Single-Cell Heterogeneity of Cutaneous T-Cell Lymphomas Revealed Using RNA-Seq Technologies


Abstract
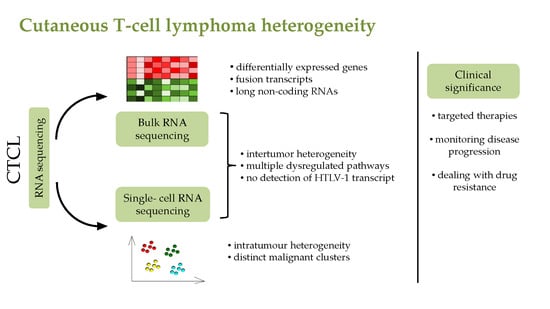
1. Introduction
2. Clinical and Immunological Features of CTCLs
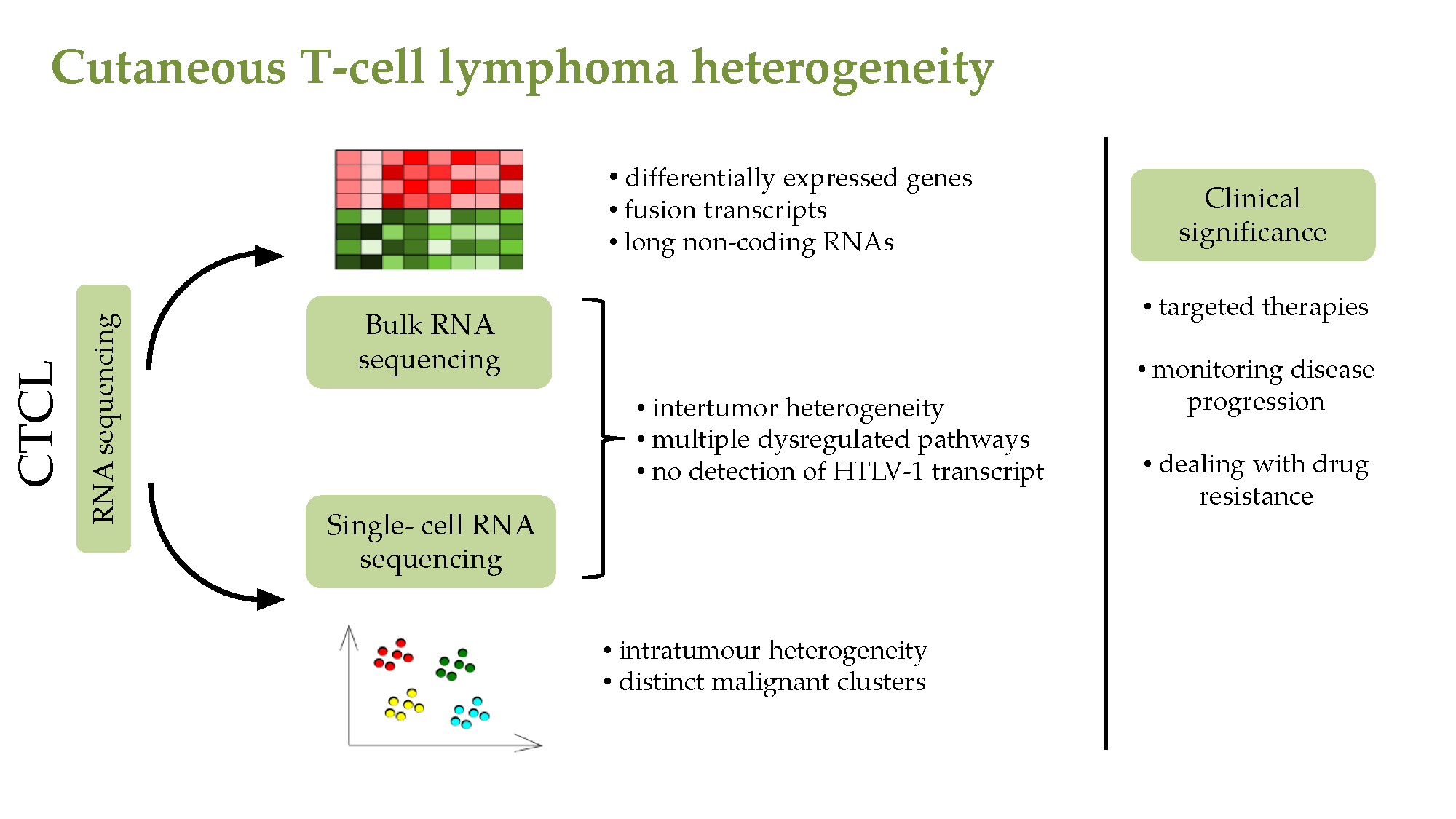
3. High-Throughput RNA Sequencing Techniques
4. RNA Seq Analysis of CTCL Patients
5. Identification of Long Non-Coding RNAs in CTCL
6. Single-Cell RNA Seq in CTCL Studies
6.1. The Population of Malignant Sézary Cells can be Divided into Distinct Subpopulations
6.2. Heterogeneity of Malignant Population
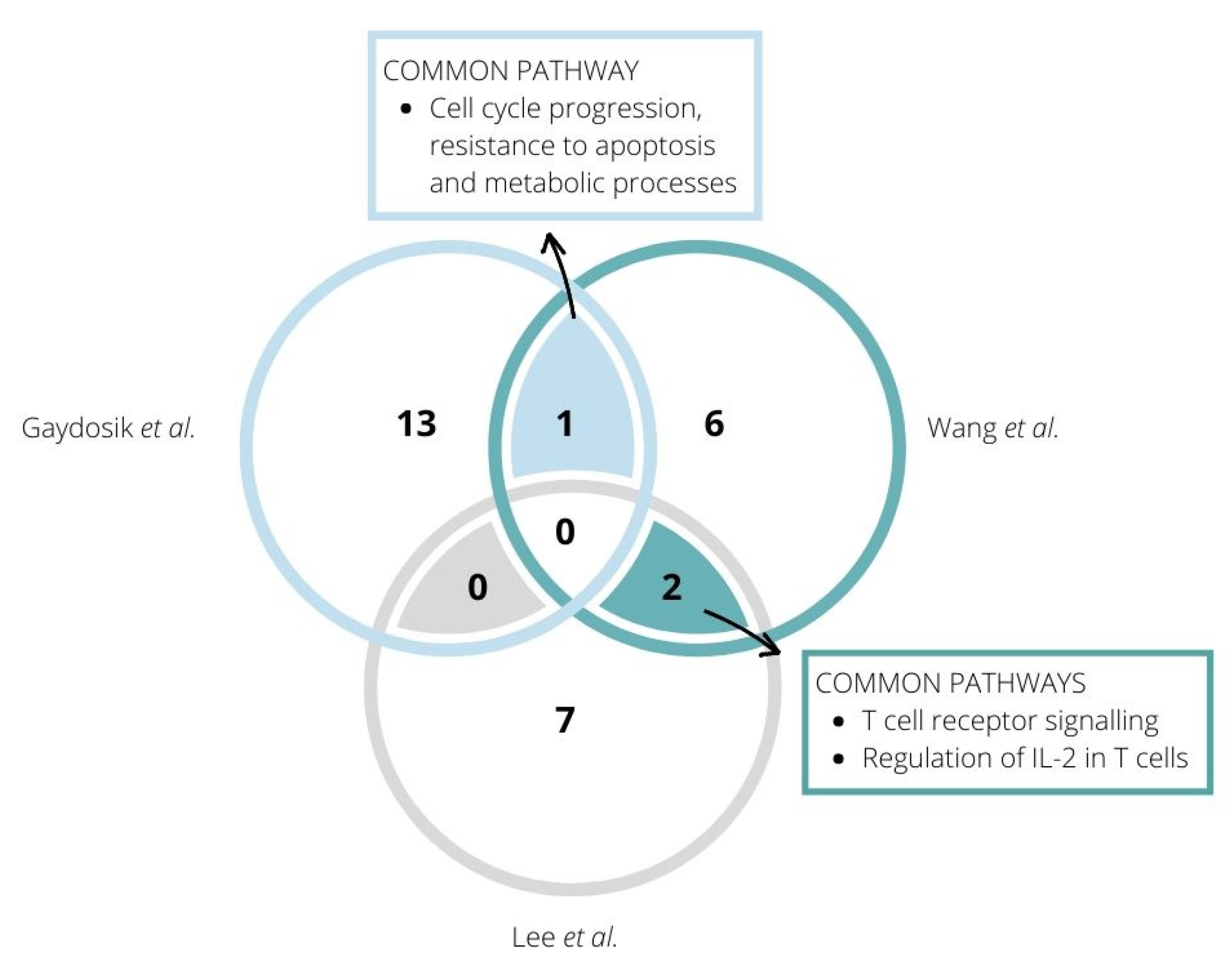
7. Dysregulated Signalling Pathways Revealed in CTCL Patients by RNA Sequencing
8. Role of Infectious Agents in Disease Onset and Progression
9. Clinical Significance of Novel Single-Cell RNA Sequencing Technologies
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Xu, J.; Huang, H.; Wang, S.; Chen, Y.; Yin, X.; Zhang, X.; Zhang, Y. Molecular profiling of TOX-deficient neoplastic cells in cutaneous T cell lymphoma. Arch. Dermatol. Res. 2019, 312, 513–525. [Google Scholar] [CrossRef]
- Bastidas Torres, A.N.; Najidh, S.; Tensen, C.P.; Vermeer, M.H. Molecular advances in cutaneous T-cell lymphoma. Semin. Cutan. Med. Surg. 2018, 37, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Cerroni, L. Mycosis fungoides-clinical and histopathologic features, differential diagnosis, and treatment. Semin. Cutan. Med. Surg. 2018, 37, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Spicknall, K.E. Sezary syndrome-clinical and histopathologic features, differential diagnosis, and treatment. Semin. Cutan. Med. Surg. 2018, 37, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Vakiti, A.; Padala, S.A.; Singh, D. Sezary Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Agar, N.; Morris, S.; Russell-Jones, R.; Hawk, J.; Whittaker, S. Case report of four patients with erythrodermic cutaneous T-cell lymphoma and severe photosensitivity mimicking chronic actinic dermatitis. Br. J. Dermatol. 2009, 160, 698–703. [Google Scholar] [CrossRef]
- Jawed, S.I.; Myskowski, P.L.; Horwitz, S.; Moskowitz, A.; Querfeld, C. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sezary syndrome): Part I. Diagnosis: Clinical and histopathologic features and new molecular and biologic markers. J. Am. Acad. Dermatol. 2014, 70, 205 e1–205 e16. [Google Scholar] [CrossRef]
- Saulite, I.; Hoetzenecker, W.; Weidinger, S.; Cozzio, A.; Guenova, E.; Wehkamp, U. Sezary Syndrome and Atopic Dermatitis: Comparison of Immunological Aspects and Targets. Biom. Res. Int. 2016, 2016, 9717530. [Google Scholar] [CrossRef]
- Roelens, M.; Delord, M.; Ram-Wolff, C.; Marie-Cardine, A.; Alberdi, A.; Maki, G.; Homyrda, L.; Bensussan, A.; Bagot, M.; Toubert, A.; et al. Circulating and skin-derived Sezary cells: Clonal but with phenotypic plasticity. Blood 2017, 130, 1468–1471. [Google Scholar] [CrossRef]
- Buus, T.B.; Willerslev-Olsen, A.; Fredholm, S.; Blumel, E.; Nastasi, C.; Gluud, M.; Hu, T.; Lindahl, L.M.; Iversen, L.; Fogh, H.; et al. Single-cell heterogeneity in Sezary syndrome. Blood Adv. 2018, 2, 2115–2126. [Google Scholar] [CrossRef]
- Gaydosik, A.M.; Tabib, T.; Geskin, L.J.; Bayan, C.A.; Conway, J.F.; Lafyatis, R.; Fuschiotti, P. Single-Cell Lymphocyte Heterogeneity in Advanced Cutaneous T-cell Lymphoma Skin Tumors. Clin. Cancer Res. 2019, 25, 4443–4454. [Google Scholar] [CrossRef]
- Borcherding, N.; Voigt, A.P.; Liu, V.; Link, B.K.; Zhang, W.; Jabbari, A. Single-Cell Profiling of Cutaneous T-Cell Lymphoma Reveals Underlying Heterogeneity Associated with Disease Progression. Clin. Cancer Res. 2019, 25, 2996–3005. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Miyagaki, T.; Sugaya, M. Immunological milieu in mycosis fungoides and Sezary syndrome. J. Dermatol. 2014, 41, 11–18. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ma, Y.; Yin, K.; Li, W.; Chen, W.; Zhang, Y.; Zhu, C.; Li, T.; Han, B.; Liu, X.; et al. Long non-coding and coding RNA profiling using strand-specific RNA-seq in human hypertrophic cardiomyopathy. Sci. Data 2019, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Uphoff, C.C.; Pommerenke, C.; Denkmann, S.A.; Drexler, H.G. Screening human cell lines for viral infections applying RNA-Seq data analysis. PLoS ONE 2019, 14, e0210404. [Google Scholar] [CrossRef] [PubMed]
- Picelli, S. Single-cell RNA-sequencing: The future of genome biology is now. RNA Biol. 2017, 14, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 96. [Google Scholar] [CrossRef]
- Olsen, T.K.; Baryawno, N. Introduction to Single-Cell RNA Sequencing. Curr. Protoc. Mol. Biol. 2018, 122, e57. [Google Scholar] [CrossRef]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef]
- Litvinov, I.V.; Tetzlaff, M.T.; Thibault, P.; Gangar, P.; Moreau, L.; Watters, A.K.; Netchiporouk, E.; Pehr, K.; Prieto, V.G.; Rahme, E.; et al. Gene expression analysis in Cutaneous T-Cell Lymphomas (CTCL) highlights disease heterogeneity and potential diagnostic and prognostic indicators. Oncoimmunology 2017, 6, e1306618. [Google Scholar] [CrossRef] [PubMed]
- Morimura, S.; Sugaya, M.; Suga, H.; Miyagaki, T.; Ohmatsu, H.; Fujita, H.; Asano, Y.; Tada, Y.; Kadono, T.; Sato, S. TOX expression in different subtypes of cutaneous lymphoma. Arch. Dermatol. Res. 2014, 306, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Luo, Y.; Liu, J.; Liu, Y.; Sun, Q. TOX acts an oncological role in mycosis fungoides. PLoS ONE 2015, 10, e0117479. [Google Scholar] [CrossRef] [PubMed]
- Van Kester, M.S.; Borg, M.K.; Zoutman, W.H.; Out-Luiting, J.J.; Jansen, P.M.; Dreef, E.J.; Vermeer, M.H.; van Doorn, R.; Willemze, R.; Tensen, C.P. A meta-analysis of gene expression data identifies a molecular signature characteristic for tumor-stage mycosis fungoides. J. Investig. Dermatol. 2012, 132, 2050–2059. [Google Scholar] [CrossRef]
- Kopp, K.L.; Ralfkiaer, U.; Gjerdrum, L.M.; Helvad, R.; Pedersen, I.H.; Litman, T.; Jonson, L.; Hagedorn, P.H.; Krejsgaard, T.; Gniadecki, R.; et al. STAT5-mediated expression of oncogenic miR-155 in cutaneous T-cell lymphoma. Cell Cycle 2013, 12, 1939–1947. [Google Scholar] [CrossRef]
- Lefrancois, P.; Tetzlaff, M.T.; Moreau, L.; Watters, A.K.; Netchiporouk, E.; Provost, N.; Gilbert, M.; Ni, X.; Sasseville, D.; Duvic, M.; et al. TruSeq-Based Gene Expression Analysis of Formalin-Fixed Paraffin-Embedded (FFPE) Cutaneous T-Cell Lymphoma Samples: Subgroup Analysis Results and Elucidation of Biases from FFPE Sample Processing on the TruSeq Platform. Front. Med. 2017, 4, 153. [Google Scholar] [CrossRef]
- Prasad, A.; Rabionet, R.; Espinet, B.; Zapata, L.; Puiggros, A.; Melero, C.; Puig, A.; Sarria-Trujillo, Y.; Ossowski, S.; Garcia-Muret, M.P.; et al. Identification of Gene Mutations and Fusion Genes in Patients with Sezary Syndrome. J. Investig. Dermatol. 2016, 136, 1490–1499. [Google Scholar] [CrossRef]
- Izykowska, K.; Przybylski, G.K.; Gand, C.; Braun, F.C.; Grabarczyk, P.; Kuss, A.W.; Olek-Hrab, K.; Bastidas Torres, A.N.; Vermeer, M.H.; Zoutman, W.H.; et al. Genetic rearrangements result in altered gene expression and novel fusion transcripts in Sezary syndrome. Oncotarget 2017, 8, 39627–39639. [Google Scholar] [CrossRef]
- Wang, L.; Ni, X.; Covington, K.R.; Yang, B.Y.; Shiu, J.; Zhang, X.; Xi, L.; Meng, Q.; Langridge, T.; Drummond, J.; et al. Genomic profiling of Sezary syndrome identifies alterations of key T cell signaling and differentiation genes. Nat. Genet. 2015, 47, 1426–1434. [Google Scholar] [CrossRef]
- Chase, A.; Ernst, T.; Fiebig, A.; Collins, A.; Grand, F.; Erben, P.; Reiter, A.; Schreiber, S.; Cross, N.C. TFG, a target of chromosome translocations in lymphoma and soft tissue tumors, fuses to GPR128 in healthy individuals. Haematologica 2010, 95, 20–26. [Google Scholar] [CrossRef]
- Yoo, H.Y.; Kim, P.; Kim, W.S.; Lee, S.H.; Kim, S.; Kang, S.Y.; Jang, H.Y.; Lee, J.E.; Kim, J.; Kim, S.J.; et al. Frequent CTLA4-CD28 gene fusion in diverse types of T-cell lymphoma. Haematologica 2016, 101, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Liang, W.S.; Tembe, W.; Izatt, T.; Kruglyak, S.; Kiefer, J.A.; Cuyugan, L.; Zismann, V.; Legendre, C.; Pittelkow, M.R.; et al. Personalized treatment of Sezary syndrome by targeting a novel CTLA4:CD28 fusion. Mol. Genet. Genom. Med. 2015, 3, 130–136. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Smyth, M.J.; Teng, M.W.L. PD1 functions by inhibiting CD28-mediated co-stimulation. Clin. Transl. Immunol. 2017, 6, e138. [Google Scholar] [CrossRef] [PubMed]
- Khodadoust, M.S.; Rook, A.H.; Porcu, P.; Foss, F.; Moskowitz, A.J.; Shustov, A.; Shanbhag, S.; Sokol, L.; Fling, S.P.; Ramchurren, N.; et al. Pembrolizumab in Relapsed and Refractory Mycosis Fungoides and Sezary Syndrome: A Multicenter Phase II Study. J. Clin. Oncol. 2020, 38, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Suga, H.; Sugaya, M.; Miyagaki, T.; Kawaguchi, M.; Fujita, H.; Asano, Y.; Tada, Y.; Kadono, T.; Sato, S. The role of IL-32 in cutaneous T-cell lymphoma. J. Investig. Dermatol. 2014, 134, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.C.R.; Acuna, S.M.; Aoki, J.I.; Floeter-Winter, L.M.; Muxel, S.M. Long Non-Coding RNAs in the Regulation of Gene Expression: Physiology and Disease. Noncoding RNA 2019, 5, 17. [Google Scholar] [CrossRef]
- Chi, Y.; Wang, D.; Wang, J.; Yu, W.; Yang, J. Long Non-Coding RNA in the Pathogenesis of Cancers. Cells 2019, 8, 1015. [Google Scholar] [CrossRef]
- DiStefano, J.K. The Emerging Role of Long Noncoding RNAs in Human Disease. Methods Mol. Biol. 2018, 1706, 91–110. [Google Scholar] [CrossRef]
- Salamon, I.; Saccani Jotti, G.; Condorelli, G. The long noncoding RNA landscape in cardiovascular disease: A brief update. Curr. Opin. Cardiol. 2018, 33, 282–289. [Google Scholar] [CrossRef]
- Galamb, O.; Bartak, B.K.; Kalmar, A.; Nagy, Z.B.; Szigeti, K.A.; Tulassay, Z.; Igaz, P.; Molnar, B. Diagnostic and prognostic potential of tissue and circulating long non-coding RNAs in colorectal tumors. World J. Gastroenterol. 2019, 25, 5026–5048. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, Y.; Du, L.; Jiang, X.; Yan, S.; Duan, W.; Li, J.; Zhan, Y.; Wang, L.; Zhang, S.; et al. Circulating long noncoding RNA act as potential novel biomarkers for diagnosis and prognosis of non-small cell lung cancer. Mol. Oncol. 2018, 12, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Corey, D.R. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 2017, 16, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.; Chakraborty, P.; Varadwaj, P.K. Unraveling long non-coding RNAs through analysis of high-throughput RNA-sequencing data. Noncoding RNA Res. 2017, 2, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Ungewickell, A.; Bhaduri, A.; Qu, K.; Webster, D.E.; Armstrong, R.; Weng, W.K.; Aros, C.J.; Mah, A.; Chen, R.O.; et al. Transcriptome sequencing in Sezary syndrome identifies Sezary cell and mycosis fungoides-associated lncRNAs and novel transcripts. Blood 2012, 120, 3288–3297. [Google Scholar] [CrossRef]
- Wong, H.K.; Mishra, A.; Hake, T.; Porcu, P. Evolving insights in the pathogenesis and therapy of cutaneous T-cell lymphoma (mycosis fungoides and Sezary syndrome). Br. J. Haematol. 2011, 155, 150–166. [Google Scholar] [CrossRef]
- Pomerantz, R.G.; Mirvish, E.D.; Erdos, G.; Falo, L.D., Jr.; Geskin, L.J. Novel approach to gene expression profiling in Sezary syndrome. Br. J. Dermatol. 2010, 163, 1090–1094. [Google Scholar] [CrossRef] [PubMed]
- Booken, N.; Gratchev, A.; Utikal, J.; Weiss, C.; Yu, X.; Qadoumi, M.; Schmuth, M.; Sepp, N.; Nashan, D.; Rass, K.; et al. Sezary syndrome is a unique cutaneous T-cell lymphoma as identified by an expanded gene signature including diagnostic marker molecules CDO1 and DNM3. Leukemia 2008, 22, 393–399. [Google Scholar] [CrossRef]
- Kamstrup, M.R.; Gjerdrum, L.M.; Biskup, E.; Lauenborg, B.T.; Ralfkiaer, E.; Woetmann, A.; Odum, N.; Gniadecki, R. Notch1 as a potential therapeutic target in cutaneous T-cell lymphoma. Blood 2010, 116, 2504–2512. [Google Scholar] [CrossRef]
- Di, L.; Srivastava, S.; Zhdanova, O.; Ding, Y.; Li, Z.; Wulff, H.; Lafaille, M.; Skolnik, E.Y. Inhibition of the K+ channel KCa3.1 ameliorates T cell-mediated colitis. Proc. Natl. Acad. Sci. USA 2010, 107, 1541–1546. [Google Scholar] [CrossRef]
- Siwiec, A.; Majdan, M. The role of the PD-1 protein in pathogenesis of autoimmune diseases, with particular consideration of rheumatoid arthritis and systemic lupus erythematosus. Postepy Hig. Med. Dosw. 2015, 69, 534–542. [Google Scholar] [CrossRef]
- Loo, D.T.; Mather, J.P. Antibody-based identification of cell surface antigens: Targets for cancer therapy. Curr. Opin. Pharmacol. 2008, 8, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma, K.; Dang, N.H.; Morimoto, C. Revisiting an old acquaintance: CD26 and its molecular mechanisms in T cell function. Trends Immunol. 2008, 29, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.; Fullen, D.; Carlson, J.A. Low CD7 expression in benign and malignant cutaneous lymphocytic infiltrates: Experience with an antibody reactive with paraffin-embedded tissue. Am. J. Dermatopathol. 2002, 24, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Goh, G.; Walradt, T.; Hong, B.S.; Bunick, C.G.; Chen, K.; Bjornson, R.D.; Maman, Y.; Wang, T.; Tordoff, J.; et al. Genomic landscape of cutaneous T cell lymphoma. Nat. Genet. 2015, 47, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Almeida, A.C.; Abate, F.; Khiabanian, H.; Martinez-Escala, E.; Guitart, J.; Tensen, C.P.; Vermeer, M.H.; Rabadan, R.; Ferrando, A.; Palomero, T. The mutational landscape of cutaneous T cell lymphoma and Sezary syndrome. Nat. Genet. 2015, 47, 1465–1470. [Google Scholar] [CrossRef]
- Izban, K.F.; Ergin, M.; Qin, J.Z.; Martinez, R.L.; Pooley, R.J.; Saeed, S.; Alkan, S. Constitutive expression of NF-kappa B is a characteristic feature of mycosis fungoides: Implications for apoptosis resistance and pathogenesis. Hum. Pathol. 2000, 31, 1482–1490. [Google Scholar] [CrossRef]
- Ungewickell, A.; Bhaduri, A.; Rios, E.; Reuter, J.; Lee, C.S.; Mah, A.; Zehnder, A.; Ohgami, R.; Kulkarni, S.; Armstrong, R.; et al. Genomic analysis of mycosis fungoides and Sezary syndrome identifies recurrent alterations in TNFR2. Nat. Genet. 2015, 47, 1056–1060. [Google Scholar] [CrossRef]
- Mirvish, J.J.; Pomerantz, R.G.; Falo, L.D., Jr.; Geskin, L.J. Role of infectious agents in cutaneous T-cell lymphoma: Facts and controversies. Clin. Dermatol. 2013, 31, 423–431. [Google Scholar] [CrossRef]
- Abrams, J.T.; Balin, B.J.; Vonderheid, E.C. Association between Sezary T cell-activating factor, Chlamydia pneumoniae, and cutaneous T cell lymphoma. Ann. N. Y. Acad. Sci. 2001, 941, 69–85. [Google Scholar] [CrossRef]
- Nedoszytko, B.; Wierzbicki, P.; Karenko, L.; Maciejewska-Radomska, A.; Stachewicz, P.; Zablotna, M.; Glen, J.; Vakeva, L.; Nowicki, R.J.; Sokolowska-Wojdylo, M. Presence of Chlamydophila pneumoniae DNA in blood cells is a frequent event in patients with the late stage of primary cutaneous lymphomas and with atopic dermatitis. Postepy Dermatol. Alergol. 2018, 35, 274–279. [Google Scholar] [CrossRef]
- Ponzoni, M.; Ferreri, A.J.; Mappa, S.; Pasini, E.; Govi, S.; Facchetti, F.; Fanoni, D.; Tucci, A.; Vino, A.; Doglioni, C.; et al. Prevalence of Borrelia burgdorferi infection in a series of 98 primary cutaneous lymphomas. Oncologist 2011, 16, 1582–1588. [Google Scholar] [CrossRef] [PubMed]
- Tothova, S.M.; Bonin, S.; Trevisan, G.; Stanta, G. Mycosis fungoides: Is it a Borrelia burgdorferi-associated disease? Br. J. Cancer 2006, 94, 879–883. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.; Huggins, R.H.; Lertsburapa, T.; Bauer, K.; Rademaker, A.; Gerami, P.; Guitart, J. Cutaneous T-cell lymphoma and Staphylococcus aureus colonization. J. Am. Acad. Dermatol. 2008, 59, 949–952. [Google Scholar] [CrossRef] [PubMed]
- Talpur, R.; Bassett, R.; Duvic, M. Prevalence and treatment of Staphylococcus aureus colonization in patients with mycosis fungoides and Sezary syndrome. Br. J. Dermatol. 2008, 159, 105–112. [Google Scholar] [CrossRef]
- Lindahl, L.M.; Willerslev-Olsen, A.; Gjerdrum, L.M.R.; Nielsen, P.R.; Blumel, E.; Rittig, A.H.; Celis, P.; Herpers, B.; Becker, J.C.; Stausbol-Gron, B.; et al. Antibiotics inhibit tumor and disease activity in cutaneous T-cell lymphoma. Blood 2019, 134, 1072–1083. [Google Scholar] [CrossRef]
- Pancake, B.A.; Zucker-Franklin, D.; Coutavas, E.E. The cutaneous T cell lymphoma, mycosis fungoides, is a human T cell lymphotropic virus-associated disease. A study of 50 patients. J. Clin. Investig. 1995, 95, 547–554. [Google Scholar] [CrossRef]
- Zucker-Franklin, D.; Coutavas, E.E.; Rush, M.G.; Zouzias, D.C. Detection of human T-lymphotropic virus-like particles in cultures of peripheral blood lymphocytes from patients with mycosis fungoides. Proc. Natl. Acad. Sci. USA 1991, 88, 7630–7634. [Google Scholar] [CrossRef]
- Zucker-Franklin, D.; Hooper, W.C.; Evatt, B.L. Human lymphotropic retroviruses associated with mycosis fungoides: Evidence that human T-cell lymphotropic virus type II (HTLV-II) as well as HTLV-I may play a role in the disease. Blood 1992, 80, 1537–1545. [Google Scholar] [CrossRef]
- Bazarbachi, A.; Saal, F.; Laroche, L.; Flageul, B.; Peries, J.; de The, H. HTLV-1 provirus and mycosis fungoides. Science 1993, 259, 1470–1471. [Google Scholar] [CrossRef]
- Bazarbachi, A.; Soriano, V.; Pawson, R.; Vallejo, A.; Moudgil, T.; Matutes, E.; Peries, J.; Molina, A.; de The, H.; Schulz, T.F.; et al. Mycosis fungoides and Sezary syndrome are not associated with HTLV-I infection: An international study. Br. J. Haematol. 1997, 98, 927–933. [Google Scholar] [CrossRef]
- Boni, R.; Davis-Daneshfar, A.; Burg, G.; Fuchs, D.; Wood, G.S. No detection of HTLV-I proviral DNA in lesional skin biopsies from Swiss and German patients with cutaneous T-cell lymphoma. Br. J. Dermatol. 1996, 134, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Courgnaud, V.; Duthanh, A.; Guillot, B.; Sitbon, M.; Dereure, O. Absence of HTLV-related sequences in skin lesions and peripheral blood of cutaneous T-cell lymphomas. J. Investig. Dermatol. 2009, 129, 2520–2522. [Google Scholar] [CrossRef] [PubMed]
- Pawlaczyk, M.; Filas, V.; Sobieska, M.; Gozdzicka-Jozefiak, A.; Wiktorowicz, K.; Breborowicz, J. No evidence of HTLV-I infection in patients with mycosis fungoides and Sezary syndrome. Neoplasma 2005, 52, 52–55. [Google Scholar]
- Poiesz, B.; Dube, D.; Dube, S.; Love, J.; Papsidero, L.; Uner, A.; Hutchinson, R. HTLV-II-associated cutaneous T-cell lymphoma in a patient with HIV-1 infection. N. Engl. J. Med. 2000, 342, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Bachelez, H.; Hadida, F.; Gorochov, G. Massive infiltration of the skin by HIV-specific cytotoxic CD8+ T cells. N. Engl. J. Med. 1996, 335, 61–62. [Google Scholar] [CrossRef]
- Wilkins, K.; Turner, R.; Dolev, J.C.; LeBoit, P.E.; Berger, T.G.; Maurer, T.A. Cutaneous malignancy and human immunodeficiency virus disease. J. Am. Acad. Dermatol. 2006, 54, 189–206. [Google Scholar] [CrossRef]
- Gahongayire, F. Mycosis fungoides and Sezary syndrome against a human immunodeficiency virus-positive background: Case report. Int. J. Dermatol. 2007, 46, 32–35. [Google Scholar] [CrossRef]
- Haverkos, B.M.; Gru, A.A.; Geyer, S.M.; Bingman, A.K.; Hemminger, J.A.; Mishra, A.; Wong, H.K.; Pancholi, P.; Freud, A.G.; Caligiuri, M.A.; et al. Increased Levels of Plasma Epstein Barr Virus DNA Identify a Poor-Risk Subset of Patients with Advanced Stage Cutaneous T-Cell Lymphoma. Clin. Lymphoma Myeloma Leuk. 2016, 16, S181–S190. [Google Scholar] [CrossRef]
- Novelli, M.; Merlino, C.; Ponti, R.; Bergallo, M.; Quaglino, P.; Cambieri, I.; Comessatti, A.; Sidoti, F.; Costa, C.; Corino, D.; et al. Epstein-Barr virus in cutaneous T-cell lymphomas: Evaluation of the viral presence and significance in skin and peripheral blood. J. Investig. Dermatol. 2009, 129, 1556–1561. [Google Scholar] [CrossRef]
- Park, S.; Lee, D.Y.; Kim, W.S.; Ko, Y.H. Primary cutaneous Epstein-Barr virus-associated T-cell lymphoproliferative disorder-2 cases with unusual, prolonged clinical course. Am. J. Dermatopathol. 2010, 32, 832–836. [Google Scholar] [CrossRef]
- Tournadre, A.; D’Incan, M.; Dubost, J.J.; Franck, F.; Dechelotte, P.; Souteyrand, P.; Soubrier, M. Cutaneous lymphoma associated with Epstein-Barr virus infection in 2 patients treated with methotrexate. Mayo Clin. Proc. 2001, 76, 845–848. [Google Scholar] [CrossRef]
- Brice, S.L.; Jester, J.D.; Friednash, M.; Golitz, L.E.; Leahy, M.A.; Stockert, S.S.; Weston, W.L. Examination of cutaneous T-cell lymphoma for human herpesviruses by using the polymerase chain reaction. J. Cutan. Pathol. 1993, 20, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Erkek, E.; Senturk, N.; Dincer, I.; Olut, A.I.; Kocagoz, T.; Bukulmez, G.; Sahin, S. Identification of herpes simplex virus DNA and lack of human herpesvirus-8 DNA in mycosis fungoides. Acta Derm. Venereol. 2002, 82, 214–216. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, A.; Bischoff, S.; Skrygan, M.; Wieland, U.; Brockmeyer, N.H.; Stucker, M.; Altmeyer, P.; Gambichler, T. High association of human herpesvirus 8 in large-plaque parapsoriasis and mycosis fungoides. Arch. Dermatol. 2008, 144, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Ponti, R.; Bergallo, M.; Costa, C.; Quaglino, P.; Fierro, M.T.; Comessatti, A.; Stroppiana, E.; Sidoti, F.; Merlino, C.; Novelli, M.; et al. Human herpesvirus 7 detection by quantitative real time polymerase chain reaction in primary cutaneous T-cell lymphomas and healthy subjects: Lack of a pathogenic role. Br. J. Dermatol. 2008, 159, 1131–1137. [Google Scholar] [CrossRef]
- Bergallo, M.; Dapra, V.; Fava, P.; Ponti, R.; Calvi, C.; Montanari, P.; Novelli, M.; Quaglino, P.; Galliano, I.; Fierro, M.T. DNA from Human Polyomaviruses, MWPyV, HPyV6, HPyV7, HPyV9 and HPyV12 in Cutaneous T-cell Lymphomas. Anticancer Res. 2018, 38, 4111–4114. [Google Scholar] [CrossRef]
- Kreuter, A.; Silling, S.; Dewan, M.; Stucker, M.; Wieland, U. Evaluation of 4 recently discovered human polyomaviruses in primary cutaneous B-cell and T-cell lymphoma. Arch. Dermatol. 2011, 147, 1449–1451. [Google Scholar] [CrossRef]
- Miertusova, S.; Bonin, S.; Trevisan, G.; Stanta, G. Mycosis fungoides is not associated with hepatitis C virus infection. Br. J. Dermatol. 2004, 151, 1108–1110. [Google Scholar] [CrossRef]
- Netchiporouk, E.; Gantchev, J.; Tsang, M.; Thibault, P.; Watters, A.K.; Hughes, J.M.; Ghazawi, F.M.; Woetmann, A.; Odum, N.; Sasseville, D.; et al. Analysis of CTCL cell lines reveals important differences between mycosis fungoides/Sezary syndrome vs. HTLV-1(+) leukemic cell lines. Oncotarget 2017, 8, 95981–95998. [Google Scholar] [CrossRef]
- Schmidt, A.N.; Robbins, J.B.; Greer, J.P.; Zic, J.A. Conjugal transformed mycosis fungoides: The unknown role of viral infection and environmental exposures in the development of cutaneous T-cell lymphoma. J. Am. Acad. Dermatol. 2006, 54, S202–S205. [Google Scholar] [CrossRef]
- Weder, P.; Anliker, M.; Itin, P.; Bargetzi, M. Familial cutaneous mycosis fungoides: Successful treatment with a combination of gemcitabine and alemtuzumab. Dermatology 2004, 208, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Lozano, A.; Duvic, M. Cutaneous T-cell lymphoma in non-blood-related family members: Report of an additional case. J. Am. Acad. Dermatol. 2007, 56, 521. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.M.; Schmidt, J.A.; Carson, K.R.; Musiek, A.C.; Mehta-Shah, N.; Payton, J.E. Novel cell adhesion/migration pathways are predictive markers of HDAC inhibitor resistance in cutaneous T cell lymphoma. EBioMedicine 2019, 46, 170–183. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Features | Single-Cell RNA Sequencing | RNA Sequencing |
|---|---|---|
| Transcriptome analysis | Unique transcriptome expression of each of many diverse cell types | Average transcriptome expression of many cells |
| Heterogeneity of the population | Cell to cell variability | Cells from tissues considered to be homogeneous |
| Populations of cells | Identifying rare populations | - |
| Conditions of cells | Cells from one condition are generally captured and sequenced | Compares differentially expressed genes under multiple conditions |
| Statistical power | Increased (capturing thousands of cells in one condition) | - |
| Cells uniqueness | Revealing latent changes, new cell types, cells subpopulations | - |
| Generated data | Noisier, more variable data | Less background noise, less variable |
| Capture efficiency | Low | High |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rassek, K.; Iżykowska, K. Single-Cell Heterogeneity of Cutaneous T-Cell Lymphomas Revealed Using RNA-Seq Technologies. Cancers 2020, 12, 2129. https://doi.org/10.3390/cancers12082129
Rassek K, Iżykowska K. Single-Cell Heterogeneity of Cutaneous T-Cell Lymphomas Revealed Using RNA-Seq Technologies. Cancers. 2020; 12(8):2129. https://doi.org/10.3390/cancers12082129
Chicago/Turabian StyleRassek, Karolina, and Katarzyna Iżykowska. 2020. "Single-Cell Heterogeneity of Cutaneous T-Cell Lymphomas Revealed Using RNA-Seq Technologies" Cancers 12, no. 8: 2129. https://doi.org/10.3390/cancers12082129
APA StyleRassek, K., & Iżykowska, K. (2020). Single-Cell Heterogeneity of Cutaneous T-Cell Lymphomas Revealed Using RNA-Seq Technologies. Cancers, 12(8), 2129. https://doi.org/10.3390/cancers12082129