Extracellular Vesicle Transfer from Endothelial Cells Drives VE-Cadherin Expression in Breast Cancer Cells, Thereby Causing Heterotypic Cell Contacts

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
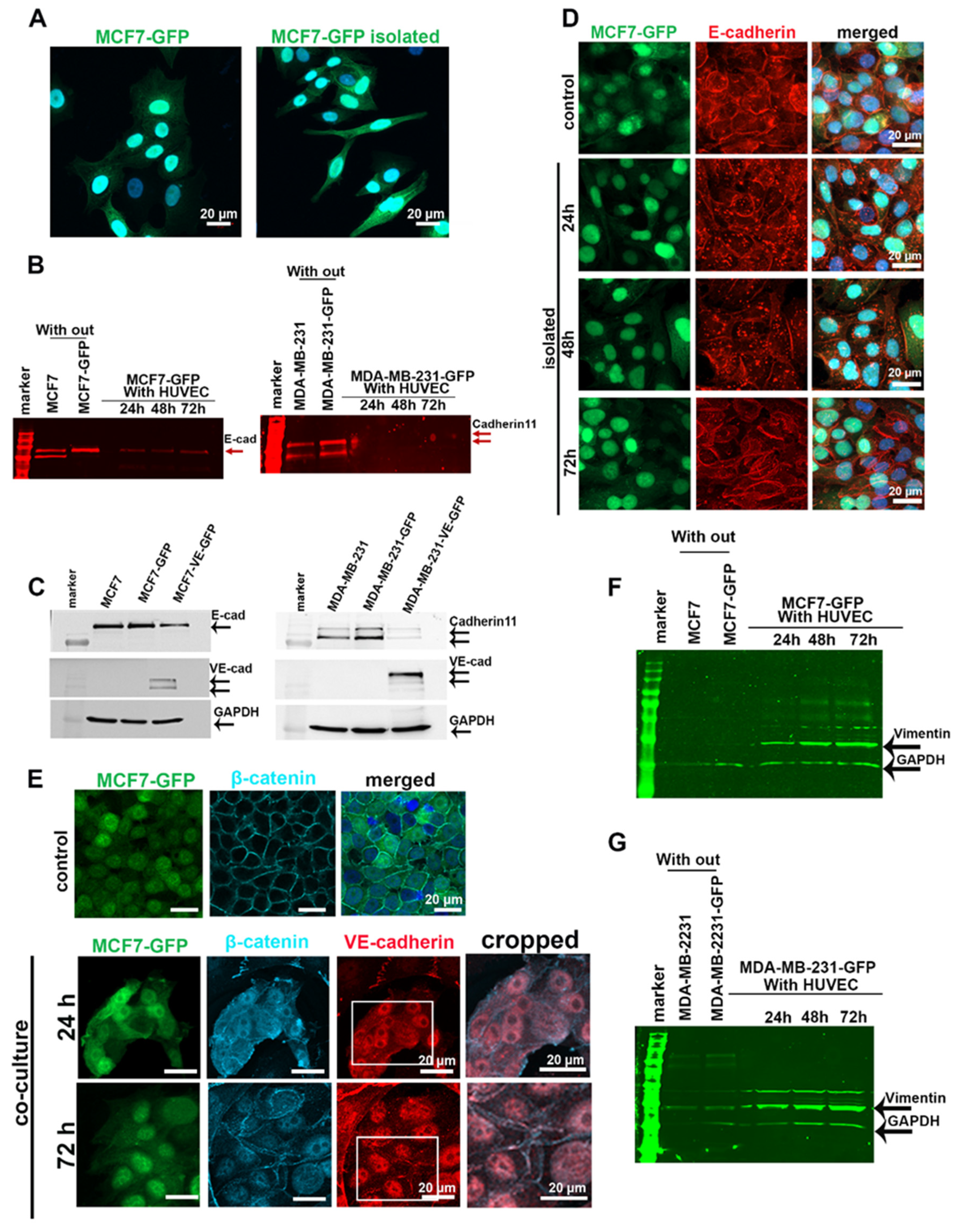
2.1. Expression of VE-Cadherin, VEGFRI, and VEGFRII Is Induced in Breast Cancer Cells during Co-Culture with Endothelial Cells
2.2. EC-Induced VE-Cadherin Expression Also Occurred in Breast Cancer Cells In Vivo
2.3. EC-Induced VE-Cadherin Expression in Tumor Cells Resulted in Downregulation of Endogenous Cadherins and Dislocation of β-Catenin from Cell Junctions
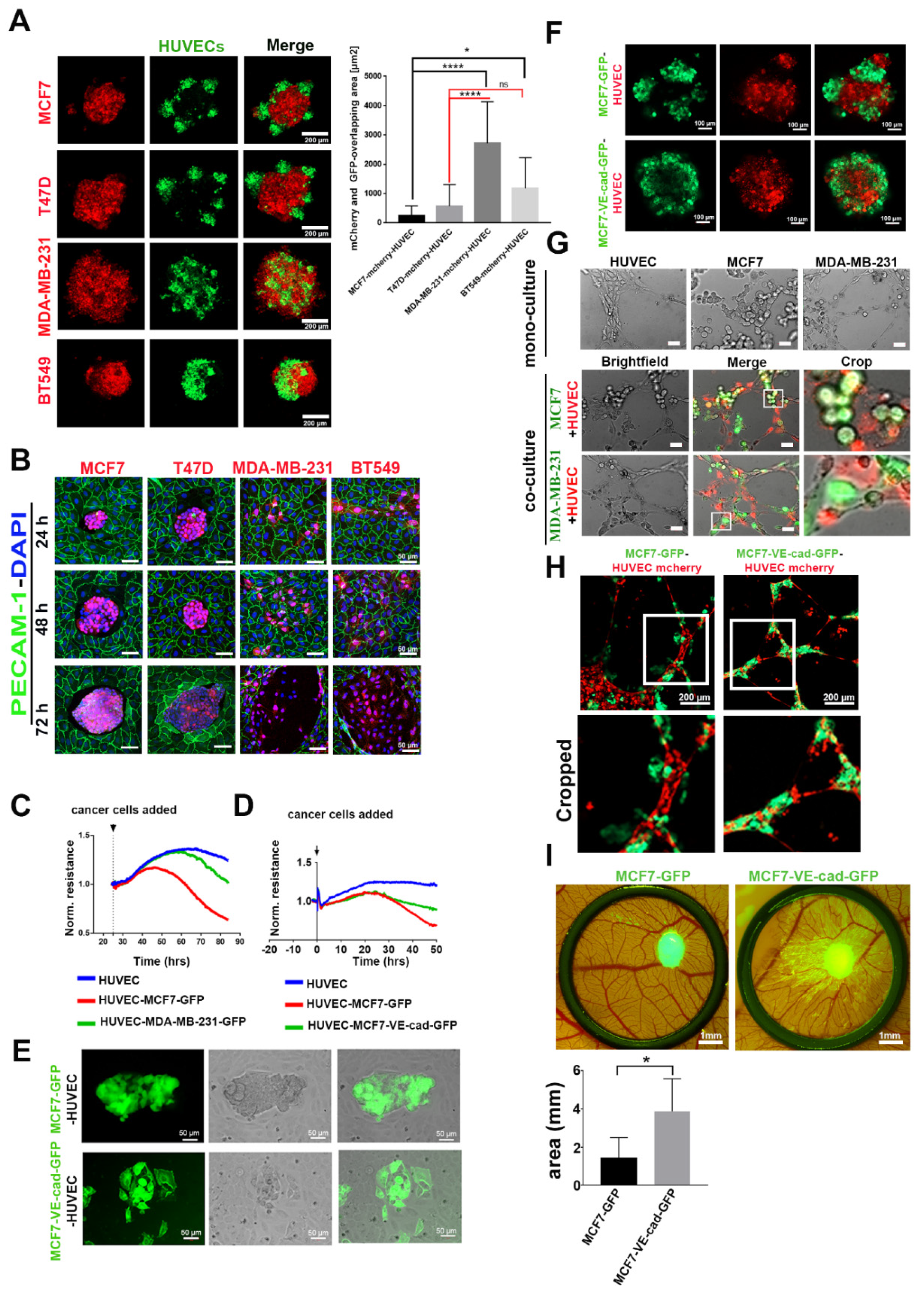
2.4. VE-Cadherin Expression in Breast Cancer Cells Promoted their Association with ECs and Integration into Endothelial Tubes
2.5. Extracellular Vesicles (EVs) Were Involved in the Mechanism of EC-Induced VE-Cadherin Expression in Breast Cancer Cells
2.6. Cancer Cells Phagocytosed Extracellular Vesicles from ECs
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. PiggyBac Transposon-Based Reporter Expression in MCF7 Cells
4.3. Lentiviral Transduction of Target Cells and Generation of Stable Cell Lines
4.4. Analysis of Cell–Cell Interaction at Single-Cell Resolution in a 3D Environment
4.5. Homo- and Heterospheroid Formation
4.6. Chick Chorioallantoic Membrane (CAM) Assay with MCF7 and MDA-MB-231 Cells
4.7. Immunofluorescence Staining of Cells and Frozen Tumor Sections
4.8. Immunoblot Analysis
4.9. RNA Isolation and Reverse Transcription PCR Analysis
4.10. Electric Cell-Substrate Impedance Sensing
4.11. Endothelial Tube Formation
4.12. Flow Cytometry
4.13. Apoptosis Assays
4.14. Exosome Isolation from Cell Culture Medium (CCM)
4.15. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CAM | chick chorioallantoic membrane |
| GFP | green fluorescent protein |
| HUVECs | human umbilical vein endothelial cells |
| ECs | endothelial cells |
| ECM | extracellular matrix |
| EMT | epithelial-to-mesenchymal transition |
| EVs | extracellular vesicles |
| FACS | fluorescence-activated cell sorting |
| sVE-cadherin | soluble VE-cadherin |
| TCs | tumor cells |
| TER | transendothelial electrical resistance |
| TME | tumor microenvironment |
| VE-cadherin | vascular endothelial cadherin |
| VEGFR | vascular endothelial growth factor receptor |
References
- Leckband, D.; Prakasam, A. Mechanism and dynamics of cadherin adhesion. Annu. Rev. Biomed. Eng. 2006, 8, 259–287. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E.; Orsenigo, F.; Lampugnani, M.G. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 2008, 121, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Yap, A.S.; Kovacs, E.M. Direct cadherin-activated cell signaling: A view from the plasma membrane. J. Cell Biol. 2003, 160, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; Taylor, M.R.; Williams, M.E. The classic cadherins in synaptic specificity. Cell Adh. Migr. 2015, 9, 193–201. [Google Scholar] [CrossRef]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Cardiff, R.D. Epithelial to Mesenchymal Transition Tumors: Fallacious or Snail’s Pace? Clin. Cancer Res. 2005, 11, 8534–8537. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef]
- Perl, A.K.; Wilgenbus, P.; Dahl, U.; Semb, H.; Christofori, G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 1998, 392, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Christofori, G.; Semb, H. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends Biochem. Sci. 1999, 24, 73–76. [Google Scholar] [CrossRef]
- Gilles, C.; Polette, M.; Mestdagt, M.; Nawrocki-Raby, B.; Ruggeri, P.; Birembaut, P.; Foidart, J.M. Transactivation of vimentin by b-catenin in human breast cancer cells. Cancer Res. 2003, 63, 2658–2664. [Google Scholar] [CrossRef] [PubMed]
- Auersperg, N.; Pan, J.; Grove, B.D.; Peterson, T.; Fisher, J.; Maines-Bandiera, S.; Somasiri, A.; Roskelley, C.D. E-cadherin induces mesenchymal-to-epithelial transition in human ovarian surface epithelium. Proc. Natl. Acad. Sci. USA 1999, 96, 6249–6254. [Google Scholar] [CrossRef] [PubMed]
- Hay, E.D.; Zuk, A. Transformations between epithelium and mesenchyme: Normal, pathological, and experimentally induced. Am. J. Kidney Dis. 1995, 26, 678–690. [Google Scholar] [CrossRef]
- Vleminckx, K.; Vakaet, L., Jr.; Mareel, M.; Fiers, W.; van Roy, F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell 1991, 66, 107–119. [Google Scholar] [CrossRef]
- Takeichi, M. Cadherins in cancer: Implications for invasion and metastasis. Curr. Opin. Cell Biol. 1993, 5, 806–811. [Google Scholar] [CrossRef]
- Nieman, M.T.; Prudoff, R.S.; Johnson, K.R.; Wheelock, M.J. N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J. Cell Biol. 1999, 147, 631–644. [Google Scholar] [CrossRef]
- Vestweber, D. VE-cadherin: The major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arter. Thromb. Vasc. Biol. 2008, 28, 223–232. [Google Scholar] [CrossRef]
- Rezaei, M.; Cao, J.; Friedrich, K.; Kemper, B.; Brendel, O.; Grosser, M.; Adrian, M.; Baretton, G.; Breier, G.; Schnittler, H.J. The expression of VE-cadherin in breast cancer cells modulates cell dynamics as a function of tumor differentiation and promotes tumor-endothelial cell interactions. Histochem. Cell Biol. 2018, 149, 15–30. [Google Scholar] [CrossRef]
- Labelle, M.; Schnittler, H.J.; Aust, D.E.; Friedrich, K.; Baretton, G.; Vestweber, D.; Breier, G. Vascular endothelial cadherin promotes breast cancer progression via transforming growth factor b signaling. Cancer Res. 2008, 68, 1388–1397. [Google Scholar] [CrossRef]
- Cavaco, A.; Rezaei, M.; Niland, S.; Eble, J.A. Collateral Damage Intended-Cancer-Associated Fibroblasts and Vasculature Are Potential Targets in Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 2355. [Google Scholar] [CrossRef]
- Eble, J.A.; Niland, S. The extracellular matrix in tumor progression and metastasis. Clin. Exp. Metastasis 2019, 36, 171–198. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Yoshioka, Y.; Prieto-Vila, M.; Ochiya, T. Involvement of Extracellular Vesicles in Vascular-Related Functions in Cancer Progression and Metastasis. Int. J. Mol. Sci. 2019, 20, 2584. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yuan, X.; Shi, H.; Wu, L.; Qian, H.; Xu, W. Exosomes in cancer: Small particle, big player. J. Hematol. Oncol. 2015, 8, 83. [Google Scholar] [CrossRef]
- Feng, D.; Zhao, W.L.; Ye, Y.Y.; Bai, X.C.; Liu, R.Q.; Chang, L.F.; Zhou, Q.; Sui, S.F. Cellular internalization of exosomes occurs through phagocytosis. Traffic 2010, 11, 675–687. [Google Scholar] [CrossRef]
- Aga, M.; Bentz, G.L.; Raffa, S.; Torrisi, M.R.; Kondo, S.; Wakisaka, N.; Yoshizaki, T.; Pagano, J.S.; Shackelford, J. Exosomal HIF1alpha supports invasive potential of nasopharyngeal carcinoma-associated LMP1-positive exosomes. Oncogene 2014, 33, 4613–4622. [Google Scholar] [CrossRef]
- Maji, S.; Chaudhary, P.; Akopova, I.; Nguyen, P.M.; Hare, R.J.; Gryczynski, I.; Vishwanatha, J.K. Exosomal Annexin II Promotes Angiogenesis and Breast Cancer Metastasis. Mol. Cancer Res. 2017, 15, 93–105. [Google Scholar] [CrossRef]
- Zeng, Y.; Yao, X.; Liu, X.; He, X.; Li, L.; Liu, X.; Yan, Z.; Wu, J.; Fu, B.M. Anti-angiogenesis triggers exosomes release from endothelial cells to promote tumor vasculogenesis. J. Extracell. Vesicles 2019, 8, 1629865. [Google Scholar] [CrossRef]
- Maishi, N.; Annan, D.A.; Kikuchi, H.; Hida, Y.; Hida, K. Tumor Endothelial Heterogeneity in Cancer Progression. Cancers (Basel) 2019, 11, 1511. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Chang, Y.S.; di Tomaso, E.; McDonald, D.M.; Jones, R.; Jain, R.K.; Munn, L.L. Mosaic blood vessels in tumors: Frequency of cancer cells in contact with flowing blood. Proc. Natl. Acad. Sci. USA 2000, 97, 14608–14613. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.; Zheng, G. E-cadherin/b-catenin complex and the epithelial barrier. J. Biomed. Biotechnol. 2011, 2011, 567305. [Google Scholar] [CrossRef] [PubMed]
- Ramos, T.L.; Sanchez-Abarca, L.I.; Muntion, S.; Preciado, S.; Puig, N.; Lopez-Ruano, G.; Hernandez, A.; Redondo, A.; Ortega, R.; Rodriguez, C.; et al. MSC surface markers (CD44, CD73, and CD90) can identify human MSC-derived extracellular vesicles by conventional flow cytometry. Cell Commun. Signal. 2016, 14, 2. [Google Scholar] [CrossRef] [PubMed]
- Viaud, M.; Ivanov, S.; Vujic, N.; Duta-Mare, M.; Aira, L.E.; Barouillet, T.; Garcia, E.; Orange, F.; Dugail, I.; Hainault, I.; et al. Lysosomal Cholesterol Hydrolysis Couples Efferocytosis to Anti-Inflammatory Oxysterol Production. Circ. Res. 2018, 122, 1369–1384. [Google Scholar] [CrossRef] [PubMed]
- Serizier, S.B.; McCall, K. Scrambled Eggs: Apoptotic Cell Clearance by Non-Professional Phagocytes in the Drosophila Ovary. Front. Immunol. 2017, 8, 1642. [Google Scholar] [CrossRef]
- Fornetti, J.; Flanders, K.C.; Henson, P.M.; Tan, A.C.; Borges, V.F.; Schedin, P. Mammary epithelial cell phagocytosis downstream of TGF-b3 is characterized by adherens junction reorganization. Cell Death Differ. 2016, 23, 185–196. [Google Scholar] [CrossRef]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef]
- Navarro, P.; Ruco, L.; Dejana, E. Differential localization of VE- and N-cadherins in human endothelial cells: VE-cadherin competes with N-cadherin for junctional localization. J. Cell Biol. 1998, 140, 1475–1484. [Google Scholar] [CrossRef]
- Sigurdsson, V.; Hilmarsdottir, B.; Sigmundsdottir, H.; Fridriksdottir, A.J.; Ringner, M.; Villadsen, R.; Borg, A.; Agnarsson, B.A.; Petersen, O.W.; Magnusson, M.K.; et al. Endothelial induced EMT in breast epithelial cells with stem cell properties. PLoS ONE 2011, 6, e23833. [Google Scholar] [CrossRef]
- Maeda, M.; Johnson, E.; Mandal, S.H.; Lawson, K.R.; Keim, S.A.; Svoboda, R.A.; Caplan, S.; Wahl, J.K., 3rd; Wheelock, M.J.; Johnson, K.R. Expression of inappropriate cadherins by epithelial tumor cells promotes endocytosis and degradation of E-cadherin via competition for p120(ctn). Oncogene 2006, 25, 4595–4604. [Google Scholar] [CrossRef] [PubMed]
- Brasch, J.; Katsamba, P.S.; Harrison, O.J.; Ahlsen, G.; Troyanovsky, R.B.; Indra, I.; Kaczynska, A.; Kaeser, B.; Troyanovsky, S.; Honig, B.; et al. Homophilic and Heterophilic Interactions of Type II Cadherins Identify Specificity Groups Underlying Cell-Adhesive Behavior. Cell Rep. 2018, 23, 1840–1852. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Bellido, D.; Serrano-Saenz, S.; Fernandez-Cortes, M.; Oliver, F.J. Vasculogenic mimicry signaling revisited: Focus on non-vascular VE-cadherin. Mol. Cancer 2017, 16, 65. [Google Scholar] [CrossRef] [PubMed]
- O’Loghlen, A. Role for extracellular vesicles in the tumour microenvironment. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20160488. [Google Scholar] [CrossRef]
- Margolis, L.; Sadovsky, Y. The biology of extracellular vesicles: The known unknowns. PLoS Biol. 2019, 17, e3000363. [Google Scholar] [CrossRef]
- Tong, Y.; Zhou, Y.L.; Wang, Y.X.; Zhao, P.Q.; Wang, Z.Y. Retinal pigment epithelium cell-derived exosomes: Possible relevance to CNV in wet-age related macular degeneration. Med. Hypotheses 2016, 97, 98–101. [Google Scholar] [CrossRef]
- Strilic, B.; Yang, L.; Albarran-Juarez, J.; Wachsmuth, L.; Han, K.; Muller, U.C.; Pasparakis, M.; Offermanns, S. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 2016, 536, 215–218. [Google Scholar] [CrossRef]
- Flemming, S.; Burkard, N.; Renschler, M.; Vielmuth, F.; Meir, M.; Schick, M.A.; Wunder, C.; Germer, C.T.; Spindler, V.; Waschke, J.; et al. Soluble VE-cadherin is involved in endothelial barrier breakdown in systemic inflammation and sepsis. Cardiovasc. Res. 2015, 107, 32–44. [Google Scholar] [CrossRef]
- Kleine-Bruggeney, H.; van Vliet, L.D.; Mulas, C.; Gielen, F.; Agley, C.C.; Silva, J.C.R.; Smith, A.; Chalut, K.; Hollfelder, F. Long-Term Perfusion Culture of Monoclonal Embryonic Stem Cells in 3D Hydrogel Beads for Continuous Optical Analysis of Differentiation. Small 2019, 15, e1804576. [Google Scholar] [CrossRef]
- Kleine-Brueggeney, H.; Zorzi, G.K.; Fecker, T.; El Gueddari, N.E.; Moerschbacher, B.M.; Goycoolea, F.M. A rational approach towards the design of chitosan-based nanoparticles obtained by ionotropic gelation. Colloids Surf. B Biointerfaces 2015, 135, 99–108. [Google Scholar] [CrossRef]
- Cavaco, A.C.M.; Rezaei, M.; Caliandro, M.F.; Lima, A.M.; Stehling, M.; Dhayat, S.A.; Haier, J.; Brakebusch, C.; Eble, J.A. The Interaction between Laminin-332 and α3β1 Integrin Determines Differentiation and Maintenance of CAFs, and Supports Invasion of Pancreatic Duct Adenocarcinoma Cells. Cancers (Basel) 2018, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, M.; Cavaco, A.C.; Seebach, J.; Niland, S.; Zimmermann, J.; Hanschmann, E.M.; Hallmann, R.; Schillers, H.; Eble, J.A. Signals of the Neuropilin-1-MET Axis and Cues of Mechanical Force Exertion Converge to Elicit Inflammatory Activation in Coherent Endothelial Cells. J. Immunol. 2019, 202, 1559–1572. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Lok, J.; Liu, Y.; Hayakawa, K.; Leung, W.; Xing, C.; Ji, X.; Lo, E.H. Assays to examine endothelial cell migration, tube formation, and gene expression profiles. Methods Mol. Biol. 2014, 1135, 393–402. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rezaei, M.; Martins Cavaco, A.C.; Stehling, M.; Nottebaum, A.; Brockhaus, K.; Caliandro, M.F.; Schelhaas, S.; Schmalbein, F.; Vestweber, D.; Eble, J.A. Extracellular Vesicle Transfer from Endothelial Cells Drives VE-Cadherin Expression in Breast Cancer Cells, Thereby Causing Heterotypic Cell Contacts. Cancers 2020, 12, 2138. https://doi.org/10.3390/cancers12082138
Rezaei M, Martins Cavaco AC, Stehling M, Nottebaum A, Brockhaus K, Caliandro MF, Schelhaas S, Schmalbein F, Vestweber D, Eble JA. Extracellular Vesicle Transfer from Endothelial Cells Drives VE-Cadherin Expression in Breast Cancer Cells, Thereby Causing Heterotypic Cell Contacts. Cancers. 2020; 12(8):2138. https://doi.org/10.3390/cancers12082138
Chicago/Turabian StyleRezaei, Maryam, Ana C. Martins Cavaco, Martin Stehling, Astrid Nottebaum, Katrin Brockhaus, Michele F. Caliandro, Sonja Schelhaas, Felix Schmalbein, Dietmar Vestweber, and Johannes A. Eble. 2020. "Extracellular Vesicle Transfer from Endothelial Cells Drives VE-Cadherin Expression in Breast Cancer Cells, Thereby Causing Heterotypic Cell Contacts" Cancers 12, no. 8: 2138. https://doi.org/10.3390/cancers12082138
APA StyleRezaei, M., Martins Cavaco, A. C., Stehling, M., Nottebaum, A., Brockhaus, K., Caliandro, M. F., Schelhaas, S., Schmalbein, F., Vestweber, D., & Eble, J. A. (2020). Extracellular Vesicle Transfer from Endothelial Cells Drives VE-Cadherin Expression in Breast Cancer Cells, Thereby Causing Heterotypic Cell Contacts. Cancers, 12(8), 2138. https://doi.org/10.3390/cancers12082138