Natural Born Killers: NK Cells in Cancer Therapy



Abstract
1. Introduction
2. Natural Killer Cells
NK Cell Cytotoxicity
3. Endogenous NK Cells
3.1. Inhibitory Receptor Blocking
3.2. Cytokines to Activate Endogenous NK cells
3.3. Immunogenic Modulation
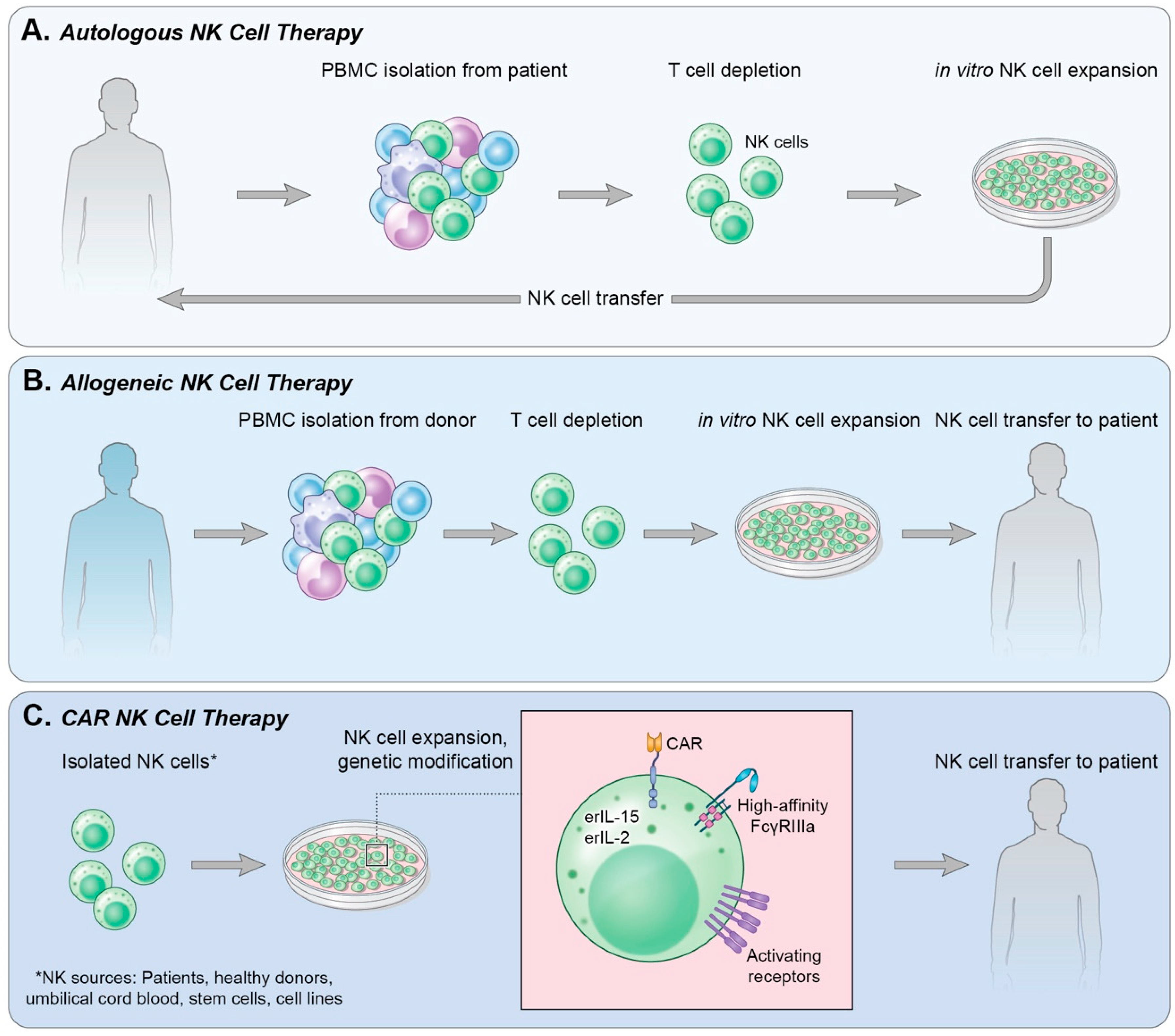
4. Adoptive Cell Transfer
4.1. Autologous NK Cells
4.2. Allogeneic NK Cells
4.3. Immortalized NK Cells
5. Chimeric Antigen Receptor-Expressing NK Cells (CAR-NK)
5.1. Hematologic Malignancies
5.2. Solid Tumors
5.3. Early Phase I Trials
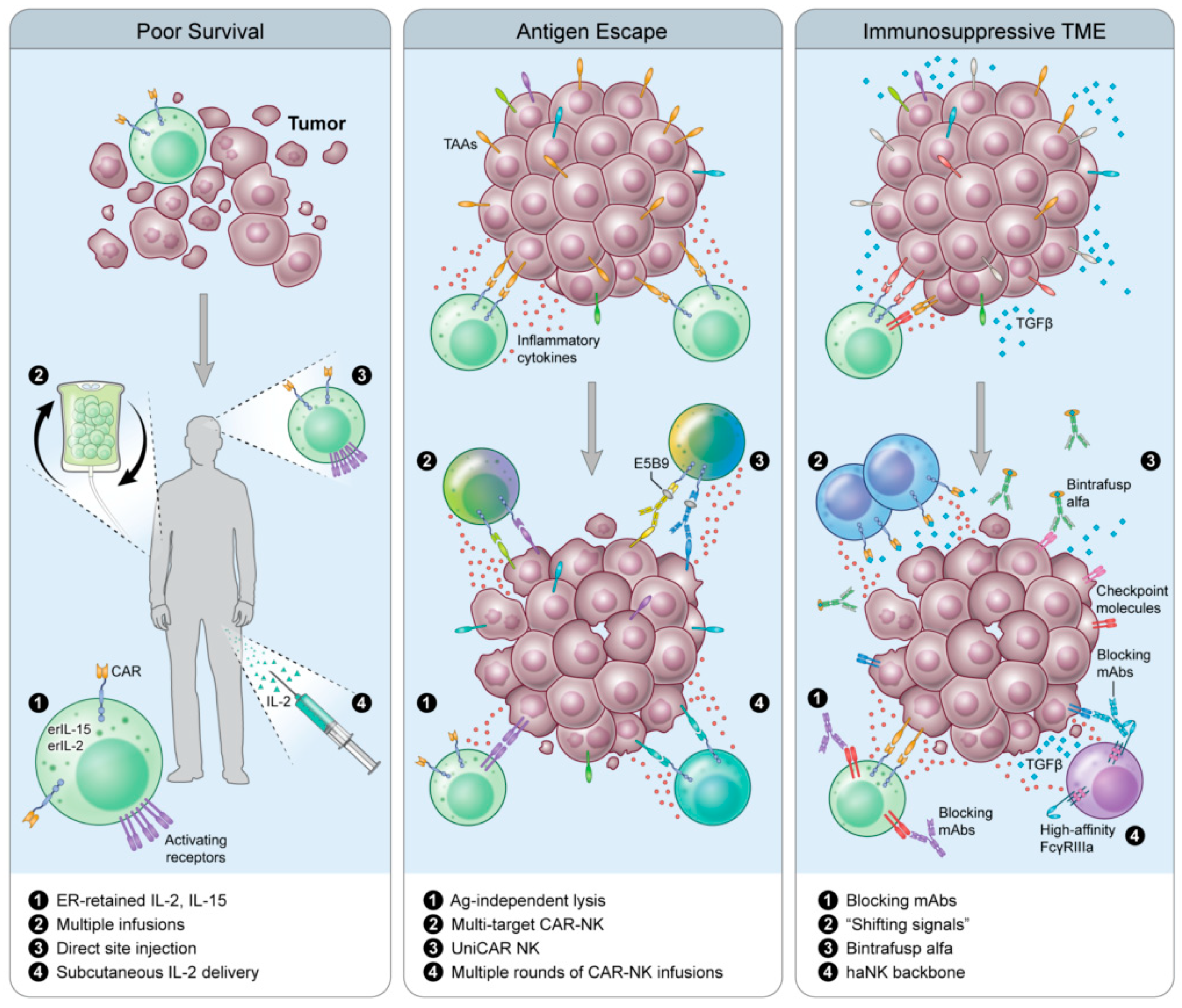
6. Next-Next-Generation CAR-NKs
6.1. Universal CAR (UniCAR)
6.2. Shifting Signals
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, J.E.; Hsu, K.C. Natural killer cell education and the response to infection and cancer therapy: Stay tuned. Trends Immunol. 2018, 39, 222–239. [Google Scholar] [CrossRef]
- Osinska, I.; Popko, K.; Demkow, U. Perforin: An important player in immune response. Cent. Eur. J. Immunol. 2014, 39, 109–115. [Google Scholar] [CrossRef]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef]
- Lo Nigro, C.; Macagno, M.; Sangiolo, D.; Bertolaccini, L.; Aglietta, M.; Merlano, M.C. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: Biological evidence and clinical perspectives. Ann. Transl. Med. 2019, 7, 105. [Google Scholar] [CrossRef]
- Benson, D.M., Jr.; Cohen, A.D.; Jagannath, S.; Munshi, N.C.; Spitzer, G.; Hofmeister, C.C.; Efebera, Y.A.; Andre, P.; Zerbib, R.; Caligiuri, M.A. A phase I trial of the anti-KIR antibody IPH2101 and lenalidomide in patients with relapsed/refractory multiple myeloma. Clin. Cancer Res. 2015, 21, 4055–4061. [Google Scholar] [CrossRef]
- Sola, C.; Andre, P.; Lemmers, C.; Fuseri, N.; Bonnafous, C.; Blery, M.; Wagtmann, N.R.; Romagne, F.; Vivier, E.; Ugolini, S. Genetic and antibody-mediated reprogramming of natural killer cell missing-self recognition in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 12879–12884. [Google Scholar] [CrossRef]
- Carlsten, M.; Korde, N.; Kotecha, R.; Reger, R.; Bor, S.; Kazandjian, D.; Landgren, O.; Childs, R.W. Checkpoint inhibition of KIR2D with the monoclonal antibody IPH2101 induces contraction and hyporesponsiveness of NK cells in patients with myeloma. Clin. Cancer Res. 2016, 22, 5211–5222. [Google Scholar] [CrossRef] [PubMed]
- Vey, N.; Karlin, L.; Sadot-Lebouvier, S.; Broussais, F.; Berton-Rigaud, D.; Rey, J.; Charbonnier, A.; Marie, D.; Andre, P.; Paturel, C.; et al. A phase 1 study of lirilumab (antibody against killer immunoglobulin-like receptor antibody KIR2D; IPH2102) in patients with solid tumors and hematologic malignancies. Oncotarget 2018, 9, 17675–17688. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, E.M.; Mele, J.M.; Cheney, C.; Timmerman, E.A.; Fiazuddin, F.; Strattan, E.J.; Mo, X.; Byrd, J.C.; Muthusamy, N.; Awan, F.T. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. Oncoimmunology 2016, 5, 1226720. [Google Scholar] [CrossRef] [PubMed]
- Tinker, A.V.; Hirte, H.W.; Provencher, D.; Butler, M.; Ritter, H.; Tu, D.; Azim, H.A., Jr.; Paralejas, P.; Grenier, N.; Hahn, S.A.; et al. Dose-ranging and cohort-expansion study of monalizumab (IPH2201) in patients with advanced gynecologic malignancies: A trial of the Canadian Cancer Trials Group (CCTG): IND221. Clin. Cancer Res. 2019, 25, 6052–6060. [Google Scholar] [CrossRef]
- Khan, M.; Arooj, S.; Wang, H. NK cell-based immune checkpoint inhibition. Front. Immunol. 2020, 11, 167. [Google Scholar] [CrossRef]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer immunotherapy based on natural killer cells: Current progress and new opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef]
- Floros, T.; Tarhini, A.A. Anticancer cytokines: Biology and clinical effects of interferon-alpha2, interleukin (IL)-2, IL-15, IL-21, and IL-12. Semin. Oncol. 2015, 42, 539–548. [Google Scholar] [CrossRef]
- Domingues, B.; Lopes, J.M.; Soares, P.; Populo, H. Melanoma treatment in review. Immunotargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef]
- Kim, P.S.; Kwilas, A.R.; Xu, W.; Alter, S.; Jeng, E.K.; Wong, H.C.; Schlom, J.; Hodge, J.W. IL-15 superagonist/IL-15RalphaSushi-Fc fusion complex (IL-15SA/IL-15RalphaSu-Fc; ALT-803) markedly enhances specific subpopulations of NK and memory CD8+ T cells, and mediates potent anti-tumor activity against murine breast and colon carcinomas. Oncotarget 2016, 7, 16130–16145. [Google Scholar] [CrossRef]
- Wrangle, J.M.; Velcheti, V.; Patel, M.R.; Garrett-Mayer, E.; Hill, E.G.; Ravenel, J.G.; Miller, J.S.; Farhad, M.; Anderton, K.; Lindsey, K.; et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: A non-randomised, open-label, phase 1b trial. Lancet Oncol. 2018, 19, 694–704. [Google Scholar] [CrossRef]
- Fallon, J.; Tighe, R.; Kradjian, G.; Guzman, W.; Bernhardt, A.; Neuteboom, B.; Lan, Y.; Sabzevari, H.; Schlom, J.; Greiner, J.W. The immunocytokine NHS-IL12 as a potential cancer therapeutic. Oncotarget 2014, 5, 1869–1884. [Google Scholar] [CrossRef] [PubMed]
- Hodge, J.W.; Kwilas, A.; Ardiani, A.; Gameiro, S.R. Attacking malignant cells that survive therapy: Exploiting immunogenic modulation. Oncoimmunology 2013, 2, e26937. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hodge, J.W.; Ardiani, A.; Farsaci, B.; Kwilas, A.R.; Gameiro, S.R. The tipping point for combination therapy: Cancer vaccines with radiation, chemotherapy, or targeted small molecule inhibitors. Semin. Oncol. 2012, 39, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Garnett, C.T.; Schlom, J.; Hodge, J.W. Combination of docetaxel and recombinant vaccine enhances T-cell responses and antitumor activity: Effects of docetaxel on immune enhancement. Clin. Cancer Res. 2008, 14, 3536–3544. [Google Scholar] [CrossRef] [PubMed]
- Hodge, J.W.; Garnett, C.T.; Farsaci, B.; Palena, C.; Tsang, K.Y.; Ferrone, S.; Gameiro, S.R. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int. J. Cancer 2013, 133, 624–636. [Google Scholar] [CrossRef]
- Zingoni, A.; Fionda, C.; Borrelli, C.; Cippitelli, M.; Santoni, A.; Soriani, A. Natural killer cell response to chemotherapy-stressed cancer cells: Role in tumor immunosurveillance. Front. Immunol. 2017, 8, 1194. [Google Scholar] [CrossRef]
- Ardiani, A.; Gameiro, S.R.; Kwilas, A.R.; Donahue, R.N.; Hodge, J.W. Androgen deprivation therapy sensitizes prostate cancer cells to T-cell killing through androgen receptor dependent modulation of the apoptotic pathway. Oncotarget 2014, 5, 9335–9348. [Google Scholar] [CrossRef]
- Kwilas, A.R.; Gameiro, S.R.; Kim, P.S.; Malamas, A.S.; Hodge, J.W. Improving clinical benefit for prostate cancer patients through the combination of androgen deprivation and immunotherapy. Oncoimmunology 2015, 4, e1009303. [Google Scholar] [CrossRef][Green Version]
- Hodge, J.W.; Guha, C.; Neefjes, J.; Gulley, J.L. Synergizing radiation therapy and immunotherapy for curing incurable cancers. Opportunities and challenges. Oncology (Williston Park) 2008, 22, 1064–1070. [Google Scholar]
- Chakraborty, M.; Abrams, S.I.; Camphausen, K.; Liu, K.; Scott, T.; Coleman, C.N.; Hodge, J.W. Irradiation of tumor cells up-regulates Fas and enhances CTL lytic activity and CTL adoptive immunotherapy. J. Immunol. 2003, 170, 6338–6347. [Google Scholar] [CrossRef]
- Chakraborty, M.; Gelbard, A.; Carrasquillo, J.A.; Yu, S.; Mamede, M.; Paik, C.H.; Camphausen, K.; Schlom, J.; Hodge, J.W. Use of radiolabeled monoclonal antibody to enhance vaccine-mediated antitumor effects. Cancer Immunol. Immunother. 2008, 57, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Kong, Q.; Wang, G.; Jin, H.; Zhou, L.; Yu, D.; Niu, C.; Han, W.; Li, W.; Cui, J. Low-dose ionizing radiation induces direct activation of natural killer cells and provides a novel approach for adoptive cellular immunotherapy. Cancer Biother. Radiopharm. 2014, 29, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Grenga, I.; Kwilas, A.R.; Donahue, R.N.; Farsaci, B.; Hodge, J.W. Inhibition of the angiopoietin/Tie2 axis induces immunogenic modulation, which sensitizes human tumor cells to immune attack. J. Immunother. Cancer 2015, 3, 52. [Google Scholar] [CrossRef] [PubMed]
- Donahue, R.N.; Duncan, B.B.; Fry, T.J.; Jones, B.; Bachovchin, W.W.; Kiritsy, C.P.; Lai, J.H.; Wu, W.; Zhao, P.; Liu, Y.; et al. A pan inhibitor of DASH family enzymes induces immunogenic modulation and sensitizes murine and human carcinoma cells to antigen-specific cytotoxic T lymphocyte killing: Implications for combination therapy with cancer vaccines. Vaccine 2014, 32, 3223–3231. [Google Scholar] [CrossRef] [PubMed]
- Fenerty, K.E.; Padget, M.; Wolfson, B.; Gameiro, S.R.; Su, Z.; Lee, J.H.; Rabizadeh, S.; Soon-Shiong, P.; Hodge, J.W. Immunotherapy utilizing the combination of natural killer- and antibody dependent cellular cytotoxicity (ADCC)-mediating agents with poly (ADP-ribose) polymerase (PARP) inhibition. J. Immunother. Cancer 2018, 6, 133. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, A.; Berg, M.; Smith, A.; Childs, R.W. Bortezomib treatment to potentiate the anti-tumor immunity of ex-vivo expanded adoptively infused autologous natural killer cells. J. Cancer 2011, 2, 383–385. [Google Scholar] [CrossRef]
- Suck, G.; Oei, V.Y.; Linn, Y.C.; Ho, S.H.; Chu, S.; Choong, A.; Niam, M.; Koh, M.B. Interleukin-15 supports generation of highly potent clinical-grade natural killer cells in long-term cultures for targeting hematological malignancies. Exp. Hematol. 2011, 39, 904–914. [Google Scholar] [CrossRef]
- Brehm, C.; Huenecke, S.; Quaiser, A.; Esser, R.; Bremm, M.; Kloess, S.; Soerensen, J.; Kreyenberg, H.; Seidl, C.; Becker, P.S.; et al. IL-2 stimulated but not unstimulated NK cells induce selective disappearance of peripheral blood cells: Concomitant results to a phase I/II study. PLoS ONE 2011, 6, e27351. [Google Scholar] [CrossRef]
- Sakamoto, N.; Ishikawa, T.; Kokura, S.; Okayama, T.; Oka, K.; Ideno, M.; Sakai, F.; Kato, A.; Tanabe, M.; Enoki, T.; et al. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J. Transl. Med. 2015, 13, 277. [Google Scholar] [CrossRef]
- Luevano, M.; Daryouzeh, M.; Alnabhan, R.; Querol, S.; Khakoo, S.; Madrigal, A.; Saudemont, A. The unique profile of cord blood natural killer cells balances incomplete maturation and effective killing function upon activation. Hum. Immunol. 2012, 73, 248–257. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Hacein-Bey, S.; de Saint Basile, G.; De Coene, C.; Selz, F.; Le Deist, F.; Fischer, A. Role of interleukin-2 (IL-2), IL-7, and IL-15 in natural killer cell differentiation from cord blood hematopoietic progenitor cells and from gamma c transduced severe combined immunodeficiency X1 bone marrow cells. Blood 1996, 88, 3901–3909. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.S.; Shpall, E.J.; Rezvani, K. Cord blood as a source of natural killer cells. Front. Med. (Lausanne) 2015, 2, 93. [Google Scholar] [CrossRef] [PubMed]
- Knorr, D.A.; Ni, Z.; Hermanson, D.; Hexum, M.K.; Bendzick, L.; Cooper, L.J.; Lee, D.A.; Kaufman, D.S. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl. Med. 2013, 2, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Dezell, S.A.; Ahn, Y.O.; Spanholtz, J.; Wang, H.; Weeres, M.; Jackson, S.; Cooley, S.; Dolstra, H.; Miller, J.S.; Verneris, M.R. Natural killer cell differentiation from hematopoietic stem cells: A comparative analysis of heparin- and stromal cell-supported methods. Biol. Blood Marrow Transplant. 2012, 18, 536–545. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Woll, P.S.; Grzywacz, B.; Tian, X.; Marcus, R.K.; Knorr, D.A.; Verneris, M.R.; Kaufman, D.S. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood 2009, 113, 6094–6101. [Google Scholar] [CrossRef] [PubMed]
- Knorr, D.A.; Kaufman, D.S. Pluripotent stem cell-derived natural killer cells for cancer therapy. Transl. Res. 2010, 156, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Burns, L.J.; Weisdorf, D.J.; DeFor, T.E.; Vesole, D.H.; Repka, T.L.; Blazar, B.R.; Burger, S.R.; Panoskaltsis-Mortari, A.; Keever-Taylor, C.A.; Zhang, M.J.; et al. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: A phase I/II trial. Bone Marrow Transplant. 2003, 32, 177–186. [Google Scholar] [CrossRef]
- Ishikawa, E.; Tsuboi, K.; Saijo, K.; Harada, H.; Takano, S.; Nose, T.; Ohno, T. Autologous natural killer cell therapy for human recurrent malignant glioma. Anticancer Res. 2004, 24, 1861–1871. [Google Scholar]
- Krause, S.W.; Gastpar, R.; Andreesen, R.; Gross, C.; Ullrich, H.; Thonigs, G.; Pfister, K.; Multhoff, G. Treatment of colon and lung cancer patients with ex vivo heat shock protein 70-peptide-activated, autologous natural killer cells: A clinical phase I trial. Clin. Cancer Res. 2004, 10, 3699–3707. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Riley, J.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin. Cancer Res. 2011, 17, 6287–6297. [Google Scholar] [CrossRef]
- Cheng, M.; Chen, Y.; Xiao, W.; Sun, R.; Tian, Z. NK cell-based immunotherapy for malignant diseases. Cell Mol. Immunol. 2013, 10, 230–252. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.; Chen, X.; Sun, X.; Zhou, M.; Liu, B.; Li, Z.; Yu, Z.; Gao, W.; Liu, T. Effect of autologous NK cell immunotherapy on advanced lung adenocarcinoma with EGFR mutations. Precis. Clin. Med. 2019, 2, 235–245. [Google Scholar] [CrossRef]
- Cooley, S.; Parham, P.; Miller, J.S. Strategies to activate NK cells to prevent relapse and induce remission following hematopoietic stem cell transplantation. Blood 2018, 131, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Bishara, A.; De Santis, D.; Witt, C.C.; Brautbar, C.; Christiansen, F.T.; Or, R.; Nagler, A.; Slavin, S. The beneficial role of inhibitory KIR genes of HLA class I NK epitopes in haploidentically mismatched stem cell allografts may be masked by residual donor-alloreactive T cells causing GVHD. Tissue Antigens 2004, 63, 204–211. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef]
- Giebel, S.; Locatelli, F.; Lamparelli, T.; Velardi, A.; Davies, S.; Frumento, G.; Maccario, R.; Bonetti, F.; Wojnar, J.; Martinetti, M.; et al. Survival advantage with KIR ligand incompatibility in hematopoietic stem cell transplantation from unrelated donors. Blood 2003, 102, 814–819. [Google Scholar] [CrossRef]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef]
- Curti, A.; Ruggeri, L.; D’Addio, A.; Bontadini, A.; Dan, E.; Motta, M.R.; Trabanelli, S.; Giudice, V.; Urbani, E.; Martinelli, G.; et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood 2011, 118, 3273–3279. [Google Scholar] [CrossRef]
- Rubnitz, J.E.; Inaba, H.; Ribeiro, R.C.; Pounds, S.; Rooney, B.; Bell, T.; Pui, C.H.; Leung, W. NKAML: A pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 955–959. [Google Scholar] [CrossRef]
- Shaffer, B.C.; Le Luduec, J.B.; Forlenza, C.; Jakubowski, A.A.; Perales, M.A.; Young, J.W.; Hsu, K.C. Phase II study of haploidentical natural killer cell infusion for treatment of relapsed or persistent myeloid malignancies following allogeneic hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 2016, 22, 705–709. [Google Scholar] [CrossRef]
- Bachanova, V.; Burns, L.J.; McKenna, D.H.; Curtsinger, J.; Panoskaltsis-Mortari, A.; Lindgren, B.R.; Cooley, S.; Weisdorf, D.; Miller, J.S. Allogeneic natural killer cells for refractory lymphoma. Cancer Immunol. Immunother. 2010, 59, 1739–1744. [Google Scholar] [CrossRef] [PubMed]
- Geller, M.A.; Cooley, S.; Judson, P.L.; Ghebre, R.; Carson, L.F.; Argenta, P.A.; Jonson, A.L.; Panoskaltsis-Mortari, A.; Curtsinger, J.; McKenna, D.; et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 2011, 13, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulou, E.G.; Kountourakis, P.; Karamouzis, M.V.; Doufexis, D.; Ardavanis, A.; Baxevanis, C.N.; Rigatos, G.; Papamichail, M.; Perez, S.A. A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunol. Immunother. 2010, 59, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Veluchamy, J.P.; Kok, N.; van der Vliet, H.J.; Verheul, H.M.W.; de Gruijl, T.D.; Spanholtz, J. The rise of allogeneic natural killer cells as a platform for cancer immunotherapy: Recent innovations and future developments. Front. Immunol. 2017, 8, 631. [Google Scholar] [CrossRef]
- Lupo, K.B.; Matosevic, S. Natural killer cells as allogeneic effectors in adoptive cancer immunotherapy. Cancers 2019, 11, 769. [Google Scholar] [CrossRef]
- Yagita, M.; Huang, C.L.; Umehara, H.; Matsuo, Y.; Tabata, R.; Miyake, M.; Konaka, Y.; Takatsuki, K. A novel natural killer cell line (KHYG-1) from a patient with aggressive natural killer cell leukemia carrying a p53 point mutation. Leukemia 2000, 14, 922–930. [Google Scholar] [CrossRef]
- Tsuchiyama, J.; Yoshino, T.; Mori, M.; Kondoh, E.; Oka, T.; Akagi, T.; Hiraki, A.; Nakayama, H.; Shibuya, A.; Ma, Y.; et al. Characterization of a novel human natural killer-cell line (NK-YS) established from natural killer cell lymphoma/leukemia associated with Epstein-Barr virus infection. Blood 1998, 92, 1374–1383. [Google Scholar] [CrossRef]
- Yodoi, J.; Teshigawara, K.; Nikaido, T.; Fukui, K.; Noma, T.; Honjo, T.; Takigawa, M.; Sasaki, M.; Minato, N.; Tsudo, M.; et al. TCGF (IL 2)-receptor inducing factor(s). I. Regulation of IL 2 receptor on a natural killer-like cell line (YT cells). J. Immunol. 1985, 134, 1623–1630. [Google Scholar]
- Yoneda, N.; Tatsumi, E.; Kawano, S.; Teshigawara, K.; Oka, T.; Fukuda, M.; Yamaguchi, N. Detection of Epstein-Barr virus genome in natural-killer-like cell line, YT. Leukemia 1992, 6, 136–141. [Google Scholar]
- Kornbluth, J.; Flomenberg, N.; Dupont, B. Cell surface phenotype of a cloned line of human natural killer cells. J. Immunol. 1982, 129, 2831–2837. [Google Scholar]
- Robertson, M.J.; Cochran, K.J.; Cameron, C.; Le, J.M.; Tantravahi, R.; Ritz, J. Characterization of a cell line, NKL, derived from an aggressive human natural killer cell leukemia. Exp. Hematol. 1996, 24, 406–415. [Google Scholar] [PubMed]
- Gong, J.H.; Maki, G.; Klingemann, H.G. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 1994, 8, 652–658. [Google Scholar] [PubMed]
- Maki, G.; Klingemann, H.G.; Martinson, J.A.; Tam, Y.K. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J. Hematother. Stem Cell Res. 2001, 10, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Tonn, T.; Becker, S.; Esser, R.; Schwabe, D.; Seifried, E. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J. Hematother. Stem Cell 2001, 10, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Meagher, R.; Swearingen, M.; Myint, H.; Rich, E.; Martinson, J.; Klingemann, H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 2008, 10, 625–632. [Google Scholar] [CrossRef]
- Boyiadzis, M.; Agha, M.; Redner, R.L.; Sehgal, A.; Im, A.; Hou, J.Z.; Farah, R.; Dorritie, K.A.; Raptis, A.; Lim, S.H.; et al. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy 2017, 19, 1225–1232. [Google Scholar] [CrossRef]
- Williams, B.A.; Law, A.D.; Routy, B.; denHollander, N.; Gupta, V.; Wang, X.H.; Chaboureau, A.; Viswanathan, S.; Keating, A. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget 2017, 8, 89256–89268. [Google Scholar] [CrossRef]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.G.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef]
- Konstantinidis, K.V.; Alici, E.; Aints, A.; Christensson, B.; Ljunggren, H.G.; Dilber, M.S. Targeting IL-2 to the endoplasmic reticulum confines autocrine growth stimulation to NK-92 cells. Exp. Hematol. 2005, 33, 159–164. [Google Scholar] [CrossRef]
- Jochems, C.; Hodge, J.W.; Fantini, M.; Fujii, R.; Morillon, Y.M., 2nd; Greiner, J.W.; Padget, M.R.; Tritsch, S.R.; Tsang, K.Y.; Campbell, K.S.; et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget 2016, 7, 86359–86373. [Google Scholar] [CrossRef]
- Jochems, C.; Hodge, J.W.; Fantini, M.; Tsang, K.Y.; Vandeveer, A.J.; Gulley, J.L.; Schlom, J. ADCC employing an NK cell line (haNK) expressing the high affinity CD16 allele with avelumab, an anti-PD-L1 antibody. Int. J. Cancer 2017, 141, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Calderon, H.; Posey, A.D., Jr.; Maus, M.V. Engineering and design of chimeric antigen receptors. Mol. Ther. Methods Clin. Dev. 2019, 12, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Walter, R.B.; Appelbaum, F.R.; Estey, E.H.; Bernstein, I.D. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood 2012, 119, 6198–6208. [Google Scholar] [CrossRef]
- Gill, S.; Tasian, S.K.; Ruella, M.; Shestova, O.; Li, Y.; Porter, D.L.; Carroll, M.; Danet-Desnoyers, G.; Scholler, J.; Grupp, S.A.; et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 2014, 123, 2343–2354. [Google Scholar] [CrossRef]
- You, F.; Wang, Y.; Jiang, L.; Zhu, X.; Chen, D.; Yuan, L.; An, G.; Meng, H.; Yang, L. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am. J. Cancer Res. 2019, 9, 64–78. [Google Scholar]
- Fabian, K.P.; Padget, M.R.; Donahue, R.N.; Solocinski, K.; Robbins, Y.; Allen, C.T.; Lee, J.H.; Rabizadeh, S.; Soon-Shiong, P.; Schlom, J.; et al. PD-L1 targeting high-affinity NK (t-haNK) cells induce direct antitumor effects and target suppressive MDSC populations. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Burstein, H.J. The distinctive nature of HER2-positive breast cancers. N. Engl. J. Med. 2005, 353, 1652–1654. [Google Scholar] [CrossRef]
- Grabsch, H.; Sivakumar, S.; Gray, S.; Gabbert, H.E.; Muller, W. HER2 expression in gastric cancer: Rare, heterogeneous and of no prognostic value—Conclusions from 924 cases of two independent series. Cell Oncol. 2010, 32, 57–65. [Google Scholar] [CrossRef]
- Fléjou, J.F.; Paraf, F.; Muzeau, F.; Fékété, F.; Hénin, D.; Jothy, S.; Potet, F. Expression of c-erbB-2 oncogene product in Barrett’s adenocarcinoma: Pathological and prognostic correlations. J. Clin. Pathol. 1994, 47, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.H.; Shi, Q.; Sukov, W.R.; Wiktor, A.E.; Khan, M.; Sattler, C.A.; Grothey, A.; Wu, T.T.; Diasio, R.B.; Jenkins, R.B.; et al. Association of HER2/ErbB2 expression and gene amplification with pathologic features and prognosis in esophageal adenocarcinomas. Clin. Cancer Res. 2012, 18, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Berchuck, A.; Kamel, A.; Whitaker, R.; Kerns, B.; Olt, G.; Kinney, R.; Soper, J.T.; Dodge, R.; Clarke-Pearson, D.L.; Marks, P.; et al. Overexpression of HER-2/neu is associated with poor survival in advanced epithelial ovarian cancer. Cancer Res. 1990, 50, 4087–4091. [Google Scholar] [PubMed]
- Rolitsky, C.D.; Theil, K.S.; McGaughy, V.R.; Copeland, L.J.; Niemann, T.H. HER-2/neu amplification and overexpression in endometrial carcinoma. Int. J. Gynecol. Pathol. 1999, 18, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, J.; Stumm, G.; Brändle, K.; Merdes, A.; Mechtersheimer, G.; Hynes, N.E.; Kiessling, M. Amplification and differential expression of members of the erbB-gene family in human glioblastoma. J. NeuroOncol. 1994, 22, 201–207. [Google Scholar] [CrossRef]
- Press, M.F.; Cordon-Cardo, C.; Slamon, D.J. Expression of the HER-2/neu proto-oncogene in normal human adult and fetal tissues. Oncogene 1990, 5, 953–962. [Google Scholar]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef]
- Sugawa, N.; Ekstrand, A.J.; James, C.D.; Collins, V.P. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc. Natl. Acad. Sci. USA 1990, 87, 8602–8606. [Google Scholar] [CrossRef]
- Chen, X.; Han, J.; Chu, J.; Zhang, L.; Zhang, J.; Chen, C.; Chen, L.; Wang, Y.; Wang, H.; Yi, L.; et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget 2016, 7, 27764–27777. [Google Scholar] [CrossRef] [PubMed]
- Genssler, S.; Burger, M.C.; Zhang, C.; Oelsner, S.; Mildenberger, I.; Wagner, M.; Steinbach, J.P.; Wels, W.S. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology 2016, 5, e1119354. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Chu, J.; Keung Chan, W.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.H.; Grandi, P.; et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci. Rep. 2015, 5, 11483. [Google Scholar] [CrossRef] [PubMed]
- Gendler, S.J. MUC1, the renaissance molecule. J. Mammary Gland Biol. Neoplasia 2001, 6, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.F.; Zhao, H.L.; Phillips, J.; Greenburg, G. The epithelial mucin, MUC1, is expressed on resting T lymphocytes and can function as a negative regulator of T cell activation. Cell Immunol. 2000, 201, 83–88. [Google Scholar] [CrossRef]
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef]
- Finn, O.J.; Gantt, K.R.; Lepisto, A.J.; Pejawar-Gaddy, S.; Xue, J.; Beatty, P.L. Importance of MUC1 and spontaneous mouse tumor models for understanding the immunobiology of human adenocarcinomas. Immunol. Res. 2011, 50, 261–268. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Y.; Lin, M.; Xia, L.; Bao, Y.; Sun, X.; Yang, L. Abstract A014: Phase I clinical trial with PD-1/MUC1 CAR-pNK92 immunotherapy. Cancer Immunol. Res. 2019, 7, A014. [Google Scholar] [CrossRef]
- Spear, P.; Wu, M.R.; Sentman, M.L.; Sentman, C.L. NKG2D ligands as therapeutic targets. Cancer Immun. 2013, 13, 8. [Google Scholar]
- Duan, S.; Guo, W.; Xu, Z.; He, Y.; Liang, C.; Mo, Y.; Wang, Y.; Xiong, F.; Guo, C.; Li, Y.; et al. Natural killer group 2D receptor and its ligands in cancer immune escape. Mol. Cancer 2019, 18, 29. [Google Scholar] [CrossRef]
- Ruocco, M.G.; Pilones, K.A.; Kawashima, N.; Cammer, M.; Huang, J.; Babb, J.S.; Liu, M.; Formenti, S.C.; Dustin, M.L.; Demaria, S. Suppressing T cell motility induced by anti-CTLA-4 monotherapy improves antitumor effects. J. Clin. Investig. 2012, 122, 3718–3730. [Google Scholar] [CrossRef]
- Hassan, R.; Thomas, A.; Alewine, C.; Le, D.T.; Jaffee, E.M.; Pastan, I. Mesothelin immunotherapy for cancer: Ready for prime time? J. Clin. Oncol. 2016, 34, 4171–4179. [Google Scholar] [CrossRef] [PubMed]
- Mitwasi, N.; Feldmann, A.; Arndt, C.; Koristka, S.; Berndt, N.; Jureczek, J.; Loureiro, L.R.; Bergmann, R.; Mathe, D.; Hegedus, N.; et al. “UniCAR”-modified off-the-shelf NK-92 cells for targeting of GD2-expressing tumour cells. Sci. Rep. 2020, 10, 2141. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Cheung, N.K. Disialoganglioside GD2 as a therapeutic target for human diseases. Expert Opin. Ther. Targets 2015, 19, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [PubMed]
- Sait, S.; Modak, S. Anti-GD2 immunotherapy for neuroblastoma. Expert Rev. Anticancer Ther. 2017, 17, 889–904. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, L.; Song, Y.; Zhang, Y.; Lin, D.; Hu, B.; Mei, Y.; Sandikin, D.; Liu, H. Augmented anti-tumor activity of NK-92 cells expressing chimeric receptors of TGF-betaR II and NKG2D. Cancer Immunol. Immunother. 2017, 66, 537–548. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinical Trial | Indication | Status | NK | NK Source | Trial Phase | n | Outcomes/Key Points |
|---|---|---|---|---|---|---|---|
| NCT03056339 | B Lymphoid Malignancies | Recruiting | iC9/CAR.19/IL-15 NK | Umbilical Cord Blood | Phase I/II | 36 | Safety, relative efficacy; ORS (CR/PR), persistence of infused NK, comprehensive immune reconstitution studies |
| NCT02944162 | Relapsed and Refractory CD33+ Acute Myeloid Leukemia | Unknown | anti-CD33CAR NK | NK-92 | Phase I/II | 10 | Safety; anti-leukemia response; determine anti-NK response |
| NCT03692767 | Relapsed and Refractory B Cell Lymphoma | Not Yet Recruiting | anti-CD22CAR NK | Undisclosed | Early Phase I | 9 | Occurrence of TRAE per CTCAE v4.0 |
| NCT03690310 | Relapsed and Refractory B Cell Lymphoma | Not Yet Recruiting | anti-CD19CAR NK | Undisclosed | Early Phase I | 9 | Occurrence of TRAE per CTCAE v4.0 |
| NCT03824964 | Relapsed and Refractory B Cell Lymphoma | Not Yet Recruiting | anti-CD19/CD22 CAR NK | Undisclosed | Early Phase I | 10 | Occurrence of TRAE per CTCAE v4.0 |
| NCT02892695 | CD19+ Leukemia and Lymphoma | Unknown | anti-CD19CAR NK | NK-92 | Phase I/II | 10 | TRAEs; Objective Response Rate |
| NCT04052061 | Diffuse Large B Cell Lymphoma | Not Yet Recruiting | anti-CD19t-haNK | haNK | Phase I | 18 | MTD, HTD, RP2D, incidence of DLTs and TRAE per CTCAE v5.0; ORR, PFS, OS |
| NCT02742727 | CD7+ Leukemia and Lymphoma | Unknown | anti-CD7CAR-pNK | NK-92 | Phase I/II | 10 | Occurrence of TRAE per CTCAE v4.0; CR and persistence of NK |
| NCT03940833 | Relapsed and Refractory Multiple Myeloma | Recruiting | anti-BCMACAR NK-92 | NK-92 | Phase I/II | 20 | Occurrence of TRAE per CTCAE v4.0 |
| NCT03579927 | B Cell Non-Hodgkin Lymphoma | Withdrawn | CAR.CD19-CD28-zeta2A-iCasp9-IL15 | Umbilical Cord Blood | Phase I/II | 0 | Safety, relative efficacy; RFS, OS, persistence of infused NKs |
| Clinical Trial | Indication | Status | NK | NK Source | Trial Phase | n | Outcomes/Key Points |
|---|---|---|---|---|---|---|---|
| NCT04050709 | Locally Advanced or Metastatic Solid Cancers | Recruiting | anti-PD-L1t-haNK | haNK | Phase I | 16 | MTD, HTD, RP2D, incidence of DLTs and TRAE per CTCAE v5.0; ORR, PFS, OS |
| NCT04390399 | Locally Advanced or Metastatic Pancreatic Cancer | Not Yet Recruiting | anti-PD-L1t-haNK | hanK | Phase II | 268 | PFS per RECIST V1.1; ORR, CR, DCR per RECIST, OS, QoL by Patient Reported Outcomes |
| NCT03383978 | HER2+ Glioblastoma | Recruiting | NK-92/5.28.z | NK-92 | Phase I | 30 | Occurrence of TRAE per CTCAE v4.03, MTD/MFD, persistence of NK, cytokine profile in CSF; ORR, PFR, OS and anti-NK response |
| NCT02839954 | MUC1+ Relapsed or Refractory Solid Tumors | Unknown | anti-MUC1CAR-pNK | PlacentalHSC-derived | Phase I/II | 10 | Occurrence of TRAE per CTCAE v4.0; ORR (PR/CR) |
| NCT03415100 | Metastatic Solid Tumors | Recruiting | anti-NKG2DL CAR NK | Autologous or Allogeneic | Phase I | 30 | Number of adverse events; anti-tumor response |
| NCT03692663 | Castration-Resistant Prostate Cancer | Not Yet Recruiting | anti-PSMACAR NK | Undisclosed | Early Phase I | 9 | Occurrence of TRAE per CTCAE v4.0 |
| NCT03692637 | Epithelial Ovarian Cancer | Not Yet Recruiting | anti-Mesothelin CAR NK | Autologous | Early Phase I | 30 | Occurrence of TRAE per CTCAE v4.0 |
| NCT03940820 | Solid Tumors | Recruiting | anti-ROBO1CAR NK | Undisclosed | Phase I/II | 20 | Occurrence of TRAE per CTCAE v4.0 |
| NCT03941457 | Pancreatic Cancer | Recruiting | anti-ROBO1 BiCAR NK | Undisclosed | Phase I/II | 9 | Occurrence of TRAE per CTCAE v4.03 |
| NCT03931720 | Malignant Tumors | Recruiting | anti-ROBO1 BiCAR NK/T | Undisclosed | Phase I/II | 20 | Occurrence of TRAE per CTCAE v4.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franks, S.E.; Wolfson, B.; Hodge, J.W. Natural Born Killers: NK Cells in Cancer Therapy. Cancers 2020, 12, 2131. https://doi.org/10.3390/cancers12082131
Franks SE, Wolfson B, Hodge JW. Natural Born Killers: NK Cells in Cancer Therapy. Cancers. 2020; 12(8):2131. https://doi.org/10.3390/cancers12082131
Chicago/Turabian StyleFranks, S. Elizabeth, Benjamin Wolfson, and James W. Hodge. 2020. "Natural Born Killers: NK Cells in Cancer Therapy" Cancers 12, no. 8: 2131. https://doi.org/10.3390/cancers12082131
APA StyleFranks, S. E., Wolfson, B., & Hodge, J. W. (2020). Natural Born Killers: NK Cells in Cancer Therapy. Cancers, 12(8), 2131. https://doi.org/10.3390/cancers12082131