Manipulation of Glucose Availability to Boost Cancer Immunotherapies


Simple Summary
Abstract
1. Introduction
2. Fundamentals of Cancer Metabolism
3. Basics of T Cell Metabolism
4. Metabolism of T Regulatory Cells
5. Manipulation of Glucose Metabolism
5.1. Pharmacological Manipulation of Glucose Metabolism
5.2. Side Effects Related to Manipulation of Glucose Metabolism
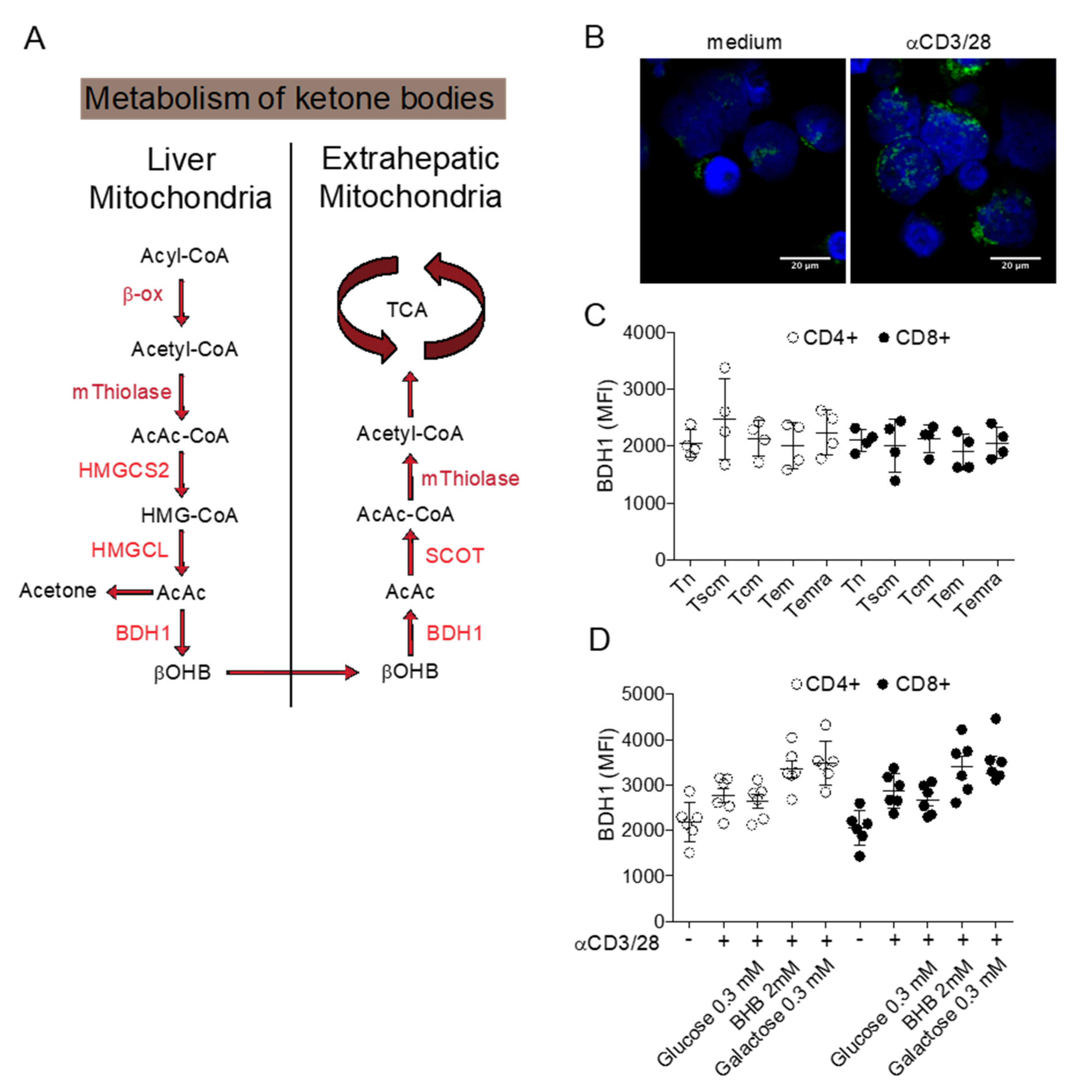
5.3. Long Term Glucose Restriction through Ketogenic Diets
6. Relevance of Immunometabolism in Cancer Immunotherapy
6.1. Manipulation of Metabolism in Immunotherapy

{kind=link}
{kind=link}
| Compound | Structure | MW | IC50 (μM) | Selectivity for Glut1 | References |
|---|---|---|---|---|---|
STF-31 |  | 423.53 | 1 | Low | [46,47,50] |
WZB-117 |  | 368.31 | 0.5 | Low | [48,49,71] |
| BAY 876 |  | 496.42 | 0.002 | High | [51,52] |
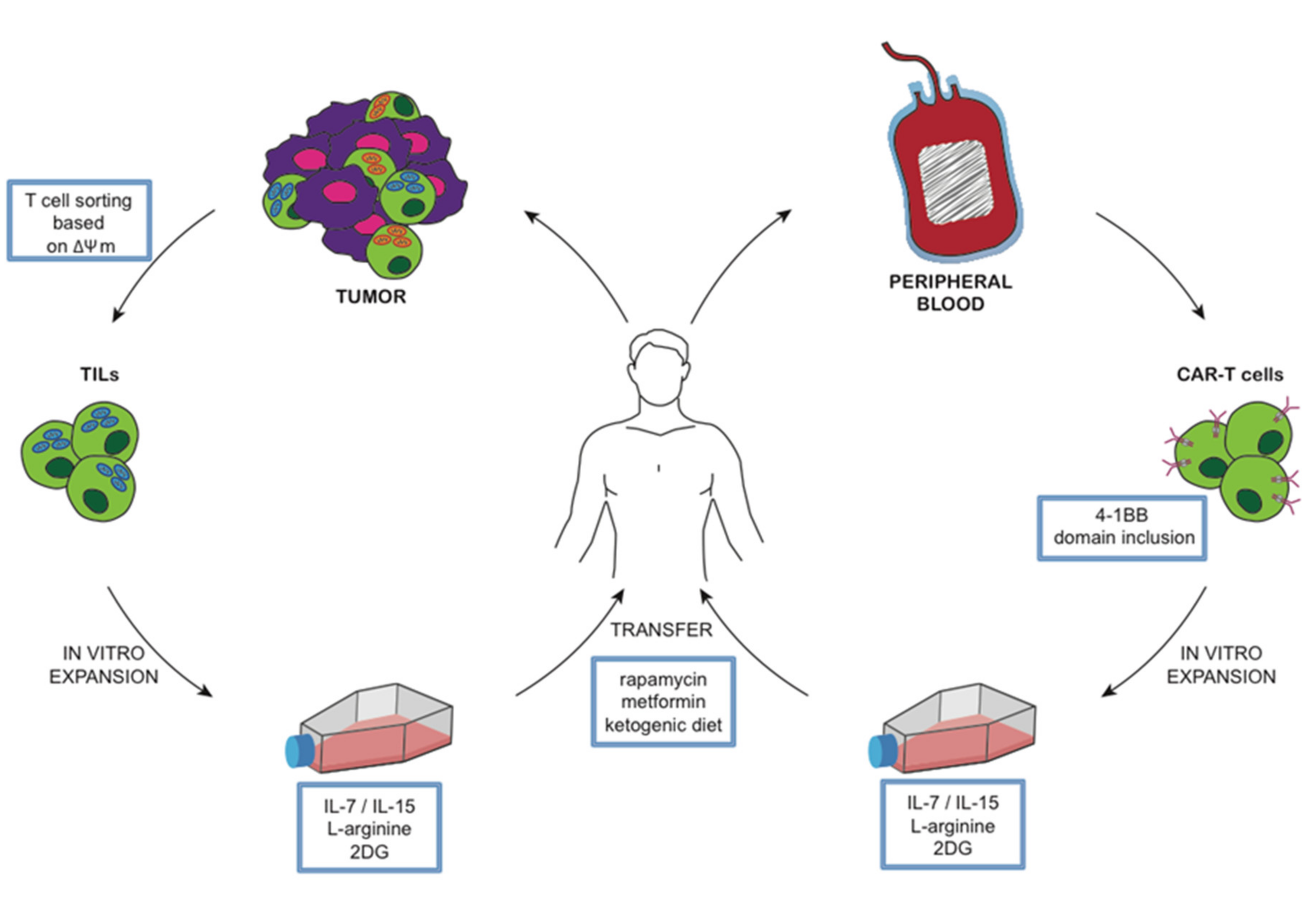
6.2. A Model to Integrate Metabolic Interventions and Cancer Immunotherapy
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.; Fendt, S.; Ho, P. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Green, D.R. Metabolic checkpoints in activated T cells. Nat. Immunol. 2012, 13, 907–915. [Google Scholar] [CrossRef]
- Kishton, R.J.; Sukumar, M.; Restifo, N.P. Review Metabolic Regulation of T Cell Longevity and Function in Tumor Immunotherapy. Cell Metab. 2017, 26, 94–109. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Di Dedda, C.; Vignali, D.; Piemonti, L.; Monti, P. Pharmacological Targeting of GLUT1 to Control Autoreactive T Cell Responses. Int. J. Mol. Sci. 2019, 20, 4962. [Google Scholar] [CrossRef] [PubMed]
- Soldati, L.; Di Renzo, L.; Jirillo, E.; Ascierto, P.A.; Marincola, F.M.; De Lorenzo, A. The influence of diet on anti—Cancer immune responsiveness. J. Transl. Med. 2018, 16, 1–18. [Google Scholar] [CrossRef]
- Turbitt, W.J.; Demark-Wahnefried, W.; Peterson, C.M.; Norian, L.A. Targeting Glucose Metabolism to Enhance Immunotherapy: Emerging Evidence on Intermittent Fasting and Calorie Restriction Mimetics. Front Immunol. 2019, 10, 1402. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.; Tardito, S.; Pillozzi, S.; Arcangeli, A.; Armento, A.; Uggeri, J.; Missale, G.; Bianchi, M.G.; Barilli, A.; Dall’Asta, V.; et al. Glutamine depletion by crisantaspase hinders the growth of human hepatocellular carcinoma xenografts. Br. J. Cancer. 2014, 111, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, A.; Kamphorst, J.J.; Markert, E.K.; Schug, Z.T.; Tardito, S.; Eyal Gottlieb, E. Cancer metabolism at a glance. J. Cell Sci. 2016, 129, 3367–3373. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer. 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Pollak, M.N. Investigating metformin for cancer prevention and treatment: The end of the beginning. Cancer Discov. 2012, 2, 778–790. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer. 2014, 14, 709–721. [Google Scholar] [CrossRef]
- Lipp, M.; Sallusto, F.; Lenig, D.; Fo, R.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials. Nature 1999, 401, 708–712. [Google Scholar]
- Gattinoni, L.; Zhong, X.-S.; Palmer, D.; Ji, Y.; Hinrichs, C.S.; Yu, Z.; Wrzesinski, C.; Boni, A.; Cassad, L.; Garvin, L.M.; et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med. 2009, 15, 808–813. [Google Scholar] [CrossRef]
- Lugli, E.; Dominguez, M.H.; Gattinoni, L.; Chattopadhyay, P.K.; Bolton, D.L.; Song, K.; Klatt, N.R.; Brenchley, J.M.; Vaccari, M.; Gostick, E.; et al. Brief report Superior T memory stem cell persistence supports long-lived T cell memory. J. Clin. Invest. 2013, 123, 594–599. [Google Scholar]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell—Like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P.; Gattinoni, L. Lineage relationship of effector and memory T cells. Curr. Opin. Immunol. 2013, 25, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Schluns, K.S.; Kieper, W.C.; Jameson, S.C.; Lefrançois, L. Interleukin-7 mediates the homeostasis of naïve and memory CD8 T cells in vivo. Nat. Immunol. 2000, 1, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Wofford, J.A.; Wieman, H.L.; Jacobs, S.R.; Zhao, Y.; Rathmell, J.C. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood 2008, 111, 2101–2111. [Google Scholar] [CrossRef] [PubMed]
- Chehtane, M.; Khaled, A.R. Interleukin-7 mediates glucose utilization in lymphocytes through transcriptional regulation of the hexokinase II gene. Am. J. Physiol. Cell Physiol. 2010, 298, C1560–C1571. [Google Scholar] [CrossRef]
- Rathmell, J.C.; Vander Heiden, M.G.; Harris, M.H.; Frauwirth, K.A.; Thompson, C.B. In the Absence of Extrinsic Signals, Nutrient Utilization by Lymphocytes Is Insufficient to Maintain Either Cell Size or Viability. Mol Cell 2000, 6, 683–692. [Google Scholar] [CrossRef]
- Van der Windt, G.J.W.; Everts, B.; Chang, C.-H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial Respiratory Capacity Is a Critical Regulator of CD8+ T Cell Memory Development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef]
- Cui, G.; Staron, M.M.; Gray, S.M.; Ho, P.C.; Amezquita, R.A.; Wu, J.; Kaech, S.M. IL-7-induced glycerol transport and TAG synthesis promotes memory CD8+ T cell longevity. Cell 2015, 161, 750–761. [Google Scholar] [CrossRef]
- Vrisekoop, N.; Den Braber, I.; De Boer, A.B.; Ruiter, A.F.C.; Van Der Crabben, S.N.; Schrijver, E.H.R.; Spierenburg, G.; Sauerwein, H.P.; Hazenberg, M.D.; Borghans, A.M.; et al. Sparse production but preferential incorporation of recently produced naive T cells in the human peripheral pool. Blood 2008, 105, 6115–6120. [Google Scholar] [CrossRef]
- Jacobs, S.R.; Herman, C.E.; Maciver, N.J.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C.; Jacobs, S.R.; Herman, C.E.; Maciver, N.J.; et al. CD28-Mediated Akt-Dependent and Independent Pathways 1. J. Immunol. 2008, 180, 4476–4486. [Google Scholar] [CrossRef]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 Signaling Pathway Regulates Glucose Metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef]
- Cammann, C.; Rath, A.; Reichl, U.; Lingel, H.; Brunner-weinzierl, M.; Simeoni, L.; Schraven, B.; Lindquist, J.A. Early changes in the metabolic profile of activated CD8 + T cells. BMC Cell Biol. 2016, 1, 28. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8 + T cell memory and antitumor function. J. Clin. Invest. 2013, 123, 4479–4488. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.W.; et al. Mitochondrial Dynamics Controls T Cell Fate Through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef]
- DeLeeuw, R.J.; Kost, S.E.; Kakal, J.A.; Nelson, B.H. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: A critical review of the literature. Clin. Cancer Res. 2012, 18, 3022–3029. [Google Scholar] [CrossRef]
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J. Clin. Oncol. 2009, 27, 186–192. [Google Scholar] [CrossRef]
- Cavalleri, T.; Bianchi, P.; Basso, G.; Celesti, G.; Grizzi, F.; Bossi, P.; Greco, L.; Pitrone, C.; Valtorta, E.; Mauri, G.; et al. Combined low densities of FOXP3þ and CD3þ tumor-infiltrating lymphocytes identify stage II colorectal cancer at high risk of progression. Cancer Immunol. Res. 2019, 7, 751–758. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S. Targeting Treg cells in cancer immunotherapy. Eur. J. Immunol. 2019, 49, 1140–1146. [Google Scholar] [CrossRef]
- Porcellini, A.; Procaccini, C.; De Rosa, V.; Galgani, M.; Abanni, L.; Calı, G.; Carbone, F.; Fontana, S.; Horvath, T.L.; La Cava, A.; et al. An Oscillatory Switch in mTOR Kinase Activity Sets Regulatory T Cell Responsiveness. Immunity 2010, 33, 929–941. [Google Scholar]
- Kishore, M.; Cheung, K.C.P.; Fu, H.; Bonacina, F.; Wang, G.; Coe, D.; Ward, E.J.; Colamatteo, A.; Jangani, M.; Baragetti, A.; et al. Regulatory T Cell Migration Is Dependent on Article Regulatory T Cell Migration Is Dependent on Glucokinase-Mediated Glycolysis. Immunity 2017, 47, 875–889. [Google Scholar] [CrossRef]
- Miska, J.; Lee-chang, C.; Rashidi, A.; Muroski, M.E.; Chang, A.L.; Lopez-rosas, A.; Zhang, P.; Panek, W.K.; Cordero, A.; Han, Y.; et al. The Role of Tregs in Glioma-Mediated Immunosuppression: Potential Target for Intervention. Neurosurg. Clin. N. Am. 2010, 21, 125–137. [Google Scholar]
- De Rosa, V.; Galgani, M.; Porcellini, A.; Colamatteo, A.; Santopaolo, M.; Zuchegna, C.; Romano, A.; De Simone, S.; Procaccini, C.; La Rocca, C.; et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat. Immunol. 2015, 16, 1174–1184. [Google Scholar] [CrossRef] [PubMed]
- Dupage, M.; Chopra, G.; Marson, A.; Bluestone, J.A.; Dupage, M.; Chopra, G.; Quiros, J.; Rosenthal, W.L.; Morar, M.M.; Holohan, D.; et al. The Chromatin-Modifying Enzyme Ezh2 Is Critical for the Maintenance of Regulatory T Cell Identity after Article The Chromatin-Modifying Enzyme Ezh2 Is Critical for the Maintenance of Regulatory T Cell Identity after Activation. Immunity 2015, 42, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Ralser, M.; Wamelink, M.M.; Struys, E.A.; Joppich, C.; Krobitsch, S.; Jakobs, C.; Lehrach, H. A catabolic block does not sufficiently explain how 2-deoxy-D-glucose inhibits cell growth. Proc. Natl. Acad. Sci. USA 2008, 105, 17807–17811. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.A.; Sutphin, P.D.; Nguyen, P.; Turcotte, S.; Lai, E.W.; Banh, A.; Reynolds, G.E.; Chi, J.-T.; Wu, J.; Solow-Cordero, D.E.; et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl. Med. 2011, 3, 94ra70. [Google Scholar] [CrossRef]
- Williamson, M.K.; Coombes, N.; Juszczak, F.; Athanasopoulos, M.; Khan, M.B.; Eykyn, T.R.; Srenathan, U.; Id, L.S.T.; Zeidler, J.D.; Da, A.T.; et al. Upregulation of Glucose Uptake and Hexokinase Activity of Primary Human CD4 + T Cells in Response to Infection with HIV-1. Viruses 2018, 10, 114. [Google Scholar] [CrossRef]
- Ojelabi, O.A.; Lloyd, K.P.; Simon, A.H.; De Zutter, J.K.; Carruthers, A. WZB117 (2-fluoro-6-(m-hydroxybenzoyloxy) Phenyl m-Hydroxybenzoate) inhibits GLUT1-mediated sugar transport by binding reversibly at the exofacial sugar binding site. J. Biol. Chem. 2016, 291, 26762–26772. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef]
- Adams, D.J.; Ito, D.; Rees, M.G.; Seashore-Ludlow, B.; Puyang, X.; Ramos, A.H.; Cheah, J.H.; Clemons, P.A.; Warmuth, M.; Zhu, P.; et al. NAMPT is the cellular target of STF-31-like small-molecule probes. ACS Chem. Biol. 2014, 9, 2247–2254. [Google Scholar] [CrossRef]
- Siebeneicher, H.; Cleve, A.; Rehwinkel, H.; Neuhaus, R.; Heisler, I.; Müller, T.; Bauser, M.; Buchmann, B. Identification and Optimization of the First Highly Selective GLUT1 Inhibitor BAY-876. ChemMedChem 2016, 11, 2261–2271. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, W.; Idowu, M.O.; Oh, U.; Wang, X.; Temkin, S.M.; Fang, X. Ovarian Cancer Relies on Glucose Transporter 1 to Fuel Glycolysis and Growth: Anti-Tumor Activity of BAY-876. Cancers 2018, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Kinet, S.; Manel, N.; Prohaska, R.; Battini, J.; Delaunay, J.; Sitbon, M.; Taylor, N. Erythrocyte Glut1 Triggers Dehydroascorbic Acid Uptake in Mammals Unable to Synthesize Vitamin C. Cell 2008, 132, 1039–1048. [Google Scholar]
- Pingitore, A.; Ruz-Maldonado, I.; Liu, B.; Huang, G.C.; Choudhary, P.; Persaud, S.J. Dynamic Profiling of Insulin Secretion and ATP Generation in Isolated Human and Mouse Islets Reveals Differential Glucose Sensitivity. Cell. Physiol. Biochem. 2017, 44, 1352–1359. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.S.; Akman, C.; Hinton, V.J.; Engelstad, K.; De Vivo, D.C. Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS). Curr. Neurol. Neurosci. Rep. 2013, 13, 1–9. [Google Scholar] [CrossRef]
- Leen, W.G.; Taher, M.; Verbeek, M.M.; Kamsteeg, E.J.; Van De Warrenburg, B.P.; Willemsen, M.A. GLUT1 deficiency syndrome into adulthood: A follow-up study. J. Neurol. 2014, 261, 589–599. [Google Scholar] [CrossRef]
- Branco, A.F.; Ferreira, A.; Simões, R.F.; Magalhães-Novais, S.; Zehowski, C.; Cope, E.; Silva, A.M.; Pereira, D.; Sardão, V.A.; Cunha-Oliveira, T. Ketogenic diets: From cancer to mitochondrial diseases and beyond. Eur. J. Clin. Invest. 2016, 46, 285–298. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Morscher, R.J.; Aminzadeh-Gohari, S.; Feichtinger, R.; Mayr, J.A.; Lang, R.; Neureiter, D.; Sperl, W.; Kofler, B. Inhibition of neuroblastoma tumor growth by ketogenic diet and/or calorie restriction in a CD1-nu mouse model. PLoS ONE 2015, 10, e0129802. [Google Scholar] [CrossRef]
- Weber, D.D.; Kofler, B. Ketogenic diet in cancer therapy. Aging 2018, 10, 164–165. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell. 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Galluzzi, L.; Chan, T.A.; Kroemer, G.; Wolchok, J.D.; López-Soto, A. The hallmarks of successful anticancer immunotherapy. Sci. Transl. Med. 2018, 10, 1–15. [Google Scholar] [CrossRef]
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Chamoto, K.; Chowdhury, P.S.; Kumar, A.; Sonomura, K.; Matsuda, F.; Fagarasan, S.; Honjo, T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E761–E770. [Google Scholar] [CrossRef]
- Weide, B.; Martens, A.; Hassel, J.C.; Berking, C.; Postow, M.A.; Bisschop, K.; Simeone, E.; Mangana, J.; Schilling, B.; Di Giacomo, A.M.; et al. Baseline biomarkers for outcome of melanoma patients treated with pembrolizumab. Clin. Cancer Res. 2016, 22, 5487–5496. [Google Scholar] [CrossRef]
- Kelderman, S.; Heemskerk, B.; Van Tinteren, H.; Van Den Brom, R.R.H.; Hospers, G.A.P.; Van Den Eertwegh, A.J.M.; Kapiteijn, E.W.; De Groot, J.W.B.; Soetekouw, P.; Jansen, R.L.; et al. Lactate dehydrogenase as a selection criterion for ipilimumab treatment in metastatic melanoma. Cancer Immunol. Immunother. 2014, 63, 449–458. [Google Scholar] [CrossRef]
- Martens, A.; Wistuba-Hamprecht, K.; Foppen, M.G.; Yuan, J.; Postow, M.A.; Wong, P.; Romano, E.; Khammari, A.; Dreno, B.; Capone, M.; et al. Baseline peripheral blood biomarkers associated with clinical outcome of advanced melanoma patients treated with ipilimumab. Clin. Cancer Res. 2016, 22, 2908–2918. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, M.L.; McMiller, T.L.; Berger, A.E.; Danilova, L.; Anders, R.A.; Netto, G.J.; Xu, H.; Pritchard, T.S.; Fan, J.; Cheadle, C.; et al. The intratumoral balance between metabolic and immunologic gene expression is associated with anti-PD-1 response in patients with renal cell carcinoma. Cancer Immunol. Res. 2016, 4, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Cortese, N.; Capretti, G.; Barbagallo, M.; Rigamonti, A.; Takis, P.G.; Castino, G.F.; Vignali, D.; Maggi, G.; Gavazzi, F.; Ridolfi, C.; et al. Metabolome of Pancreatic Juice Delineates Distinct Clinical Profiles of Pancreatic Cancer and Reveals a Link between Glucose Metabolism and PD-1+ Cells. Cancer Immunol. Res. 2020, 8, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Hatae, R.; Chamoto, K.; Kim, Y.H.; Sonomura, K.; Taneishi, K.; Kawaguchi, S.; Yoshida, H.; Ozasa, H.; Sakamori, Y.; Akrami, M.; et al. Combination of host immune metabolic biomarkers for the PD-1 blockade cancer immunotherapy. JCI Insight. 2020, 5, e133501. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, M.; Liu, J.; Mehta, G.U.; Patel, S.J.; Roychoudhuri, R.; Crompton, J.G.; Klebanoff, C.A.; Ji, Y.; Li, P.; Yu, Z.; et al. Mitochondrial Membrane Potential Identifies Cells with Enhanced Stemness for Cellular Therapy. Cell Metab. 2016, 23, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Crompton, J.G.; Sukumar, M.; Roychoudhuri, R.; Clever, D.; Gros, A.; Eil, R.L.; Tran, E.; Hanada, K.I.; Yu, Z.; Palmer, D.C.; et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res. 2015, 75, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Kawalekar, O.U.; O’ Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef]
- Cieri, N.; Camisa, B.; Cocchiarella, F.; Forcato, M.; Oliveira, G.; Provasi, E.; Bondanza, A.; Bordignon, C.; Peccatori, J.; Ciceri, F.; et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 2013, 121, 573–584. [Google Scholar] [CrossRef]
- Heninger, A.-K.; Monti, P.; Wilhelm, C.; Schwaiger, P.; Kuehn, D.; Ziegler, A.-G.; Bonifacio, E. Activation of Islet Autoreactive Naive T Cells in Infants Is Influenced by Homeostatic Mechanisms and Antigen-Presenting Capacity. Diabetes 2013, 62, 2059–2066. [Google Scholar] [CrossRef]
- Vignali, D.; Cantarelli, E.; Bordignon, C.; Canu, A.; Citro, A.; Piemonti, L.; Monti, P. Detection and characterization of CD8 + autoreactive memory stem T cells in patients with type 1 diabetes. Diabetes 2018, 67, 936–945. [Google Scholar] [CrossRef]
- Monti, P.; Brigatti, C.; Heninger, a.K.; Scirpoli, M.; Bonifacio, E. Disengaging the IL-2 receptor with daclizumab enhances IL-7-mediated proliferation of CD4(+) and CD8(+) T cells. Am. J. Transplant. 2009, 9, 2727–2735. [Google Scholar] [CrossRef] [PubMed]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.a.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Reni, M.; Dugnani, E.; Cereda, S.; Belli, C.; Balzano, G.; Nicoletti, R.; Liberati, D.; Pasquale, V.; Scavini, M.; Maggiora, P.; et al. (Ir)relevance of metformin treatment in patients with metastatic pancreatic cancer: An open-label, randomized phase II trial. Clin. Cancer Res. 2016, 22, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Eikawa, S.; Nishida, M.; Mizukami, S.; Yamazaki, C.; Nakayama, E.; Udono, H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc. Natl. Acad. Sci. USA 2015, 112, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Petruzzelli, M.; Wagner, E.F. Mechanisms of metabolic dysfunction in cancer-associated cachexia. Genes Dev. 2014, 30, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Tonouchi, H.; Sasayama, A.; Ashida, K. A Ketogenic Formula Prevents Tumor Progression and Cancer Cachexia by Attenuating Systemic Inflammation in Colon 26 Tumor-Bearing Mice. Nutrients 2018, 10, 206. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.C.; Borland, W.; Preston, T.; Tisdale, M.J.; Shenkin, A.; Calman, K.C. Cancer cachexia: Influence of systemic ketosis on substrate levels and nitrogen metabolism. Am. J. Clin. Nutr. 1988, 47, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.M.; Yun, B.; Kim, M.; Song, M.; Kim, Y.; Lee, S.H.; Lee, H.; Lee, S.M.; Lee, S.M. Postoperative serum metabolites of patients on a low carbohydrate ketogenic diet after pancreatectomy for pancreatobiliary cancer: A nontargeted metabolomics pilot study. Sci. Rep. 2019, 9, 16820. [Google Scholar] [CrossRef]
- Oliveira, A.G.; Gomes-Marcondes, M.C.C. Metformin treatment modulates the tumour-induced wasting effects in muscle protein metabolism minimising the cachexia in tumour-bearing rats. BMC Cancer 2016, 16, 41. [Google Scholar] [CrossRef]
- Pigna, E.; Berardi, E.; Aulino, P.; Rizzuto, E.; Zampieri, S.; Carraro, U.; Kern, H.; Merigliano, S.; Gruppo, M.; Mericskay, M.; et al. Aerobic Exercise and Pharmacological Treatments Counteract Cachexia by Modulating Autophagy in Colon Cancer. Sci. Rep. 2016, 6, 26991. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchesi, F.; Vignali, D.; Manini, B.; Rigamonti, A.; Monti, P. Manipulation of Glucose Availability to Boost Cancer Immunotherapies. Cancers 2020, 12, 2940. https://doi.org/10.3390/cancers12102940
Marchesi F, Vignali D, Manini B, Rigamonti A, Monti P. Manipulation of Glucose Availability to Boost Cancer Immunotherapies. Cancers. 2020; 12(10):2940. https://doi.org/10.3390/cancers12102940
Chicago/Turabian StyleMarchesi, Federica, Debora Vignali, Beatrice Manini, Alessandra Rigamonti, and Paolo Monti. 2020. "Manipulation of Glucose Availability to Boost Cancer Immunotherapies" Cancers 12, no. 10: 2940. https://doi.org/10.3390/cancers12102940
APA StyleMarchesi, F., Vignali, D., Manini, B., Rigamonti, A., & Monti, P. (2020). Manipulation of Glucose Availability to Boost Cancer Immunotherapies. Cancers, 12(10), 2940. https://doi.org/10.3390/cancers12102940