Molecular Biology of Osteosarcoma

 ,
,  , , , , and
, , , , and
Abstract
1. Introduction
2. Molecular Abnormalities in Pediatric Osteosarcoma
2.1. Hereditary Syndromes
2.1.1. Osteosarcoma in Li–Fraumeni syndrome—TP53
2.1.2. Retinoblastoma Syndrome
2.1.3. RECQ Disorders
2.1.4. Diamond–Blackfan Anemia
2.2. The Most Frequently Mutated Genes in Pediatric Osteosarcoma
2.2.1. TP53
2.2.2. RB
2.2.3. CDK
2.2.4. c-Myc
2.2.5. TGFB
3. Molecular Abnormalities in Adult Osteosarcoma
3.1. Gene Mutations and Potential Biomarkers
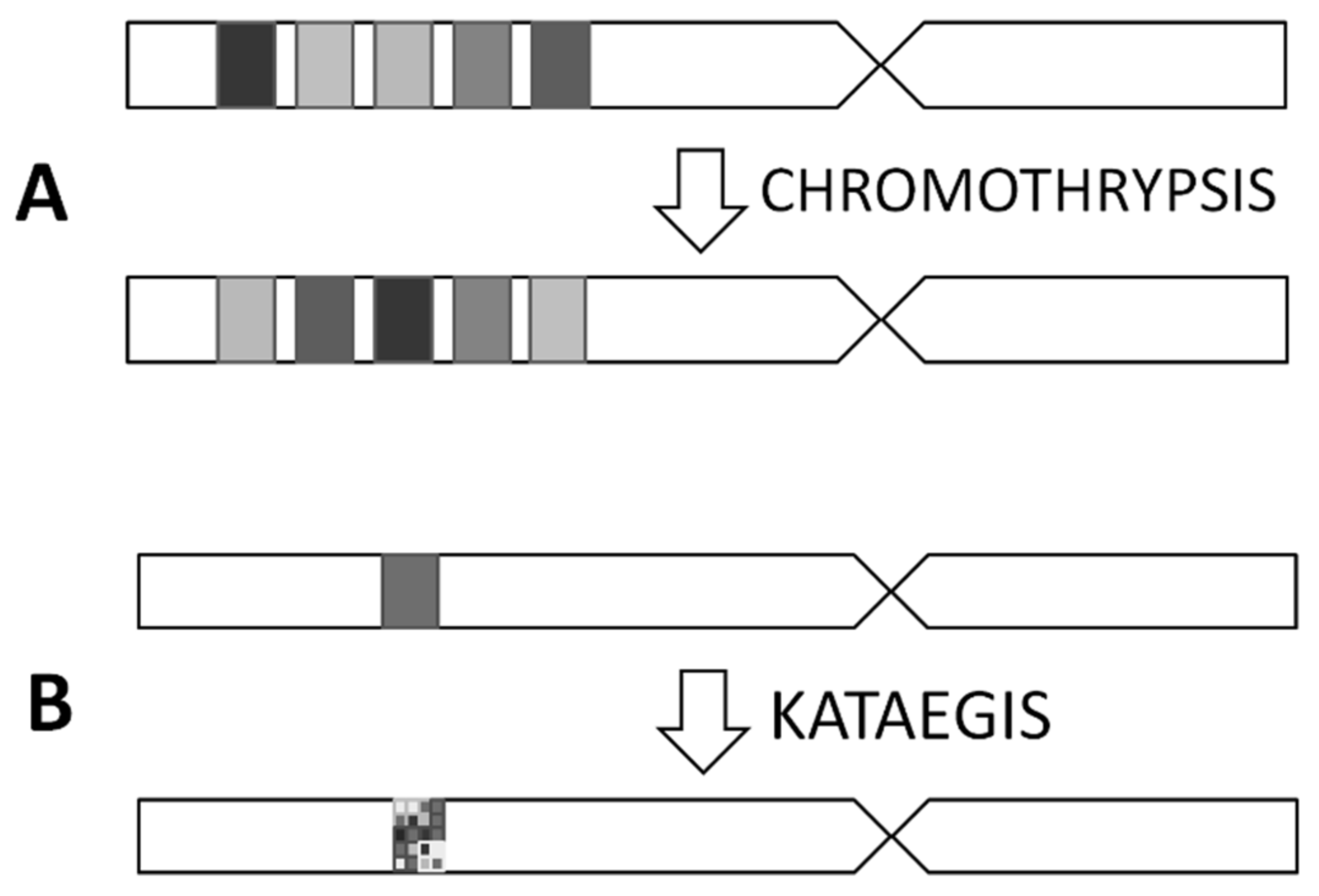
3.2. Chromothripsis and Kataegis
4. Small RNAs in Osteosarcoma
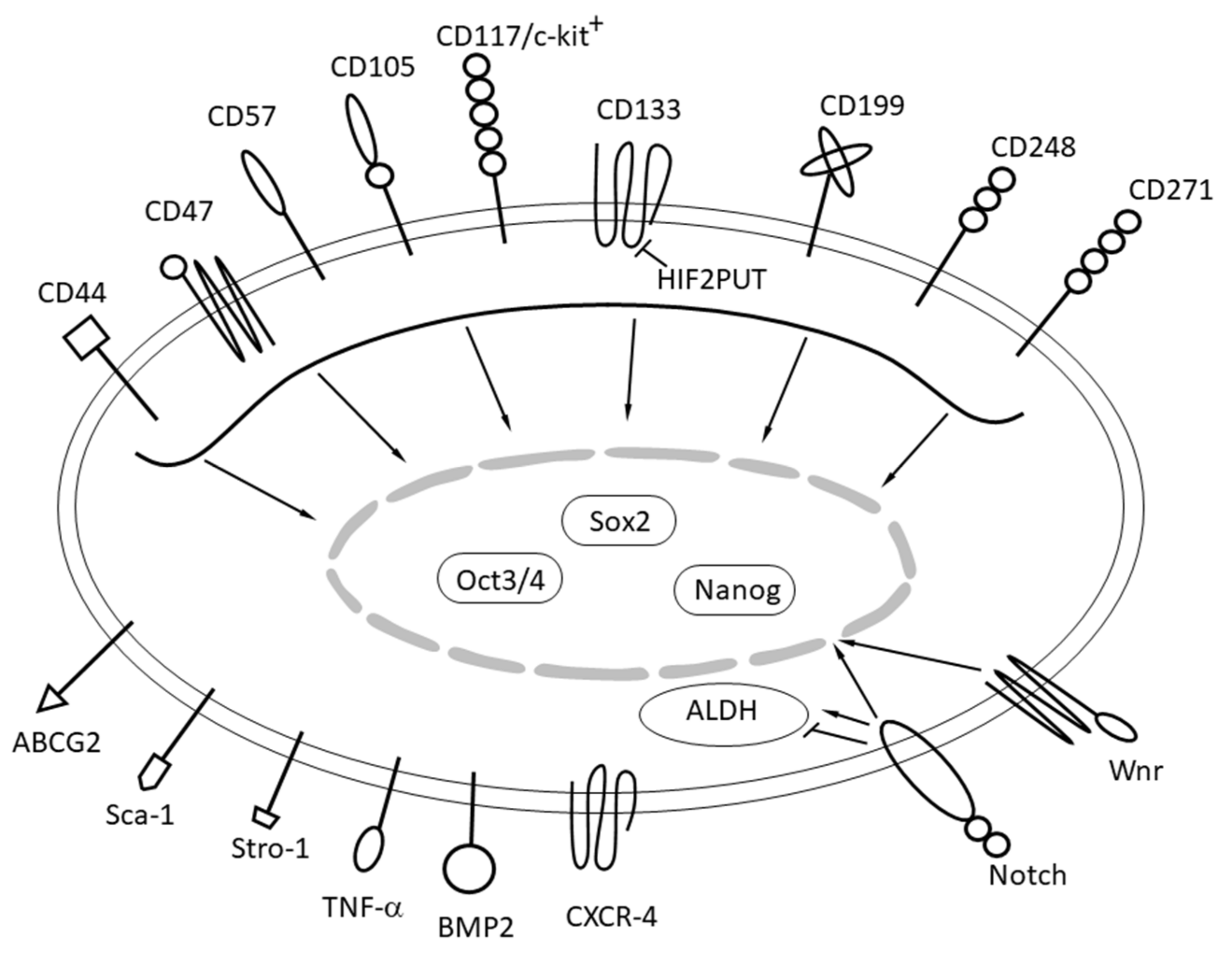
5. Role of Tumor Initiating Cells in Osteosarcoma
6. Role of Cell–Cell Interactions in Osteosarcoma
7. Molecular Signature of Osteosarcoma Metastases
8. Molecular Imaging and Theranostic Strategies for Diagnosis and Tracking of Treatment Efficacy in Osteosarcoma
9. Targeted Therapies for Osteosarcoma
9.1. Cabozantinib in Osteosarcoma
9.2. Pazopanib in Osteosarcoma
9.3. Sorafenib in Osteosarcoma
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Surveillance, Epidemiology, and End Results (SEER) Program, SEER*Stat Database: Incidence—SEER Research Data, 9 Registries, Nov 2019 Sub (1975–2017). Available online: https://seer.cancer.gov/ (accessed on 25 July 2020).
- Mirabello, L.; Troisi, R.J.; Savage, S.A. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. Int. J. Cancer 2009, 125, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Savage, S.A.; Mirabello, L. Using epidemiology and genomics to understand osteosarcoma etiology. Sarcoma 2011, 2011, 548151. [Google Scholar] [CrossRef] [PubMed]
- Rickel, K.; Fang, F.; Tao, J. Molecular genetics of osteosarcoma. Bone 2017, 102, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Scranton, P.E.J.; DeCicco, F.A.; Totten, R.S.; Yunis, E.J. Prognostic factors in osteosarcoma. A review of 20 year’s experience at the university of pittsburgh health center hospitals. Cancer 1975, 36, 2179–2191. [Google Scholar] [CrossRef] [PubMed]
- Fiedorowicz, M.; Bartnik, E.; Sobczuk, P.; Teterycz, P.; Czarnecka, A.M. Molecular biology of sarcoma. Oncol. Clin. Pr. 2018, 14, 307–330. [Google Scholar] [CrossRef]
- Huvos, A.G. Osteogenic sarcoma of bones and soft tissues in older persons. a clinicopathologic analysis of 117 patients older than 60 years. Cancer 1986, 57, 1442–1449. [Google Scholar] [CrossRef]
- Rani, A.S.; Kumar, S. Transformation of non-tumorigenic osteoblast-like human osteosarcoma cells by hexavalent chromates: Alteration of morphology, induction of anchorage-independence and proteolytic function. Carcinogenesis 1992, 13, 2021–2027. [Google Scholar] [CrossRef]
- Dutra, F.R.; Largent, E.J. Osteosarcoma induced by beryllium oxide. Am. J. Pathol. 1950, 26, 197–209. [Google Scholar]
- Mazabraud, A. Experimental production of bone sarcomas in the rabbit by a single local injection of beryllium. Bull. Cancer 1975, 62, 49–58. [Google Scholar]
- Casali, P.G.; Bielack, S.; Abecassis, N.; Aro, H.T.; Bauer, S.; Biagini, R.; Bonvalot, S.; Boukovinas, I.; Bovee, J.; Brennan, B.; et al. Bone sarcomas: ESMO-PaedCan-EURACAN clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv79–iv95. [Google Scholar] [CrossRef]
- Parkin, D.M.; Stiller, C.A.; Nectoux, J. International variations in the incidence of childhood bone tumours. Int. J. Cancer 1993, 53, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, B.A.; Markel, J.E.; Kleinerman, E.S. Osteosarcoma overview. Rheumatol 2017, 4, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Picci, P. Osteosarcoma (osteogenic sarcoma). Orphanet J. Rare Dis. 2007, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, J.; Zhao, N.; Wang, C.; Kamar, S.; Zhou, Y.; He, Z.; Yang, J.; Sun, B.; Shi, X.; et al. Progress in the chemotherapeutic treatment of osteosarcoma (Review). Oncol. Lett. 2018, 15, 6228–6237. [Google Scholar] [CrossRef]
- Lugowska, I.; Pienkowski, A.; Cieckiewicz, A.S.; Paterczyk, H.K.; Teterycz, P.; Glogowski, M.; Kozak, K.; Klimczak, A.; Falkowski, S.; Rutkowski, P. The long-term treatment outcomes of adult osteosarcoma. Pol. Merkur. Lek. 2017, 42, 158–164. [Google Scholar]
- Bousquet, M.; Noirot, C.; Accadbled, F.; Gauzy, J.D.S.; Castex, M.P.; Brousset, P.; Brouchet, A.G. Whole-exome sequencing in osteosarcoma reveals important heterogeneity of genetic alterations. Ann. Oncol. 2016, 27, 738–744. [Google Scholar] [CrossRef]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef]
- Ho, X.D.; Phung, P.; Le, V.Q.; Nguyen, V.H.; Reimann, E.; Prans, E.; Kõks, G.; Maasalu, K.; Le, N.T.; Trinh, L.H.; et al. Whole transcriptome analysis identifies differentially regulated networks between osteosarcoma and normal bone samples. Exp. Biol. Med. 2017, 242, 1802–1811. [Google Scholar] [CrossRef]
- Calvert, G.T.; Randall, R.L.; Jones, K.B.; Albright, L.C.; Lessnick, S.; Schiffman, J.D. At-risk populations for osteosarcoma: The syndromes and beyond. Sarcoma 2012, 2012, 152382. [Google Scholar] [CrossRef]
- Li, F.P.; Fraumeni, J.F.J. Soft-tissue sarcomas, breast cancer, and other neoplasms. a familial syndrome? Ann. Intern. Med. 1969, 71, 747–752. [Google Scholar] [CrossRef]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F.J.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A.; et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Zou, Z.Q.; Pirollo, K.; Blattner, W.; Chang, E.H. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature 1990, 348, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Li, F.P.; Fraumeni, J.F.J.; Mulvihill, J.J.; Blattner, W.A.; Dreyfus, M.G.; Tucker, M.A.; Miller, R.W. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988, 48, 5358–5362. [Google Scholar] [PubMed]
- Birch, J.M.; Hartley, A.L.; Tricker, K.J.; Prosser, J.; Condie, A.; Kelsey, A.M.; Harris, M.; Jones, P.H.; Binchy, A.; Crowther, D.; et al. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res. 1994, 54, 1298–1304. [Google Scholar] [PubMed]
- Chompret, A.; Abel, A.; Stoppa-Lyonnet, D.; Brugieres, L.; Pages, S.; Feunteun, J.; Bonaiti-Pellie, C. Sensitivity and predictive value of criteria for p53 germline mutation screening. J. Med. Genet. 2001, 38, 43–47. [Google Scholar] [CrossRef]
- Gianferante, D.M.; Mirabello, L.; Savage, S.A. Germline and somatic genetics of osteosarcoma—Connecting aetiology, biology and therapy. Nat. Rev. Endocrinol. 2017, 13, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Hameed, M.; Mandelker, D. Tumor syndromes predisposing to osteosarcoma. Adv. Anat. Pathol. 2018, 25, 217–222. [Google Scholar] [CrossRef]
- Mirabello, L.; Yeager, M.; Mai, P.L.; Gastier-Foster, J.M.; Gorlick, R.; Khanna, C.; Patino-Garcia, A.; Sierrasesumaga, L.; Lecanda, F.; Andrulis, I.L.; et al. Germline TP53 variants and susceptibility to osteosarcoma. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Gonzalez, K.D.; Noltner, K.A.; Buzin, C.H.; Gu, D.; Fong, C.Y.W.; Nguyen, V.Q.; Han, J.H.; Lowstuter, K.; Longmate, J.; Sommer, S.S.; et al. Beyond Li fraumeni syndrome: Clinical characteristics of families with p53 germline mutations. J. Clin. Oncol. 2009, 27, 1250–1256. [Google Scholar] [CrossRef]
- Bougeard, G.; Sesboue, R.; Desurmont, S.B.; Vasseur, S.; Martin, C.; Tinat, J.; Brugieres, L.; Chompret, A.; Paillerets, B.d.B.; Lyonnet, D.S.; et al. Molecular basis of the Li-Fraumeni syndrome: An update from the French LFS families. J. Med. Genet. 2008, 45, 535–538. [Google Scholar] [CrossRef]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Ribi, S.; Baumhoer, D.; Lee, K.; Edison, T.A.S.; Madan, B.; Zhang, K.; Kohlmann, W.K.; Yao, F.; Lee, W.H.; Hoi, Q.; et al. TP53 intron 1 hotspot rearrangements are specific to sporadic osteosarcoma and can cause Li-Fraumeni syndrome. Oncotarget 2015, 6, 7727–7740. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.W.; Varley, J.M.; Szydlo, T.E.; Kang, D.H.; Wahrer, D.C.; Shannon, K.E.; Lubratovich, M.; Verselis, S.J.; Isselbacher, K.J.; Fraumeni, J.F.; et al. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science 1999, 286, 2528–2531. [Google Scholar] [CrossRef]
- Giacomazzi, J.; Graudenz, M.S.; Osorio, C.A.; Santos, P.K.; Palmero, E.I.; Oliveira, M.Z.; Michelli, R.A.; Neto, C.S.; Fernandes, G.C.; Achatz, M.I.; et al. Prevalence of the TP53 p.R337H mutation in breast cancer patients in Brazil. PLoS ONE 2014, 9, e99893. [Google Scholar] [CrossRef]
- Mai, P.L.; Malkin, D.; Garber, J.E.; Schiffman, J.D.; Weitzel, J.N.; Strong, L.C.; Wyss, O.; Locke, L.; Means, V.; Achatz, M.I.; et al. Li-Fraumeni syndrome: Report of a clinical research workshop and creation of a research consortium. Cancer Genet. 2012, 205, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.L.; Boice, J.D.J.; Abramson, D.H.; Tarone, R.E.; Kleinerman, R.A.; Stovall, M.; Goldman, M.B.; Seddon, J.M.; Tarbell, N.; Fraumeni, J.F.J.; et al. Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA 1997, 278, 1262–1267. [Google Scholar] [CrossRef]
- Ottaviani, G.; Jaffe, N. The etiology of osteosarcoma. Cancer Treat. Res. 2009, 152, 15–32. [Google Scholar] [CrossRef]
- Imbert-Bouteille, M.; Gauthier-Villars, M.; Leroux, D.; Meunier, I.; Aerts, I.; Lumbroso-Le Rouic, L.; Lejeune, S.; Delnatte, C.; Abadie, C.; Pujol, P.; et al. Osteosarcoma without prior retinoblastoma related to RB1 low-penetrance germline pathogenic variants: A novel type of RB1-related hereditary predisposition syndrome? Mol. Genet. Genom. Med. 2019, 7, e913. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Gannavarapu, A.; Kozinetz, C.A.; Levy, M.L.; Lewis, R.A.; Chintagumpala, M.M.; Ruiz-Maldanado, R.; Ruiz, J.C.; Cunniff, C.; Erickson, R.P.; et al. Association between osteosarcoma and deleterious mutations in the RECQL4 gene in rothmund-thomson syndrome. J. Natl. Cancer Inst. 2003, 95, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Salih, A.; Inoue, S.; Onwuzurike, N. Rothmund-Thomson syndrome (RTS) with osteosarcoma due to RECQL4 mutation. BMJ Case Rep. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Stinco, G.; Governatori, G.; Mattighello, P.; Patrone, P. Multiple cutaneous neoplasms in a patient with rothmund-thomson syndrome: Case report and published work review. J. Derm. 2008, 35, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Siitonen, H.A.; Sotkasiira, J.; Biervliet, M.; Benmansour, A.; Capri, Y.; Daire, V.C.; Crandall, B.; Jouppi, K.; Hennekam, R.K.; Herzog, D.; et al. The mutation spectrum in RECQL4 diseases. Eur. J. Hum. Genet. 2009, 17, 151–158. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Miller, R.W.; Machinami, R.; Sugano, H.; Goto, M. Atypical osteosarcomas in werner syndrome (adult progeria). Jpn. J. Cancer Res. 2000, 91, 1345–1349. [Google Scholar] [CrossRef]
- Ellis, N.A.; Groden, J.; Ye, T.Z.; Straughen, J.; Lennon, D.J.; Ciocci, S.; Proytcheva, M.; German, J. The bloom’s syndrome gene product is homologous to RecQ helicases. Cell 1995, 83, 655–666. [Google Scholar] [CrossRef]
- Li, L.; Eng, C.; Desnick, R.J.; German, J.; Ellis, N.A. Carrier frequency of the bloom syndrome blmAsh mutation in the Ashkenazi Jewish population. Mol. Genet. Metab 1998, 64, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Vlachos, A.; Rosenberg, P.S.; Atsidaftos, E.; Alter, B.P.; Lipton, J.M. Incidence of neoplasia in Diamond Blackfan anemia: A report from the diamond blackfan anemia registry. Blood 2012, 119, 3815–3819. [Google Scholar] [CrossRef]
- Lee, R.S.; Higgs, D.; Haddo, O.; Pringle, J.; Briggs, T.W. Osteosarcoma associated with diamond-blackfan anaemia: A case of a child receiving growth hormone therapy. Sarcoma 2004, 8, 47–49. [Google Scholar] [CrossRef] [PubMed]
- Kleinerman, R.A.; Schonfeld, S.J.; Tucker, M.A. Sarcomas in hereditary retinoblastoma. Clin. Sarcoma Res. 2012, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.A.; Hart, M.L.; Busi, S.; Parker, T.; Goerndt, A.; Jones, K.; Landgraf, J.M.A.; Bryda, E.C. Fischer-344 Tp53-knockout rats exhibit a high rate of bone and brain neoplasia with frequent metastasis. Dis. Model. Mech. 2016, 9, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Bonizzi, G.; Cicalese, A.; Insinga, A.; Pelicci, P.G. The emerging role of p53 in stem cells. Trends Mol. Med. 2012, 18, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Pfeifer, G.P. Somatic TP53 mutations in the era of genome sequencing. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, K. Centrosome amplification, chromosome instability and cancer development. Cancer Lett. 2005, 230, 6–19. [Google Scholar] [CrossRef]
- Murphy, K.L.; Dennis, A.P.; Rosen, J.M. A gain of function p53 mutant promotes both genomic instability and cell survival in a novel p53-null mammary epithelial cell model. FASEB J. 2000, 14, 2291–2302. [Google Scholar] [CrossRef] [PubMed]
- Hizawi, S.E.; Lagowski, J.P.; Martin, M.K.; Albor, A. Induction of gene amplification as a gain-of-function phenotype of mutant p53 proteins. Cancer Res. 2002, 62, 3264–3270. [Google Scholar] [PubMed]
- Blandino, G.; Levine, A.J.; Oren, M. Mutant p53 gain of function: Differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 1999, 18, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Sayles, L.C.; Breese, M.R.; Koehne, A.L.; Leung, S.G.; Lee, A.G.; Liu, H.Y.; Spillinger, A.; Shah, A.T.; Tanasa, B.; Straessler, K.; et al. Genome-informed targeted therapy for osteosarcoma. Cancer Discov. 2019, 9, 46–63. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Miller, C.; Koeffler, H.P.; Battifora, H.; Cline, M.J. Rearrangement of the p53 gene in human osteogenic sarcomas. Proc. Natl. Acad. Sci. USA 1987, 84, 7716–7719. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.W.; Aslo, A.; Tsay, C.; Slamon, D.; Ishizaki, K.; Toguchida, J.; Yamamuro, T.; Lampkin, B.; Koeffler, H.P. Frequency and structure of p53 rearrangements in human osteosarcoma. Cancer Res. 1990, 50, 7950–7954. [Google Scholar]
- Chandar, N.; Billig, B.; McMaster, J.; Novak, J. Inactivation of p53 gene in human and murine osteosarcoma cells. Br. J. Cancer 1992, 65, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Gokgoz, N.; Wunder, J.S.; Mousses, S.; Eskandarian, S.; Bell, R.S.; Andrulis, I.L. Comparison of p53 mutations in patients with localized osteosarcoma and metastatic osteosarcoma. Cancer 2001, 92, 2181–2189. [Google Scholar] [CrossRef]
- Overholtzer, M.; Rao, P.H.; Favis, R.; Lu, X.Y.; Elowitz, M.B.; Barany, F.; Ladanyi, M.; Gorlick, R.; Levine, A.J. The presence of p53 mutations in human osteosarcomas correlates with high levels of genomic instability. Proc. Natl. Acad. Sci. USA 2003, 100, 11547–11552. [Google Scholar] [CrossRef]
- Lozano, G. Mouse models of p53 functions. Cold Spring Harb. Perspect. Biol. 2010, 2, a001115. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T.; Wiman, K.G. TP53: An oncogene in disguise. Cell Death Differ. 2015, 22, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.; Williford, C.L.B.; Franklin, C.L.; Weinstein, E.J.; Cui, X. Creation and preliminary characterization of a Tp53 knockout rat. Dis. Model. Mech. 2013, 6, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Boxtel, R.V.; Kuiper, R.V.; Toonen, P.W.; Heesch, S.V.; Hermsen, R.; Bruin, A.D.; Cuppen, E. Homozygous and heterozygous p53 knockout rats develop metastasizing sarcomas with high frequency. Am. J. Pathol. 2011, 179, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Smyczyńska, U.; Strzemecki, D.; Czarnecka, A.M.; Fendler, W.; Fiedorowicz, M.; Kamińska, M.W.; Guzowska, M.; Synoradzki, K.; Cheda, Ł.; Rogulski, Z.; et al. TP53-deficient angiosarcoma expression profiling in rat model. Cancers 2020, 12, 1525. [Google Scholar] [CrossRef] [PubMed]
- Ragland, B.D.; Bell, W.C.; Lopez, R.R.; Siegal, G.P. Cytogenetics and molecular biology of osteosarcoma. Lab. Investig. 2002, 82, 365–373. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Feugeas, O.; Guriec, N.; Boilletot, A.B.; Marcellin, L.; Simon, P.; Babin, S.; Thyss, A.; Hofman, P.; Terrier, P.; Kalifa, C.; et al. Loss of heterozygosity of the RB gene is a poor prognostic factor in patients with osteosarcoma. J. Clin. Oncol. 1996, 14, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Capodano, A. Bone: Osteosarcoma. Atlas Genet. Cytogenet. Oncol. Haematol. 2003, 7, 44–47. [Google Scholar] [CrossRef]
- Belchis, D.; Gocke, C.; Geradts, J. Alterations in the RB, p16, and Cyclin D1 cell cycle control pathway in osteosarcomas. Fetal Pediatr. Pathol. 2000, 19, 377–389. [Google Scholar] [CrossRef]
- Walkley, C.R.; Qudsi, R.; Sankaran, V.G.; Perry, J.A.; Gostissa, M.; Roth, S.I.; Rodda, S.J.; Snay, E.; Dunning, P.; Fahey, F.H.; et al. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev. 2008, 22, 1662–1676. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, J.W.V.D.; Fernandes, T.A.A.M.; Fernandes, J.V.J.; Azevedo, J.C.V.D.; Lanza, D.C.F.; Bezerra, C.M.; Andrade, V.S.; Araujo, J.M.G.D.; Fernandes, J.V. Biology and pathogenesis of human osteosarcoma. Oncol. Lett. 2020, 19, 1099–1116. [Google Scholar] [CrossRef] [PubMed]
- Kohlmeyer, J.L.; Gordon, D.J.; Tanas, M.R.; Monga, V.; Dodd, R.D.; Quelle, D.E. CDKs in sarcoma: Mediators of disease and emerging therapeutic targets. Int. J. Mol. Sci. 2020, 21, 3018. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Shen, J.K.; Yu, Z.; Hornicek, F.J.; Kan, Q.; Duan, Z. Expression and therapeutic implications of cyclin-dependent kinase 4 (CDK4) in osteosarcoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1573–1582. [Google Scholar] [CrossRef]
- Ueda, T.; Healey, J.H.; Huvos, A.G.; Ladanyi, M. Amplification of the MYC gene in osteosarcoma secondary to paget’s disease of bone. Sarcoma 1997, 1, 131–134. [Google Scholar] [CrossRef]
- Han, G.; Wang, Y.; Bi, W. C-Myc overexpression promotes osteosarcoma cell invasion via activation of MEK-ERK pathway. Oncol. Res. 2012, 20, 149–156. [Google Scholar] [CrossRef]
- Chen, D.; Zhao, Z.; Huang, Z.; Chen, D.-C.; Zhu, X.-X.; Wang, Y.-Z.; Yan, Y.-W.; Tang, S.; Madhavan, S.; Ni, W.; et al. Super enhancer inhibitors suppress MYC driven transcriptional amplification and tumor progression in osteosarcoma. Bone Res. 2018, 6, 11. [Google Scholar] [CrossRef]
- Lamora, A.; Talbot, J.; Mullard, M.; Royer, B.B.-L.; Redini, F.; Verrecchia, F. TGF-β Signaling in bone remodeling and osteosarcoma progression. J. Clin. Med. 2016, 5, 96. [Google Scholar] [CrossRef]
- Roberts, A.B.; Wakefield, L.M. The two faces of transforming growth factor beta in carcinogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 8621–8623. [Google Scholar] [CrossRef]
- Huang, J.J.; Blobe, G.C. Dichotomous roles of TGF-β in human cancer. Biochem. Soc. Trans. 2016, 44, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Verrecchia, F.; Rédini, F. Transforming growth Factor-β signaling plays a pivotal role in the interplay between osteosarcoma cells and their microenvironment. Front. Oncol. 2018, 8, 133. [Google Scholar] [CrossRef] [PubMed]
- Barris, D.M.; Weiner, S.B.; Dubin, R.A.; Fremed, M.; Zhang, X.; Piperdi, S.; Zhang, W.; Maqbool, S.; Gill, J.; Roth, M.; et al. Detection of circulating tumor DNA in patients with osteosarcoma. Oncotarget 2018, 9, 12695–12704. [Google Scholar] [CrossRef] [PubMed]
- Kovac, M.; Blattmann, C.; Ribi, S.; Smida, J.; Mueller, N.S.; Engert, F.; Giner, F.C.; Weischenfeldt, J.; Kovacova, M.; Krieg, A.; et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat. Commun. 2015, 6, 8940. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S.; Haase, K.; Ye, H.; Young, M.D.; Alexandrov, L.B.; Farndon, S.J.; Collord, G.; Wedge, D.C.; Martincorena, I.; et al. Recurrent mutation of IGF signalling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nat. Commun. 2017, 8, 15936. [Google Scholar] [CrossRef] [PubMed]
- Joseph, C.G.; Hwang, H.; Jiao, Y.; Wood, L.D.; Kinde, I.; Wu, J.; Mandahl, N.; Luo, J.; Hruban, R.H.; Diaz, L.A.J.; et al. Exomic analysis of myxoid liposarcomas, synovial sarcomas, and osteosarcomas. Genes Chromosom. Cancer 2014, 53, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Jiang, M.M.; Jiang, L.; Salvo, J.S.; Zeng, H.C.; Dawson, B.; Bertin, T.K.; Rao, P.H.; Chen, R.; Donehower, L.A.; et al. Notch activation as a driver of osteogenic sarcoma. Cancer Cell 2014, 26, 390–401. [Google Scholar] [CrossRef]
- Kansara, M.; Tsang, M.; Kodjabachian, L.; Sims, N.A.; Trivett, M.K.; Ehrich, M.; Dobrovic, A.; Slavin, J.; Choong, P.F.; Simmons, P.J.; et al. Wnt inhibitory factor 1 is epigenetically silenced in human osteosarcoma, and targeted disruption accelerates osteosarcomagenesis in mice. J. Clin. Invest. 2009, 119, 837–851. [Google Scholar] [CrossRef]
- McAllister, K.A.; Houle, C.D.; Malphurs, J.; Ward, T.; Collins, N.K.; Gersch, W.; Wharey, L.; Seely, J.C.; Betz, L.; Bennett, L.M.; et al. Spontaneous and irradiation-induced tumor susceptibility in BRCA2 germline mutant mice and cooperative effects with a p53 germline mutation. Toxicol. Pathol. 2006, 34, 187–198. [Google Scholar] [CrossRef]
- Ribeiro, C.J.; Rodrigues, C.M.; Moreira, R.; Santos, M.M. Chemical variations on the p53 reactivation theme. Pharmaceuticals 2016, 9, 25. [Google Scholar] [CrossRef]
- Gemoll, T.; Epping, F.; Heinrich, L.; Fritzsche, B.; Roblick, U.J.; Szymczak, S.; Hartwig, S.; Depping, R.; Bruch, H.P.; Thorns, C.; et al. Increased cathepsin D protein expression is a biomarker for osteosarcomas, pulmonary metastases and other bone malignancies. Oncotarget 2015, 6, 16517–16526. [Google Scholar] [CrossRef]
- Li, Z.; Xiao, J.; Hu, K.; Wang, G.; Li, M.; Zhang, J.; Cheng, G. FBXW7 acts as an independent prognostic marker and inhibits tumor growth in human osteosarcoma. Int. J. Mol. Sci. 2015, 16, 2294–2306. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, B.; Hu, J.; Zhou, Y.; Jiang, M.; Wu, M.; Qin, L.; Yang, X. miR-421 is a diagnostic and prognostic marker in patients with osteosarcoma. Tumour. Biol. 2016, 37, 9001–9007. [Google Scholar] [CrossRef] [PubMed]
- Zandueta, C.; Ormazabal, C.; Perurena, N.; Canarias, S.M.; Zalacain, M.; Julian, M.S.; Grigoriadis, A.E.; Valencia, K.; Laborie, F.J.C.; Rivas, J.L.; et al. Matrix-Gla protein promotes osteosarcoma lung metastasis and associates with poor prognosis. J. Pathol. 2016, 239, 438–449. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhang, P.; Li, Q.; Zhou, D.; Liu, P. Expression of high mobility group box 1 protein predicts a poorer prognosis for patients with osteosarcoma. Oncol. Lett. 2016, 11, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Ciriano, I.C.; Lee, J.J.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.Z.; Pellman, D.S.; et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.A.; Kiezun, A.; Tonzi, P.; Allen, E.M.V.; Carter, S.L.; Baca, S.C.; Cowley, G.S.; Bhatt, A.S.; Rheinbay, E.; Pedamallu, C.S.; et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, 5564–5573. [Google Scholar] [CrossRef]
- Zainal, S.N.; Alexandrov, L.B.; Wedge, D.C.; Loo, P.V.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef]
- Evola, F.R.; Costarella, L.; Pavone, V.; Caff, G.; Cannavò, L.; Sessa, A.; Avondo, S.; Sessa, G. Biomarkers of osteosarcoma, chondrosarcoma, and ewing sarcoma. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef]
- Andersen, G.B.; Knudsen, A.; Hager, H.; Hansen, L.L.; Tost, J. MiRNA profiling identifies deregulated miRNAs associated with osteosarcoma development and time to metastasis in two large cohorts. Mol. Oncol. 2018, 12, 114–131. [Google Scholar] [CrossRef]
- Zhao, H.; Yan, P.; Wang, J.; Zhang, Y.; Zhang, M.; Wang, Z.; Fu, Q.; Liang, W. Clinical significance of tumor miR-21, miR-221, miR-143, and miR-106a as biomarkers in patients with osteosarcoma. Int. J. Biol. Markers 2019, 34, 184–193. [Google Scholar] [CrossRef]
- Ren, Z.; He, M.; Shen, T.; Wang, K.; Meng, Q.; Chen, X.; Zhou, L.; Han, Y.; Ji, C.; Liu, S.; et al. MiR-421 promotes the development of osteosarcoma by regulating MCPIP1 expression. Cancer Biol. 2020, 21, 231–240. [Google Scholar] [CrossRef]
- Huang, Y.Z.; Zhang, J.; Shao, H.Y.; Chen, J.P.; Zhao, H.Y. MicroRNA-191 promotes osteosarcoma cells proliferation by targeting checkpoint kinase 2. Tumour Biol. 2015, 36, 6095–6101. [Google Scholar] [CrossRef]
- Kushlinskii, N.E.; Fridman, M.V.; Braga, E.A. Molecular mechanisms and microRNAs in osteosarcoma pathogenesis. Biochemistry 2016, 81, 315–328. [Google Scholar] [CrossRef]
- Yan, K.; Gao, J.; Yang, T.; Ma, Q.; Qiu, X.; Fan, Q.; Ma, B. MicroRNA-34a inhibits the proliferation and metastasis of osteosarcoma cells both in vitro and in vivo. PLoS ONE 2012, 7, e33778. [Google Scholar] [CrossRef]
- Lian, F.; Cui, Y.; Zhou, C.; Gao, K.; Wu, L. Identification of a plasma four-microRNA panel as potential noninvasive biomarker for osteosarcoma. PLoS ONE 2015, 10, e0121499. [Google Scholar] [CrossRef]
- Zhang, Y.; Mai, Q.; Zhang, X.; Xie, C.; Zhang, Y. Microenvironment signals and mechanisms in the regulation of osteosarcoma. Osteosarcoma Biol. Behav. Mech. 2017. [Google Scholar] [CrossRef]
- Jones, K.B.; Salah, Z.; Mare, S.D.; Galasso, M.; Gaudio, E.; Nuovo, G.J.; Lovat, F.; LeBlanc, K.; Palatini, J.; Randall, R.L.; et al. MiRNA signatures associate with pathogenesis and progression of osteosarcoma. Cancer Res. 2012, 72, 1865–1877. [Google Scholar] [CrossRef]
- Liao, Y.-Y.; Tsai, H.-C.; Chou, P.-Y.; Wang, S.-W.; Chen, H.-T.; Lin, Y.-M.; Chiang, I.P.; Chang, T.-M.; Hsu, S.-K.; Chou, M.-C.; et al. CCL3 promotes angiogenesis by dysregulation of miR-374b/ VEGF-A axis in human osteosarcoma cells. Oncotarget 2016, 7, 4310–4325. [Google Scholar] [CrossRef]
- Sasaki, R.; Osaki, M.; Okada, F. MicroRNA-based diagnosis and treatment of metastatic human osteosarcoma. Cancers 2019, 11, 553. [Google Scholar] [CrossRef]
- Wang, L.H.; Tsai, H.C.; Cheng, Y.C.; Lin, C.Y.; Huang, Y.L.; Tsai, C.H.; Xu, G.H.; Wang, S.W.; Fong, Y.C.; Tang, C.H. CTGF promotes osteosarcoma angiogenesis by regulating miR-543/angiopoietin 2 signaling. Cancer Lett. 2017, 391, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Aurora, A.B.; Johnson, B.A.; Qi, X.; McAnally, J.; Hill, J.A.; Richardson, J.A.; Duby, R.B.; Olson, E.N. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell 2008, 15, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Lai, G.-H.; Huang, A.-L.; Zhao, Z.; Lu, X.-H.; Zu, W.-X. MicroRNA-218 promotes osteosarcoma cell apoptosis by down-regulating oncogene B lymphoma mouse moloney leukemia virus insertion region 1. J. South. Med Univ. 2018, 38, 505–510. [Google Scholar] [CrossRef]
- Xuan, C.; Jin, M.; Gao, Y.; Xu, S.; Wang, L.; Wang, Y.; Han, R.; An, Q. MiR-218 suppresses the proliferation of osteosarcoma through downregulation of E2F2. Oncol. Lett. 2019, 17, 571–577. [Google Scholar] [CrossRef]
- Lin, R.; Chen, L.; Chen, G.; Hu, C.; Jiang, S.; Sevilla, J.; Wan, Y.; Sampson, J.H.; Zhu, B.; Li, Q.-J. Targeting miR-23a in CD8+ cytotoxic T lymphocytes prevents tumor-dependent immunosuppression. J. Clin. Invest. 2014, 124, 5352–5367. [Google Scholar] [CrossRef]
- Carvalho, I.N.D.; Freitas, R.M.D.; Vargas, F.R. Translating microRNAs into biomarkers: What is new for pediatric cancer? Med. Oncol. 2016, 33, 49. [Google Scholar] [CrossRef]
- Maire, G.; Martin, J.W.; Yoshimoto, M.; MacNeill, S.C.; Zielenska, M.; Squire, J.A. Analysis of miRNA-gene expression-genomic profiles reveals complex mechanisms of microRNA deregulation in osteosarcoma. Cancer Genet. 2011, 204, 138–146. [Google Scholar] [CrossRef]
- Zhuang, M.; Qiu, X.; Cheng, D.; Zhu, C.; Chen, L. MicroRNA-524 promotes cell proliferation by down-regulating PTEN expression in osteosarcoma. Cancer Cell Int. 2018, 18, 114. [Google Scholar] [CrossRef]
- Meyerrose, T.E.; Herrbrich, P.; Hess, D.A.; Nolta, J.A. Immune-deficient mouse models for analysis of human stem cells. Biotechniques 2003, 35, 1262–1272. [Google Scholar] [CrossRef]
- Gedye, C.; Sirskyj, D.; Lobo, N.C.; Meens, J.; Hyatt, E.; Robinette, M.; Fleshner, N.; Hamilton, R.J.; Kulkarni, G.; Zlotta, A.; et al. Cancer stem cells are underestimated by standard experimental methods in clear cell renal cell carcinoma. Sci. Rep. 2016, 6, 25220. [Google Scholar] [CrossRef]
- Grotenhuis, B.A.; Wijnhoven, B.P.; van Lanschot, J.J. Cancer stem cells and their potential implications for the treatment of solid tumors. J. Surg. Oncol. 2012, 106, 209–215. [Google Scholar] [CrossRef]
- Buczek, M.; Escudier, B.; Bartnik, E.; Szczylik, C.; Czarnecka, A.M. Resistance to tyrosine kinase inhibitors in clear cell renal cell carcinoma: From the patient’s bed to molecular mechanisms. Biochim. Biophys. Acta 2014, 1845, 31–41. [Google Scholar] [CrossRef]
- Bielecka, Z.F.; Czarnecka, A.M.; Solarek, W.; Kornakiewicz, A.; Szczylik, C. Mechanisms of acquired resistance to tyrosine kinase inhibitors in clear-cell renal cell carcinoma (ccRCC). Curr. Signal. Transduct. Ther. 2014, 8, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Mohseny, A.B.; Hogendoorn, P.C. Concise review: Mesenchymal tumors: When stem cells go mad. Stem Cells 2011, 29, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Mohseny, A.B.; Hogendoorn, P.C.; Jansen, A.M.C. Mesenchymal stem cell transformation and sarcoma genesis. Clin. Sarcoma Res. 2013, 3, 10. [Google Scholar] [CrossRef]
- Genadry, K.C.; Pietrobono, S.; Rota, R.; Linardic, C.M. Soft Tissue sarcoma cancer stem cells: An overview. Front. Oncol. 2018, 8, 475. [Google Scholar] [CrossRef]
- Yan, G.-N.; Lv, Y.-F.; Guo, Q.-N. Advances in osteosarcoma stem cell research and opportunities for novel therapeutic targets. Cancer Lett. 2016, 370, 268–274. [Google Scholar] [CrossRef]
- Yang, J.; Ren, Z.; Du, X.; Hao, M.; Zhou, W. The role of mesenchymal stem/progenitor cells in sarcoma: Update and dispute. Stem Cell Investig. 2014, 1, 18. [Google Scholar] [CrossRef]
- Honoki, K. Do stem-like cells play a role in drug resistance of sarcomas? Expert Rev. Anticancer. 2010, 10, 261–270. [Google Scholar] [CrossRef]
- Fujiwara, T.; Ozaki, T. Overcoming therapeutic resistance of bone sarcomas: Overview of the molecular mechanisms and therapeutic targets for bone sarcoma stem cells. Stem Cells Int. 2016, 2016, 2603092. [Google Scholar] [CrossRef]
- Brown, H.K.; Tellez-Gabriel, M.; Heymann, D. Cancer stem cells in osteosarcoma. Cancer Lett. 2017, 386, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Tirino, V.; Desiderio, V.; Aquino, R.D.; Francesco, F.D.; Pirozzi, G.; Graziano, A.; Galderisi, U.; Cavaliere, C.; Rosa, A.D.; Papaccio, G.; et al. Detection and characterization of CD133+ cancer stem cells in human solid tumours. PLoS ONE 2008, 3, e3469. [Google Scholar] [CrossRef]
- Adhikari, A.S.; Agarwal, N.; Wood, B.M.; Porretta, C.; Ruiz, B.; Pochampally, R.R.; Iwakuma, T. CD117 and Stro-1 identify osteosarcoma tumor-initiating cells associated with metastasis and drug resistance. Cancer Res. 2010, 70, 4602–4612. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Li, X.; Si, M.; Liu, T.; Li, J. CD271+ osteosarcoma cells display stem-like properties. PLoS ONE 2014, 9, e98549. [Google Scholar] [CrossRef] [PubMed]
- Naka, N.; Takenaka, S.; Araki, N.; Miwa, T.; Hashimoto, N.; Yoshioka, K.; Joyama, S.; Hamada, K.; Tsukamoto, Y.; Tomita, Y.; et al. Synovial sarcoma is a stem cell malignancy. Stem Cells 2010, 28, 1119–1131. [Google Scholar] [CrossRef]
- Abarrategi, A.; Tornin, J.; Cruzado, L.M.; Hamilton, A.; Campos, E.M.; Rodrigo, J.P.; Gonzalez, M.V.; Baldini, N.; Castro, J.G.; Rodriguez, R. Osteosarcoma: Cells-of-origin, cancer stem cells, and targeted therapies. Stem Cells Int. 2016, 2016, 3631764. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, D.; Qi, Y.; Liu, R.; Li, S.; Zou, H.; Lan, J.; Ju, X.; Jiang, J.; Liang, W.; et al. Evaluation of expression of cancer stem cell markers and fusion gene in synovial sarcoma: Insights into histogenesis and pathogenesis. Oncol. Rep. 2017, 37, 3351–3360. [Google Scholar] [CrossRef]
- Corre, I.; Verrecchia, F.; Crenn, V.; Redini, F.; Trichet, V. The osteosarcoma microenvironment: A complex but targetable ecosystem. Cells 2020, 9, 976. [Google Scholar] [CrossRef]
- Kawano, M.; Tanaka, K.; Itonaga, I.; Iwasaki, T.; Tsumura, H. Interaction between human osteosarcoma and mesenchymal stem cells via an interleukin-8 signaling loop in the tumor microenvironment. Cell Commun. Signal. 2018, 16, 13. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, G.; Chen, R.; Hua, Y.; Cai, Z. Mesenchymal stem cells in the osteosarcoma microenvironment: Their biological properties, influence on tumor growth, and therapeutic implications. Stem Cell Res. 2018, 9, 22. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, H.; Zheng, J.; Yu, P.; Xu, L.; Jiang, P.; Gao, J.; Wang, H.; Zhang, Y. Transforming growth factor beta1 signal is crucial for dedifferentiation of cancer cells to cancer stem cells in osteosarcoma. Stem Cells 2013, 31, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Grappa, M.A.D.; Molyneux, S.D.; McKee, T.D.; Waterhouse, P.; Penninger, J.M.; Khokha, R. RANKL blockade prevents and treats aggressive osteosarcomas. Sci. Transl. Med. 2015, 7, 317ra197. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Goff, B.L.; Berreur, M.; Riet, A.; Moreau, A.; Blanchard, F.; Chevalier, C.; Marsollier, I.G.; Leger, J.; Guicheux, J.; et al. Human osteosarcoma cells express functional receptor activator of nuclear factor-kappa B. J. Pathol. 2007, 211, 555–562. [Google Scholar] [CrossRef]
- Navet, B.; Ando, K.; Franco, J.W.V.; Brion, R.; Amiaud, J.; Mori, K.; Yagita, H.; Mueller, C.G.; Verrecchia, F.; Dumars, C.; et al. The Intrinsic and Extrinsic Implications of RANKL/RANK signaling in osteosarcoma: From tumor initiation to lung metastases. Cancers 2018, 10, 398. [Google Scholar] [CrossRef]
- Pietrovito, L.; Leo, A.; Gori, V.; Lulli, M.; Parri, M.; Becherucci, V.; Piccini, L.; Bambi, F.; Taddei, M.L.; Chiarugi, P. Bone marrow-derived mesenchymal stem cells promote invasiveness and transendothelial migration of osteosarcoma cells via a mesenchymal to amoeboid transition. Mol. Oncol. 2018, 12, 659–676. [Google Scholar] [CrossRef]
- Tu, B.; Du, L.; Fan, Q.M.; Tang, Z.; Tang, T.T. STAT3 activation by IL-6 from mesenchymal stem cells promotes the proliferation and metastasis of osteosarcoma. Cancer Lett. 2012, 325, 80–88. [Google Scholar] [CrossRef]
- Tu, B.; Peng, Z.X.; Fan, Q.M.; Du, L.; Yan, W.; Tang, T.T. Osteosarcoma cells promote the production of pro-tumor cytokines in mesenchymal stem cells by inhibiting their osteogenic differentiation through the TGF-beta/Smad2/3 pathway. Exp. Cell Res. 2014, 320, 164–173. [Google Scholar] [CrossRef]
- Vallabhaneni, K.C.; Hassler, M.Y.; Abraham, A.; Whitt, J.; Mo, Y.Y.; Atfi, A.; Pochampally, R. Mesenchymal stem/stromal cells under stress increase osteosarcoma migration and apoptosis resistance via extracellular vesicle mediated communication. PLoS ONE 2016, 11, e0166027. [Google Scholar] [CrossRef]
- Jerez, S.; Araya, H.; Thaler, R.; Charlesworth, M.C.; Solis, R.L.; Kalergis, A.M.; Cespedes, P.F.; Dudakovic, A.; Stein, G.S.; Wijnen, A.J.V.; et al. Proteomic analysis of exosomes and exosome-free conditioned media from human osteosarcoma cell lines reveals secretion of proteins related to tumor progression. J. Cell. Biochem. 2017, 118, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Bonuccelli, G.; Avnet, S.; Grisendi, G.; Salerno, M.; Granchi, D.; Dominici, M.; Kusuzaki, K.; Baldini, N. Role of mesenchymal stem cells in osteosarcoma and metabolic reprogramming of tumor cells. Oncotarget 2014, 5, 7575–7588. [Google Scholar] [CrossRef] [PubMed]
- An, R.; Schmid, R.; Klausing, A.; Robering, J.W.; Weber, M.; Bauerle, T.; Detsch, R.; Boccaccini, A.R.; Horch, R.E.; Boos, A.M.; et al. Proangiogenic effects of tumor cells on endothelial progenitor cells vary with tumor type in an in vitro and in vivo rat model. FASEB J. 2018, 32, 5587–5601. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Gao, Y.; Zhang, L.; Cai, X.; Qian, Z.; Huang, H.; Huang, W. VEGF-siRNA silencing induces apoptosis, inhibits proliferation and suppresses vasculogenic mimicry in osteosarcoma in vitro. Exp. Oncol. 2008, 30, 29–34. [Google Scholar] [PubMed]
- Sorenson, L.; McEachron, T.A. Abstract 208: Transcriptional profiling of the microenvironment in pediatric osteosarcoma. In Proceedings of the American Association for Cancer Research Annual Meeting 2019, Atlanta, GA, Philadelphia, PA, USA, 29 March–3 April 2019; p. 208. [Google Scholar]
- Heymann, M.F.; Lezot, F.; Heymann, D. The contribution of immune infiltrates and the local microenvironment in the pathogenesis of osteosarcoma. Cell. Immunol. 2019, 343, 103711. [Google Scholar] [CrossRef]
- Li, M.; Jin, X.; Li, H.; Wu, G.; Wang, S.; Yang, C.; Deng, S. Key genes with prognostic values in suppression of osteosarcoma metastasis using comprehensive analysis. BMC Cancer 2020, 20, 65. [Google Scholar] [CrossRef]
- Li, B.; Wang, Z.; Wu, H.; Xue, M.; Lin, P.; Wang, S.; Lin, N.; Huang, X.; Pan, W.; Liu, M.; et al. Epigenetic regulation of CXCL12 plays a critical role in mediating tumor progression and the immune response in osteosarcoma. Cancer Res. 2018, 78, 3938. [Google Scholar] [CrossRef]
- Ren, L.; Khanna, C. Role of ezrin in osteosarcoma metastasis. In Current Advances in Osteosarcoma; Kleinerman, M.D.E.S., Ed.; Springer International Publishing: Cham, Switzerland, 2014; pp. 181–201. [Google Scholar]
- Bulut, G.; Hong, S.H.; Chen, K.; Beauchamp, E.M.; Rahim, S.; Kosturko, G.W.; Glasgow, E.; Dakshanamurthy, S.; Lee, H.S.; Daar, I.; et al. Small molecule inhibitors of ezrin inhibit the invasive phenotype of osteosarcoma cells. Oncogene 2012, 31, 269–281. [Google Scholar] [CrossRef]
- Zhao, D.-H.; Zhu, J.; Wang, W.-B.; Dong, F.; Zhang, Q.; Fan, H.-W.; Zhang, J.-Z.; Wang, Y.-M. Correlations of ezrin expression with pathological characteristics and prognosis of osteosarcoma: A meta-analysis. Sci. World J. 2014, 2014, 837543. [Google Scholar] [CrossRef]
- Liu, G.Y.Q.; Qian, Y. Loss of MicroRNA-489-3p promotes osteosarcoma metastasis by activating PAX3-MET pathway. Mol. Cancerog. 2017, 56, 1312–1321. [Google Scholar] [CrossRef]
- Moriarity, B.S.; Otto, G.M.; Rahrmann, E.P.; Rathe, S.K.; Wolf, N.K.; Weg, M.T.; Manlove, L.A.; LaRue, R.S.; Temiz, N.A.; Molyneux, S.D.; et al. A sleeping beauty forward genetic screen identifies new genes and pathways driving osteosarcoma development and metastasis. Nat. Genet. 2015, 47, 615–624. [Google Scholar] [CrossRef]
- Muff, R.; Ram Kumar, R.M.; Botter, S.M.; Born, W.; Fuchs, B. Genes regulated in metastatic osteosarcoma: Evaluation by microarray analysis in four human and two mouse cell line systems. Sarcoma 2012, 2012, 937506. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Guan, D.; Li, J. Identifying osteosarcoma metastasis associated genes by weighted gene co-expression network analysis (WGCNA). Medicine 2018, 97, e10781. [Google Scholar] [CrossRef] [PubMed]
- Raynor, W.; Houshmand, S.; Gholami, S.; Emamzadehfard, S.; Rajapakse, C.S.; Blomberg, B.A.; Werner, T.J.; Carlsen, P.F.H.; Baker, J.F.; Alavi, A. Evolving role of molecular imaging with (18)F-sodium fluoride PET as a biomarker for calcium metabolism. Curr. Osteoporos. Rep. 2016, 14, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Kairemo, K.; Rohren, E.M.; Anderson, P.M.; Ravizzini, G.; Rao, A.; Macapinlac, H.A.; Subbiah, V. Development of sodium fluoride PET response criteria for solid tumours (NAFCIST) in a clinical trial of radium-223 in osteosarcoma: From RECIST to PERCIST to NAFCIST. ESMO Open 2019, 4, e000439. [Google Scholar] [CrossRef]
- Gialleonardo, V.D.; Aldeborgh, H.N.; Miloushev, V.; Folkers, K.M.; Granlund, K.; Tap, W.D.; Lewis, J.S.; Weber, W.A.; Keshari, K.R. Multinuclear NMR and MRI reveal an early metabolic response to mTOR inhibition in sarcoma. Cancer Res. 2017, 77, 3113–3120. [Google Scholar] [CrossRef]
- Hansen, A.E.; Gutte, H.; Holst, P.; Johannesen, H.H.; Rahbek, S.; Clemmensen, A.E.; Larsen, M.M.E.; Schøier, C.; Larsen, J.A.; Klausen, T.L.; et al. Combined hyperpolarized (13)C-pyruvate MRS and (18)F-FDG PET (hyperPET) estimates of glycolysis in canine cancer patients. Eur. J. Radiol. 2018, 103, 6–12. [Google Scholar] [CrossRef]
- Guan, G.; Lu, Y.; Zhu, X.; Liu, L.; Chen, J.; Ma, Q.; Zhang, Y.; Wen, Y.; Yang, L.; Liu, T.; et al. CXCR4-targeted near-infrared imaging allows detection of orthotopic and metastatic human osteosarcoma in a mouse model. Sci. Rep. 2015, 5, 15244. [Google Scholar] [CrossRef]
- Li, X.; Huang, X.; Zhang, J.; Huang, H.; Zhao, L.; Yu, M.; Zhang, Y.; Wang, H. A novel peptide targets CD105 for tumour imaging in vivo. Oncol. Rep. 2018, 40, 2935–2943. [Google Scholar] [CrossRef]
- Lu, Y.; Li, L.; Lin, Z.; Li, M.; Hu, X.; Zhang, Y.; Peng, M.; Xia, H.; Han, G. Enhancing osteosarcoma killing and CT imaging using ultrahigh drug loading and NIR-responsive bismuth sulfide@mesoporous silica nanoparticles. Adv. Healthc. Mater. 2018, 7, e1800602. [Google Scholar] [CrossRef]
- Chaiyawat, P.; Settakorn, J.; Sangsin, A.; Teeyakasem, P.; Klangjorhor, J.; Soongkhaw, A.; Pruksakorn, D. Exploring targeted therapy of osteosarcoma using proteomics data. Onco Targets 2017, 10, 565–577. [Google Scholar] [CrossRef]
- Bishop, M.W.; Janeway, K.A.; Gorlick, R. Future directions in the treatment of osteosarcoma. Curr. Opin. Pediatr. 2016, 28, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.W.; Li, L.; Li, D.; Liu, J.; Li, X.; Li, W.; Xu, X.; Zhang, M.J.; Chandler, L.A.; Lin, H.; et al. The first approved gene therapy product for cancer Ad-p53 (Gendicine): 12 years in the clinic. Hum. Gene 2018, 29, 160–179. [Google Scholar] [CrossRef] [PubMed]
- Fioramonti, M.; Fausti, V.; Pantano, F.; Iuliani, M.; Ribelli, G.; Lotti, F.; Pignochino, Y.; Grignani, G.; Santini, D.; Tonini, G.; et al. Cabozantinib affects osteosarcoma growth through a direct effect on tumor cells and modifications in bone microenvironment. Sci. Rep. 2018, 8, 4177. [Google Scholar] [CrossRef] [PubMed]
- Grullich, C. Cabozantinib: Multi-kinase inhibitor of MET, AXL, RET, and VEGFR2. Recent Results Cancer Res. 2018, 211, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Mir, O.; Pelissier, S.M.; Penel, N.; Neumann, S.P.; Bompas, E.; Chevreau, C.; Duffaud, F.; Werle, N.E.; Saada, E.; et al. Cabozantinib in patients with advanced ewing sarcoma or osteosarcoma (CABONE): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 446–455. [Google Scholar] [CrossRef]
- Hoff, D.D.V. There are no bad anticancer agents, only bad clinical trial designs--twenty-first richard and hinda rosenthal foundation award lecture. Clin. Cancer Res. 1998, 4, 1079–1086. [Google Scholar]
- Coens, C.; Graaf, W.T.V.D.; Blay, J.Y.; Chawla, S.P.; Judson, I.; Sanfilippo, R.; Manson, S.C.; Hodge, R.A.; Marreaud, S.; Prins, J.B.; et al. Health-related quality-of-life results from PALETTE: A randomized, double-blind, phase 3 trial of pazopanib versus placebo in patients with soft tissue sarcoma whose disease has progressed during or after prior chemotherapy-a european organization for research and treatment of cancer soft tissue and bone sarcoma group global network study (EORTC 62072). Cancer 2015, 121, 2933–2941. [Google Scholar] [CrossRef]
- Longhi, A.; Paioli, A.; Palmerini, E.; Cesari, M.; Abate, M.E.; Setola, E.; Spinnato, P.; Donati, D.; Hompland, I.; Boye, K. Pazopanib in relapsed osteosarcoma patients: Report on 15 cases. Acta Oncol. 2019, 58, 124–128. [Google Scholar] [CrossRef]
- Pedersen, N.A.; Rossen, P.; Rose, H.; Safwat, A. Pazopanib in the treatment of bone sarcomas: Clinical experience. Transl. Oncol. 2020, 13, 295–299. [Google Scholar] [CrossRef]
- Umeda, K.; Kato, I.; Saida, S.; Okamoto, T.; Adachi, S. Pazopanib for second recurrence of osteosarcoma in pediatric patients. Pediatr. Int. 2017, 59, 937–938. [Google Scholar] [CrossRef]
- Seto, T.; Song, M.N.; Trieu, M.; Yu, J.; Sidhu, M.; Liu, C.M.; Sam, D.; Pan, M. Real-world experiences with pazopanib in patients with advanced soft tissue and bone sarcoma in northern california. Med. Sci. 2019, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Elete, K.R.; Albritton, K.H.; Akers, L.J.; Basha, R.; Ray, A. Response to pazopanib in patients with relapsed osteosarcoma. J. Pediatr. Hematol. Oncol. 2020, 42, e254–e257. [Google Scholar] [CrossRef] [PubMed]
- Safwat, A.; Boysen, A.; Lücke, A.; Rossen, P. Pazopanib in metastatic osteosarcoma: Significant clinical response in three consecutive patients. Acta Oncol. 2014, 53, 1451–1454. [Google Scholar] [CrossRef] [PubMed]
- Pignochino, Y.; Grignani, G.; Cavalloni, G.; Motta, M.; Tapparo, M.; Bruno, S.; Bottos, A.; Gammaitoni, L.; Migliardi, G.; Camussi, G.; et al. Sorafenib blocks tumour growth, angiogenesis and metastatic potential in preclinical models of osteosarcoma through a mechanism potentially involving the inhibition of ERK1/2, MCL-1 and ezrin pathways. Mol. Cancer 2009, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Pignochino, Y.; Aglio, C.D.; Basiricò, M.; Capozzi, F.; Soster, M.; Marchiò, S.; Bruno, S.; Gammaitoni, L.; Sangiolo, D.; Torchiaro, E.; et al. The combination of sorafenib and everolimus abrogates mTORC1 and mTORC2 upregulation in osteosarcoma preclinical models. Clin. Cancer Res. 2013, 19, 2117–2131. [Google Scholar] [CrossRef] [PubMed]
- Grignani, G.; Palmerini, E.; Dileo, P.; Asaftei, S.D.; D’Ambrosio, L.; Pignochino, Y.; Mercuri, M.; Picci, P.; Fagioli, F.; Casali, P.G.; et al. A phase II trial of sorafenib in relapsed and unresectable high-grade osteosarcoma after failure of standard multimodal therapy: An Italian Sarcoma Group study. Ann. Oncol. J. Eur. Soc. Med. Oncol. 2012, 23, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Raciborska, A.; Bilska, K. Sorafenib in patients with progressed and refractory bone tumors. Med. Oncol. 2018, 35, 126. [Google Scholar] [CrossRef]
- Grignani, G.; Palmerini, E.; Ferraresi, V.; D’Ambrosio, L.; Bertulli, R.; Asaftei, S.D.; Tamburini, A.; Pignochino, Y.; Sangiolo, D.; Marchesi, E.; et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: A non-randomised phase 2 clinical trial. Lancet Oncol. 2015, 16, 98–107. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Syndrome | % of OS | Genes Mutated | References |
|---|---|---|---|
| Li–Fraumeni syndrome | 12% | TP53 | [20,28] |
| Retinoblastoma syndrome | 7% | RB | [49] |
| Rothmund–Thomson type II (RTS II) syndrome | 32% | RECQL4 | [28] |
| RAPALIDINO syndromes | 13.3% | RECQL4 | [20] |
| Werner syndrome | WRN | ||
| Bloom syndrome | BLM | ||
| Diamond–Blackfan anemia | 5.4% 0.9% | RPS19, RPL5, RPL11, RPL35A, RPS24, RPS17, RPS7, RPS10, RPS26 | [47] [48] |
| Process | Pediatric | Adolescent/Adult |
|---|---|---|
| Control of cell cycle and apoptosis | TP53, RB1, CDKN2A, CDK4, MDM2, MYC, CARD11, CTNND1, BLM, CCNE1, COPS3, PRKCA | TP53, |
| PI3K-mTOR and RAS-signaling pathways | EGFR, GNAQ, GNAS, ALK, PDGFRA, PDGFRB, PIK3CA, AKT2, PIK3R1, PTEN, TSC2, VHL, CBL | PIK3CA, |
| Notch-signaling pathway | NOTCH1-4, MAML2, FBXW7, PDPK1, AKT1, E1F4B | AKT1, |
| DNA damage repair | BRCA1, BRCA2, MLH1, BAP1, ATM, WRN | SETD2, FBXW7 |
| Chromatin modification | ATRX, FANCE, RECQL4, ARID1A, EP300 | H3F3A |
| Regulation of transcription | Runx1, GAS7, MLLT3 | |
| Angiogenesis | TIE1 and KDR |
| Driver Genes | Synergistic Genes |
|---|---|
| TP53, NOTCH1, MYC, FOS, NF2, WIF1, BRCA2, APC, PTCH1, PRKAR1A | RB1, TWIST, PTEN, JUN |
| Downregulated miRNAs | Upregulated miRNAs |
|---|---|
| miR-16, miR-31, miR-100-5p, miR-221-3p, miR29b-1-5p, miR-125b-1-3p, miR-29a-5p, miR-370-3p, miR-299-5p, miR-493-5p, miR-409-3p, miR-30e-3p, miR-431-5p, miR-432-5p, miR-410-3p, miR-411-5p, miR-376c-3p, miR-125b-5p, miR-335-5p, miR-376a-3p, miR-382-5p, miR-154-5p, miR-222-3p, miR-137, miR-92b-3p, miR-433-5p, miR-127-3p, miR-143, miR-143-3p, miR-539, miR-539-3p, miR-218, miR-183, miR-3928, miR-140, miR-150, miR-449c | miR-27, miR-148a, miR-181a-5p, miR-181c-5p, miR-195, miR-223-3p, miR-342-3p, miR-378a-3p, miR-21, mirR-221, miR-106, mi-R-218, miR-126, miR-574-3p, |
| Protein | Potential Drug |
|---|---|
| DNMT1 (DNA (cytosine-5)-methyltransferase 1) | azacytidine (Vidaza), decitabine (Dacogen) |
| ERBB2 (receptor tyrosine–protein kinase erbB-2) | trastuzumab (Herceptin), lapatinib (Tycerb), afatinib (GIOTRIF/GILOTRIF), pertuzumab (PERJETA) |
| GSR (mitochondrial glutathione reductase | carmustine (GLIADEL® WAFER) |
| HDAC1 (histone deacetylase 1) | vorinostat (Zolinza) |
| HDAC2 (histone deacetylase 2) | romidepsin (Istodax) |
| KIT (mast/stem cell growth factor receptor kit) | imatinib (Gleevec), sorafenib (Nexavar), sunitinib (Sutent), pazopanib (Votrient), dasatinib (Sprycel), axitinib (Inlyta), nilotinib (Tasigna) |
| FGFR1 (fibroblast growth factor receptor 1) | lenvatinib (Lenvima) |
| MET (hepatocyte growth factor receptor) | cabozantinib (COMETRIQ), crizotinib (XALKORI) |
| MTOR (serine/threonine protein kinase mTOR) | temsirolimus (Torisel), everolimus (Afinitor) |
| PARP1 (poly (ADP–ribose) polymerase 1) | olaparib (AZD2281) |
| PDGFR α (platelet-derived growth factor receptor alpha) | imatinib (Gleevec), sorafenib (Nexavar), sunitinib (Sutent), pazopanib (Votrient), nilotinib (Tasigna), axitinib (Inlyta) and dasatinib (Sprycel) |
| PSMC2 (26S protease regulators subunit 7) | bortezomib (Velcade) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czarnecka, A.M.; Synoradzki, K.; Firlej, W.; Bartnik, E.; Sobczuk, P.; Fiedorowicz, M.; Grieb, P.; Rutkowski, P. Molecular Biology of Osteosarcoma. Cancers 2020, 12, 2130. https://doi.org/10.3390/cancers12082130
Czarnecka AM, Synoradzki K, Firlej W, Bartnik E, Sobczuk P, Fiedorowicz M, Grieb P, Rutkowski P. Molecular Biology of Osteosarcoma. Cancers. 2020; 12(8):2130. https://doi.org/10.3390/cancers12082130
Chicago/Turabian StyleCzarnecka, Anna M., Kamil Synoradzki, Wiktoria Firlej, Ewa Bartnik, Pawel Sobczuk, Michal Fiedorowicz, Pawel Grieb, and Piotr Rutkowski. 2020. "Molecular Biology of Osteosarcoma" Cancers 12, no. 8: 2130. https://doi.org/10.3390/cancers12082130
APA StyleCzarnecka, A. M., Synoradzki, K., Firlej, W., Bartnik, E., Sobczuk, P., Fiedorowicz, M., Grieb, P., & Rutkowski, P. (2020). Molecular Biology of Osteosarcoma. Cancers, 12(8), 2130. https://doi.org/10.3390/cancers12082130