorders. Ann. N. Y. Acad. Sci. 2018, 1417, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Garrett, W.S.; Trinchieri, G.; Wargo, J. The cancer microbiome. Nat. Rev. Cancer 2019, 19, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Khan, M.A.W.; Hermann, A.; Gopalakrishnan, V.; Wargo, J.A. The microbiome, cancer, and cancer therapy. Nat. Med. 2019, 25, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Corbett, A.J.; Eckle, S.B.; Birkinshaw, R.W.; Liu, L.; Patel, O.; Mahony, J.; Chen, Z.; Reantragoon, R.; Meehan, B.; Cao, H.; et al. T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature 2014, 509, 361–365. [Google Scholar] [CrossRef]
- Kjer-Nielsen, L.; Patel, O.; Corbett, A.J.; Le Nours, J.; Meehan, B.; Liu, L.; Bhati, M.; Chen, Z.; Kostenko, L.; Reantragoon, R.; et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 2012, 491, 717–723. [Google Scholar] [CrossRef]
- Le Bourhis, L.; Martin, E.; Peguillet, I.; Guihot, A.; Froux, N.; Core, M.; Levy, E.; Dusseaux, M.; Meyssonnier, V.; Premel, V.; et al. Antimicrobial activity of mucosal-associated invariant T cells. Nat. Immunol. 2010, 11, 701–708. [Google Scholar] [CrossRef]
- Tastan, C.; Karhan, E.; Zhou, W.; Fleming, E.; Voigt, A.Y.; Yao, X.; Wang, L.; Horne, M.; Placek, L.; Kozhaya, L.; et al. Tuning of human MAIT cell activation by commensal bacteria species and MR1-dependent T-cell presentation. Mucosal Immunol. 2018, 11, 1591–1605. [Google Scholar] [CrossRef]
- Treiner, E.; Duban, L.; Bahram, S.; Radosavljevic, M.; Wanner, V.; Tilloy, F.; Affaticati, P.; Gilfillan, S.; Lantz, O. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature 2003, 422, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Allen, S.; McDonald, E.; Das, I.; Mak, J.Y.W.; Liu, L.; Fairlie, D.P.; Meehan, B.S.; Chen, Z.; Corbett, A.J.; et al. MAIT Cells Promote Tumor Initiation, Growth, and Metastases via Tumor MR1. Cancer Discov. 2020, 10, 124–141. [Google Scholar] [CrossRef] [PubMed]
- Vacchini, A.; Chancellor, A.; Spagnuolo, J.; Mori, L.; De Libero, G. MR1-Restricted T Cells Are Unprecedented Cancer Fighters. Front Immunol. 2020, 11, 751. [Google Scholar] [CrossRef] [PubMed]
- Patel, O.; Kjer-Nielsen, L.; Le Nours, J.; Eckle, S.B.; Birkinshaw, R.; Beddoe, T.; Corbett, A.J.; Liu, L.; Miles, J.J.; Meehan, B.; et al. Recognition of vitamin B metabolites by mucosal-associated invariant T cells. Nat. Commun. 2013, 4, 2142. [Google Scholar] [CrossRef]
- Wang, Y.; Ho, C.T. Flavour chemistry of methylglyoxal and glyoxal. Chem. Soc. Rev. 2012, 41, 4140–4149. [Google Scholar] [CrossRef]
- Inoue, Y.; Kimura, A. Methylglyoxal and regulation of its metabolism in microorganisms. Adv. Microb. Physiol. 1995, 37, 177–227. [Google Scholar]
- Lee, C.; Park, C. Bacterial Responses to Glyoxal and Methylglyoxal: Reactive Electrophilic Species. Int. J. Mol. Sci. 2017, 18, 169. [Google Scholar] [CrossRef]
- Nokin, M.J.; Durieux, F.; Peixoto, P.; Chiavarina, B.; Peulen, O.; Blomme, A.; Turtoi, A.; Costanza, B.; Smargiasso, N.; Baiwir, D.; et al. Methylglyoxal, a glycolysis side-product, induces Hsp90 glycation and YAP-mediated tumor growth and metastasis. eLife 2016, 5, e19375. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Weickert, M.O.; Thornalley, P.J. Multiple roles of glyoxalase 1-mediated suppression of methylglyoxal glycation in cancer biology-Involvement in tumour suppression, tumour growth, multidrug resistance and target for chemotherapy. Semin. Cancer Biol. 2018, 49, 83–93. [Google Scholar] [CrossRef]
- Luengo, A.; Abbott, K.L.; Davidson, S.M.; Hosios, A.M.; Faubert, B.; Chan, S.H.; Freinkman, E.; Zacharias, L.G.; Mathews, T.P.; Clish, C.B.; et al. Reactive metabolite production is a targetable liability of glycolytic metabolism in lung cancer. Nat. Commun. 2019, 10, 5604. [Google Scholar] [CrossRef]
- Park, J.O.; Tanner, L.B.; Wei, M.H.; Khana, D.B.; Jacobson, T.B.; Zhang, Z.; Rubin, S.A.; Li, S.H.; Higgins, M.B.; Stevenson, D.M.; et al. Near-equilibrium glycolysis supports metabolic homeostasis and energy yield. Nat. Chem. Biol. 2019, 15, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg effect: 80 years on. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Tilloy, F.; Treiner, E.; Park, S.H.; Garcia, C.; Lemonnier, F.; de la Salle, H.; Bendelac, A.; Bonneville, M.; Lantz, O. An invariant T cell receptor alpha chain defines a novel TAP-independent major histocompatibility complex class Ib-restricted alpha/beta T cell subpopulation in mammals. J. Exp. Med. 1999, 189, 1907–1921. [Google Scholar] [CrossRef] [PubMed]
- Crowther, M.D.; Dolton, G.; Legut, M.; Caillaud, M.E.; Lloyd, A.; Attaf, M.; Galloway, S.A.E.; Rius, C.; Farrell, C.P.; Szomolay, B.; et al. Genome-wide CRISPR-Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat. Immunol. 2020, 21, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Kok, D.E.; Steegenga, W.T.; McKay, J.A. Folate and epigenetics: Why we should not forget bacterial biosynthesis. Epigenomics 2018, 10, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Legoux, F.; Bellet, D.; Daviaud, C.; El Morr, Y.; Darbois, A.; Niort, K.; Procopio, E.; Salou, M.; Gilet, J.; Ryffel, B.; et al. Microbial metabolites control the thymic development of mucosal-associated invariant T cells. Science 2019, 366, 494–499. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, J.G.; Milani, C.; de Giori, G.S.; Sesma, F.; van Sinderen, D.; Ventura, M. Bacteria as vitamin suppliers to their host: A gut microbiota perspective. Curr. Opin. Biotechnol. 2013, 24, 160–168. [Google Scholar] [CrossRef]
- Godfrey, D.I.; Koay, H.F.; McCluskey, J.; Gherardin, N.A. The biology and functional importance of MAIT cells. Nat. Immunol. 2019, 20, 1110–1128. [Google Scholar] [CrossRef]
- Keller, A.N.; Eckle, S.B.; Xu, W.; Liu, L.; Hughes, V.A.; Mak, J.Y.; Meehan, B.S.; Pediongco, T.; Birkinshaw, R.W.; Chen, Z. Drugs and drug-like molecules can modulate the function of mucosal-associated invariant T cells. Nat. Immunol. 2017, 18, 402. [Google Scholar] [CrossRef]
- Gold, M.C.; McLaren, J.E.; Reistetter, J.A.; Smyk-Pearson, S.; Ladell, K.; Swarbrick, G.M.; Yu, Y.Y.; Hansen, T.H.; Lund, O.; Nielsen, M.; et al. MR1-restricted MAIT cells display ligand discrimination and pathogen selectivity through distinct T cell receptor usage. J. Exp. Med. 2014, 211, 1601–1610. [Google Scholar] [CrossRef]
- Reantragoon, R.; Kjer-Nielsen, L.; Patel, O.; Chen, Z.; Illing, P.T.; Bhati, M.; Kostenko, L.; Bharadwaj, M.; Meehan, B.; Hansen, T.H.; et al. Structural insight into MR1-mediated recognition of the mucosal associated invariant T cell receptor. J. Exp. Med. 2012, 209, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Lepore, M.; Kalinichenko, A.; Colone, A.; Paleja, B.; Singhal, A.; Tschumi, A.; Lee, B.; Poidinger, M.; Zolezzi, F.; Quagliata, L.; et al. Parallel T-cell cloning and deep sequencing of human MAIT cells reveal stable oligoclonal TCRbeta repertoire. Nat. Commun. 2014, 5, 3866. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kong, D.; Wang, H. Mucosal-Associated Invariant T cell in liver diseases. Int. J. Biol. Sci. 2020, 16, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Gherardin, N.A.; Souter, M.N.; Koay, H.F.; Mangas, K.M.; Seemann, T.; Stinear, T.P.; Eckle, S.B.; Berzins, S.P.; d’Udekem, Y.; Konstantinov, I.E.; et al. Human blood MAIT cell subsets defined using MR1 tetramers. Immunol. Cell Biol. 2018, 96, 507–525. [Google Scholar] [CrossRef] [PubMed]
- Reantragoon, R.; Corbett, A.J.; Sakala, I.G.; Gherardin, N.A.; Furness, J.B.; Chen, Z.; Eckle, S.B.; Uldrich, A.P.; Birkinshaw, R.W.; Patel, O.; et al. Antigen-loaded MR1 tetramers define T cell receptor heterogeneity in mucosal-associated invariant T cells. J. Exp. Med. 2013, 210, 2305–2320. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lin, F.; Gao, Y.; Li, Z.; Zhang, J.; Xing, Y.; Deng, Z.; Yao, Z.; Tsun, A.; Li, B. FOXP3 and RORgammat: Transcriptional regulation of Treg and Th17. Int. Immunopharmacol. 2011, 11, 536–542. [Google Scholar] [CrossRef]
- Koay, H.-F.; Gherardin, N.A.; Xu, C.; Seneviratna, R.; Zhao, Z.; Chen, Z.; Fairlie, D.P.; McCluskey, J.; Pellicci, D.G.; Uldrich, A.P.; et al. Diverse MR1-restricted T cells in mice and humans. Nat. Commun. 2019, 10, 2243. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Goswami, S.; Shi, J.Y.; Wu, L.J.; Wang, X.Y.; Ma, J.Q.; Zhang, Z.; Shi, Y.; Ma, L.J.; Zhang, S.; et al. Activated and Exhausted MAIT Cells Foster Disease Progression and Indicate Poor Outcome in Hepatocellular Carcinoma. Clin. Cancer Res. 2019, 25, 3304–3316. [Google Scholar] [CrossRef]
- Rudak, P.T.; Choi, J.; Haeryfar, S.M.M. MAIT cell-mediated cytotoxicity: Roles in host defense and therapeutic potentials in infectious diseases and cancer. J. Leukoc. Biol. 2018, 104, 473–486. [Google Scholar] [CrossRef]
- Ling, L.; Lin, Y.; Zheng, W.; Hong, S.; Tang, X.; Zhao, P.; Li, M.; Ni, J.; Li, C.; Wang, L.; et al. Circulating and tumor-infiltrating mucosal associated invariant T (MAIT) cells in colorectal cancer patients. Sci. Rep. 2016, 6, 20358. [Google Scholar] [CrossRef]
- Won, E.J.; Ju, J.K.; Cho, Y.N.; Jin, H.M.; Park, K.J.; Kim, T.J.; Kwon, Y.S.; Kee, H.J.; Kim, J.C.; Kee, S.J.; et al. Clinical relevance of circulating mucosal-associated invariant T cell levels and their anti-cancer activity in patients with mucosal-associated cancer. Oncotarget 2016, 7, 76274–76290. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, P.; Ahlmanner, F.; Akeus, P.; Sundquist, M.; Alsen, S.; Yrlid, U.; Borjesson, L.; Sjoling, A.; Gustavsson, B.; Wong, S.B.; et al. Human Mucosa-Associated Invariant T Cells Accumulate in Colon Adenocarcinomas but Produce Reduced Amounts of IFN-gamma. J. Immunol. 2015, 195, 3472–3481. [Google Scholar] [CrossRef] [PubMed]
- Zabijak, L.; Attencourt, C.; Guignant, C.; Chatelain, D.; Marcelo, P.; Marolleau, J.P.; Treiner, E. Increased tumor infiltration by mucosal-associated invariant T cells correlates with poor survival in colorectal cancer patients. Cancer Immunol. Immunother. 2015, 64, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Gherardin, N.A.; Loh, L.; Admojo, L.; Davenport, A.J.; Richardson, K.; Rogers, A.; Darcy, P.K.; Jenkins, M.R.; Prince, H.M.; Harrison, S.J.; et al. Enumeration, functional responses and cytotoxic capacity of MAIT cells in newly diagnosed and relapsed multiple myeloma. Sci. Rep. 2018, 8, 4159. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, G.A.; Miller, G. Targeting the interleukin-17 immune axis for cancer immunotherapy. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Gorczynski, R.M. IL-17 Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1240, 47–58. [Google Scholar]
- Sundstrom, P.; Szeponik, L.; Ahlmanner, F.; Sundquist, M.; Wong, J.S.B.; Lindskog, E.B.; Gustafsson, B.; Quiding-Jarbrink, M. Tumor-infiltrating mucosal-associated invariant T (MAIT) cells retain expression of cytotoxic effector molecules. Oncotarget 2019, 10, 2810–2823. [Google Scholar] [CrossRef]
- Li, Q.; Han, Y.; Fei, G.; Guo, Z.; Ren, T.; Liu, Z. IL-17 promoted metastasis of non-small-cell lung cancer cells. Immunol. Lett. 2012, 148, 144–150. [Google Scholar] [CrossRef]
- Reppert, S.; Koch, S.; Finotto, S. IL-17A is a central regulator of lung tumor growth. Oncoimmunology 2012, 1, 783–785. [Google Scholar] [CrossRef]
- Chen, X.; Wan, J.; Liu, J.; Xie, W.; Diao, X.; Xu, J.; Zhu, B.; Chen, Z. Increased IL-17-producing cells correlate with poor survival and lymphangiogenesis in NSCLC patients. Lung Cancer 2010, 69, 348–354. [Google Scholar] [CrossRef]
- Qian, X.L.; Xu, P.; Zhang, Y.Q.; Song, Y.M.; Li, Y.Q.; Li, W.D.; Jiang, C.Y.; Shen, B.B.; Zhang, X.M.; Zhang, L.N.; et al. Increased number of intratumoral IL-17+ cells, a harbinger of the adverse prognosis of triple-negative breast cancer. Breast Cancer Res. Treat. 2020, 180, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Awaji, M.; Saxena, S.; Varney, M.L.; Sharma, B.; Singh, R.K. IL-17-CXC Chemokine Receptor 2 Axis Facilitates Breast Cancer Progression by Up-Regulating Neutrophil Recruitment. Am. J. Pathol. 2020, 190, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Sijilmassi, O. Folic acid deficiency and vision: A review. Graefes. Arch. Clin. Exp. Ophthalmol. 2019, 257, 1573–1580. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Yao, Y.; Yonezawa, A.; Imai, S.; Yoshimatsu, H.; Otani, Y.; Omura, T.; Nakagawa, S.; Nakagawa, T.; Matsubara, K. Riboflavin Transporters RFVT/SLC52A Mediate Translocation of Riboflavin, Rather than FMN or FAD, across Plasma Membrane. Biol. Pharm. Bull. 2017, 40, 1990–1995. [Google Scholar] [CrossRef]
- Lepore, M.; Kalinichenko, A.; Calogero, S.; Kumar, P.; Paleja, B.; Schmaler, M.; Narang, V.; Zolezzi, F.; Poidinger, M.; Mori, L.; et al. Functionally diverse human T cells recognize non-microbial antigens presented by MR1. eLife 2017, 6, e24476. [Google Scholar] [CrossRef]
- Ogg, G.; Cerundolo, V.; McMichael, A.J. Capturing the antigen landscape: HLA-E, CD1 and MR1. Curr. Opin. Immunol. 2019, 59, 121–129. [Google Scholar] [CrossRef]
- McWilliam, H.E.; Birkinshaw, R.W.; Villadangos, J.A.; McCluskey, J.; Rossjohn, J. MR1 presentation of vitamin B-based metabolite ligands. Curr. Opin. Immunol. 2015, 34, 28–34. [Google Scholar] [CrossRef]
- Zareie, P.; Farenc, C.; La Gruta, N.L. MHC Restriction: Where Are We Now? Viral Immunol. 2020, 33, 179–187. [Google Scholar] [CrossRef]
- Le Nours, J.; Gherardin, N.A.; Ramarathinam, S.H.; Awad, W.; Wiede, F.; Gully, B.S.; Khandokar, Y.; Praveena, T.; Wubben, J.M.; Sandow, J.J.; et al. A class of γδ T cell receptors recognize the underside of the antigen-presenting molecule MR1. Science 2019, 366, 1522–1527. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SUBSETS | Classical MAIT | Non-Classical MAIT | MR1T | γδ T |
|---|---|---|---|---|
| TRAV1-2 | TRAV1-2+ | TRAV1-2- | ||
| Frequency in blood T cells | 1%–10% | 0.001%–0.01% | 0.02% | <0.001%–0.1% |
| Presence in organs | Liver ~ 50%,gastrointestinal tract, lung, thymus, lymph nodes, spleen, kidney, skin | Unknown or absent | Liver, stomach, lung, and duodenum; disease tissues | |
| Antigen | All 5-OP-RU; subset-specific recognition of other antigens | Unknown | 5-OP-RU, Ac-6-FP | |
| TCR diversity | TRAV1-2, TRAJ33 or 12 or 20, TRBV6 or 20-1 | TRAV36, TRAJ34 or 37, TRBV28, TRBJ2-5 | TRAVX, TRAJX, TRBVX | TRGVx, TRDV1 (72%), TRDV3/5 |
| Transcription factors | PLZF+ RORγt+ | PLZF+ | PLZF- | Diverse |
| Co-receptors* | CD8αβ, CD8αα, DN, CD4, DP | CD8, DN, CD4, or DP | CD8, CD161, DN | DN, CD8α with CD161 |
| Cytokines secreted | IL-17A, TNF-α, IFN-γ, IL-2, IL-22 | Unknown | IL-17A, TNF, IFN-γ, IL-2 | IFN-γ, TNF-α, and others |
| Clone | TCRα (Accession No) | TCRα Sequence VJ | TCRβ (Accession No) | TCRβ V(D)J Sequence |
|---|---|---|---|---|
| DGB129 | TRAV29 (MF085365) | CAASLYNQGGKLIFGQGTELSVKP | TRBV12-4 (MF085366) | CASSYRGTEAFFGQGTRLTVV |
| DGB70 | TRAV5 (MF085363) | CAETWTDRGSTLGRLYFGRGTQLTVWP | TRBV28 (MF085364) | CASSLGATGANEKLFFGSGTQLSVL |
| DGA28 | TRAV25 (MF085361) | CAAAGGTSYGKLTFGQGTILTVHP | TRBV291 (MF085362) | CSVGAGQGPYTDTQYFGPGTRLTVL |
| JMA | TRAV27 (MF085369) | CAGENSGYALNFGKGTSLLVTP | TRBV25-1 (MF085370) | CASSQLYRDTSNTGELFFGEGSRLTVL |
| TC5A87 | TRAV131 (MF085371) | CAANWSPQGNEKLTFGTGTRLTIIP | TRBV25-1 (MF085372) | CASSEYIQYSGNTIYFGEGSWLTVV |
| CH9A3 | TRAV24 (MF085367) | CASGDSGYALNFGKGTSLLVTP | TRAV24 (MF085368) | CASSFDVGLPPLHFGNGTRLTVT |
| MC.7.G5 | TRAV38.2/DV8TRAJ31 (MN782533) | CAYRSAVNARLMFGDGTQLVVKP | TRBV25.1TRBJ2.3 (MN782534) | CASSEARGLAEFTDTQYFGPGTRLTVL |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores-Villanueva, P.; Sobhani, N.; Wang, X.; Li, Y. MR1-Restricted T Cells in Cancer Immunotherapy. Cancers 2020, 12, 2145. https://doi.org/10.3390/cancers12082145
Flores-Villanueva P, Sobhani N, Wang X, Li Y. MR1-Restricted T Cells in Cancer Immunotherapy. Cancers. 2020; 12(8):2145. https://doi.org/10.3390/cancers12082145
Chicago/Turabian StyleFlores-Villanueva, Pedro, Navid Sobhani, Xu Wang, and Yong Li. 2020. "MR1-Restricted T Cells in Cancer Immunotherapy" Cancers 12, no. 8: 2145. https://doi.org/10.3390/cancers12082145
APA StyleFlores-Villanueva, P., Sobhani, N., Wang, X., & Li, Y. (2020). MR1-Restricted T Cells in Cancer Immunotherapy. Cancers, 12(8), 2145. https://doi.org/10.3390/cancers12082145