Pan-Cancer Analysis of the Genomic Alterations and Mutations of the Matrisome




Abstract
1. Introduction
2. Results and Discussion
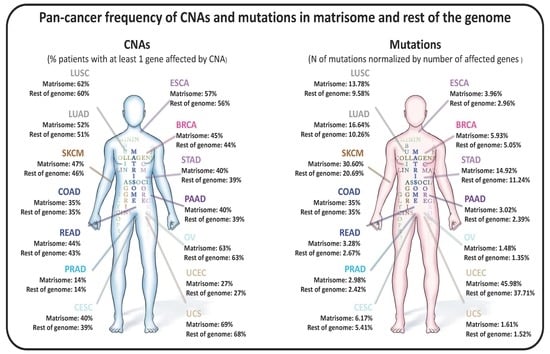
2.1. Copy Number Alterations in Matrisome Genes Are More Frequent than in the Rest of the Genome
2.2. Consequences of CNAs on Matrisome Gene Expression Levels
2.3. Matrisome Genes Are Significantly More Susceptible to Be Mutated
2.4. Mutated Protein Domains and Potential Consequences on ECM Protein Functions
2.5. Functional Consequences of Matrisome Gene Mutations
2.6. Identification of the Top 10 Most Mutated Matrisome Genes across 14 Cancer Types
2.7. Consequences of Matrisome Gene Mutations on Patient Survival
2.8. Cross-Validation Using Independent Cancer Patient Cohorts
3. Methods
3.1. Source Data
3.2. Matrisome Gene List
3.3. Gene Expression Data
3.4. Copy Number Alterations (CNAs)
3.5. Clinical Data
3.6. Pan-Cancer Purity Data
3.7. Cross-Validation Data
3.8. Statistical Analysis
3.9. Data Availability
3.10. Code Availability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bender, E. Big data in biomedicine. Nature 2015, 527, S1. [Google Scholar] [CrossRef]
- Hutter, C.; Zenklusen, J.C. The Cancer Genome Atlas: Creating Lasting Value beyond Its Data. Cell 2018, 173, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Bailey, M.H.; Porta-Pardo, E.; Thorsson, V.; Colaprico, A.; Bertrand, D.; Gibbs, D.L.; Weerasinghe, A.; Huang, K.; Tokheim, C.; et al. Perspective on Oncogenic Processes at the End of the Beginning of Cancer Genomics. Cell 2018, 173, 305–320.e10. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Yang, T.-H.O.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Pleasance, E.; Titmuss, E.; Williamson, L.; Kwan, H.; Culibrk, L.; Zhao, E.Y.; Dixon, K.; Fan, K.; Bowlby, R.; Jones, M.R.; et al. Pan-cancer analysis of advanced patient tumors reveals interactions between therapy and genomic landscapes. Nat. Cancer 2020, 1, 452–468. [Google Scholar] [CrossRef]
- DeClerck, Y.A.; Pienta, K.J.; Woodhouse, E.C.; Singer, D.S.; Mohla, S. The Tumor Microenvironment at a Turning Point Knowledge Gained Over the Last Decade, and Challenges and Opportunities Ahead: A White Paper from the NCI TME Network. Cancer Res. 2017, 77, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Polyak, K. Microenvironmental regulation of cancer development. Curr. Opin. Genet. Dev. 2008, 18, 27–34. [Google Scholar] [CrossRef]
- Brassart-Pasco, S.; Brézillon, S.; Brassart, B.; Ramont, L.; Oudart, J.-B.; Monboisse, J.C. Tumor Microenvironment: Extracellular Matrix Alterations Influence Tumor Progression. Front. Oncol. 2020, 10, 397. [Google Scholar] [CrossRef]
- Hynes, R.O.; Yamada, K.M. Extracellular Matrix Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2012. [Google Scholar]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef]
- Anastassiades, O.T.; Pryce, D.M. Fibrosis as an indication of time in infiltrating breast cancer and its importance in prognosis. Br. J. Cancer 1974, 29, 232–239. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Socovich, A.M.; Naba, A. The cancer matrisome: From comprehensive characterization to biomarker discovery. Semin. Cell Dev. Biol. 2019, 89, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Lelkes, P.I.; Cukierman, E. Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol. 2015, 33, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Taha, I.N.; Naba, A. Exploring the extracellular matrix in health and disease using proteomics. Essays Biochem. 2019, 63, 417–432. [Google Scholar] [CrossRef]
- Ricard-Blum, S.; Naba, A. The Extracellular Matrix Goes-Omics: Resources and Tools. In Extracellular Matrix Omics; Biology of Extracellular Matrix; Springer Nature: Basel, Switzerland, 2020; accepted. [Google Scholar]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The matrisome: In silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell Proteom. 2012, 11, M111.014647. [Google Scholar] [CrossRef]
- Hynes, R.O.; Naba, A. Overview of the Matrisome-An Inventory of Extracellular Matrix Constituents and Functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef]
- Tomko, L.A.; Hill, R.C.; Barrett, A.; Szulczewski, J.M.; Conklin, M.W.; Eliceiri, K.W.; Keely, P.J.; Hansen, K.C.; Ponik, S.M. Targeted matrisome analysis identifies thrombospondin-2 and tenascin-C in aligned collagen stroma from invasive breast carcinoma. Sci. Rep. 2018, 8, 12941. [Google Scholar] [CrossRef]
- Hebert, J.D.; Myers, S.A.; Naba, A.; Abbruzzese, G.; Lamar, J.; Carr, S.A.; Hynes, R.O. Proteomic profiling of the ECM of xenograft breast cancer metastases in different organs reveals distinct metastatic niches. Cancer Res. 2020, 80. [Google Scholar] [CrossRef]
- Naba, A.; Clauser, K.R.; Lamar, J.M.; Carr, S.A.; Hynes, R.O. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. eLife 2014, 3, e01308. [Google Scholar] [CrossRef]
- Gocheva, V.; Naba, A.; Bhutkar, A.; Guardia, T.; Miller, K.M.; Li, C.M.-C.; Dayton, T.L.; Sanchez-Rivera, F.J.; Kim-Kiselak, C.; Jailkhani, N.; et al. Quantitative proteomics identify Tenascin-C as a promoter of lung cancer progression and contributor to a signature prognostic of patient survival. Proc. Natl. Acad. Sci. USA 2017, 114, E5625–E5634. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Clauser, K.R.; Öhlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R.; Carr, S.A.; Tuveson, D.A.; Hynes, R.O. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19609–19618. [Google Scholar] [CrossRef] [PubMed]
- Pearce, O.M.T.; Delaine-Smith, R.M.; Maniati, E.; Nichols, S.; Wang, J.; Böhm, S.; Rajeeve, V.; Ullah, D.; Chakravarty, P.; Jones, R.R.; et al. Deconstruction of a Metastatic Tumor Microenvironment Reveals a Common Matrix Response in Human Cancers. Cancer Discov. 2018, 8, 304–319. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Maniati, E.; Berlato, C.; Gopinathan, G.; Heath, O.; Kotantaki, P.; Lakhani, A.; McDermott, J.; Pegrum, C.; Delaine-Smith, R.M.; Pearce, O.M.T.; et al. Mouse Ovarian Cancer Models Recapitulate the Human Tumor Microenvironment and Patient Response to Treatment. Cell Rep. 2020, 30, 525–540.e7. [Google Scholar] [CrossRef]
- Mitra, S.; Tiwari, K.; Podicheti, R.; Pandhiri, T.; Rusch, D.B.; Bonetto, A.; Zhang, C.; Mitra, A.K. Transcriptome Profiling Reveals Matrisome Alteration as a Key Feature of Ovarian Cancer Progression. Cancers 2019, 11, 1513. [Google Scholar] [CrossRef]
- Izzi, V.; Lakkala, J.; Devarajan, R.; Ruotsalainen, H.; Savolainen, E.-R.; Koistinen, P.; Heljasvaara, R.; Pihlajaniemi, T. An extracellular matrix signature in leukemia precursor cells and acute myeloid leukemia. Haematologica 2017, 102, e245–e248. [Google Scholar] [CrossRef]
- Lim, S.B.; Chua, M.L.K.; Yeong, J.P.S.; Tan, S.J.; Lim, W.-T.; Lim, C.T. Pan-cancer analysis connects tumor matrisome to immune response. NPJ Precis. Oncol. 2019, 3. [Google Scholar] [CrossRef]
- Izzi, V.; Lakkala, J.; Devarajan, R.; Kääriäinen, A.; Koivunen, J.; Heljasvaara, R.; Pihlajaniemi, T. Pan-Cancer analysis of the expression and regulation of matrisome genes across 32 tumor types. Matrix Biol. Plus 2019, 1, 100004. [Google Scholar] [CrossRef]
- Izzi, V.; Lakkala, J.; Devarajan, R.; Savolainen, E.-R.; Koistinen, P.; Heljasvaara, R.; Pihlajaniemi, T. Expression of a specific extracellular matrix signature is a favorable prognostic factor in acute myeloid leukemia. Leuk. Res. Rep. 2018, 9, 9–13. [Google Scholar] [CrossRef]
- Yuzhalin, A.E.; Urbonas, T.; Silva, M.A.; Muschel, R.J.; Gordon-Weeks, A.N. A core matrisome gene signature predicts cancer outcome. Br. J. Cancer 2018, 118, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Langlois, B.; Saupe, F.; Rupp, T.; Arnold, C.; van der Heyden, M.; Orend, G.; Hussenet, T. AngioMatrix, a signature of the tumor angiogenic switch-specific matrisome, correlates with poor prognosis for glioma and colorectal cancer patients. Oncotarget 2014, 5, 10529–10545. [Google Scholar] [CrossRef] [PubMed]
- Lamandé, S.R.; Bateman, J.F. Genetic Disorders of the Extracellular Matrix. Anat. Rec. 2019, 303. [Google Scholar] [CrossRef] [PubMed]
- Izzi, V.; Koivunen, J.; Rappu, P.; Heino, J.; Pihlajaniemi, T. Integration of Matrisome Omics: Towards System Biology of the Tumor Matrisome. In Extracellular Matrix Omics; Biology of Extracellular Matrix; Springer: Basel, Switzerland, 2020; Accepted. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Shao, X.; Lv, N.; Liao, J.; Long, J.; Xue, R.; Ai, N.; Xu, D.; Fan, X. Copy number variation is highly correlated with differential gene expression: A pan-cancer study. BMC Med. Genet. 2019, 20, 175. [Google Scholar] [CrossRef]
- Ofria, C.; Adami, C.; Collier, T.C. Selective pressures on genomes in molecular evolution. J. Theor. Biol. 2003, 222, 477–483. [Google Scholar] [CrossRef][Green Version]
- Metzgar, D.; Wills, C. Evidence for the Adaptive Evolution of Mutation Rates. Cell 2000, 101, 581–584. [Google Scholar] [CrossRef]
- Hibi, K.; Mizukami, H.; Saito, M.; Kigawa, G.; Nemoto, H.; Sanada, Y. FBN2 Methylation Is Detected in the Serum of Colorectal Cancer Patients with Hepatic Metastasis. Anticancer Res. 2012, 32, 4371–4374. [Google Scholar]
- Yang, Y.; Xing, Y.; Liang, C.; Hu, L.; Xu, F.; Mei, Q. An examination of the regulatory mechanism of Pxdn mutation-induced eye disorders using microarray analysis. Int. J. Mol. Med. 2016, 37, 1449–1456. [Google Scholar] [CrossRef][Green Version]
- Song, K.; Bi, J.-H.; Qiu, Z.-W.; Felizardo, R.; Girard, L.; Minna, J.D.; Gazdar, A.F. A quantitative method for assessing smoke associated molecular damage in lung cancers. Transl. Lung Cancer Res. 2018, 7, 439–449. [Google Scholar] [CrossRef]
- Efthymiou, G.; Saint, A.; Ruff, M.; Rekad, Z.; Ciais, D.; Van Obberghen-Schilling, E. Shaping Up the Tumor Microenvironment with Cellular Fibronectin. Front. Oncol 2020, 10, 641. [Google Scholar] [CrossRef] [PubMed]
- Frey, K.; Fiechter, M.; Schwager, K.; Belloni, B.; Barysch, M.J.; Neri, D.; Dummer, R. Different patterns of fibronectin and tenascin-C splice variants expression in primary and metastatic melanoma lesions. Exp. Dermatol. 2011, 20, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Binding to the Extracellular Matrix and Proteolytic Processing: Two Key Mechanisms Regulating Vascular Endothelial Growth Factor Action. Mol. Biol. Cell 2010, 21, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Mardon, H.J.; Grant, R.P.; Grant, K.E.; Harris, H. Fibronectin splice variants are differentially incorporated into the extracellular matrix of tumorigenic and non-tumorigenic hybrids between normal fibroblasts and sarcoma cells. J. Cell Sci. 1993, 104, 783–792. [Google Scholar]
- Jailkhani, N.; Ingram, J.R.; Rashidian, M.; Rickelt, S.; Tian, C.; Mak, H.; Jiang, Z.; Ploegh, H.L.; Hynes, R.O. Noninvasive imaging of tumor progression, metastasis, and fibrosis using a nanobody targeting the extracellular matrix. Proc. Natl. Acad. Sci. USA 2019, 116, 14181–14190. [Google Scholar] [CrossRef]
- Pasche, N.; Frey, K.; Neri, D. The targeted delivery of IL17 to the mouse tumor neo-vasculature enhances angiogenesis but does not reduce tumor growth rate. Angiogenesis 2012, 15, 165–169. [Google Scholar] [CrossRef]
- Hohenester, E.; Engel, J. Domain structure and organisation in extracellular matrix proteins. Matrix Biol. 2002, 21, 115–128. [Google Scholar] [CrossRef]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Jayadev, R.; Sherwood, D.R. Basement membranes. Curr. Biol. 2017, 27, R207–R211. [Google Scholar] [CrossRef]
- Kikutake, C.; Yoshihara, M.; Sato, T.; Saito, D.; Suyama, M. Intratumor heterogeneity of HMCN1 mutant alleles associated with poor prognosis in patients with breast cancer. Oncotarget 2018, 9, 33337–33347. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Je, E.M.; Yoo, N.J.; Lee, S.H. HMCN1, a cell polarity-related gene, is somatically mutated in gastric and colorectal cancers. Pathol. Oncol. Res. 2015, 21, 847–848. [Google Scholar] [CrossRef] [PubMed]
- King, R.J.; Yu, F.; Singh, P.K. Genomic alterations in mucins across cancers. Oncotarget 2017, 8, 67152–67168. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Hoersch, S.; Hynes, R.O. Towards definition of an ECM parts list: An advance on GO categories. Matrix Biol. 2012, 31, 371–372. [Google Scholar] [CrossRef] [PubMed]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.F.; Parker, J.S.; Reynolds, S.M.; Silva, T.C.; Wang, L.-B.; Zhou, W.; Akbani, R.; Bailey, M.; Balu, S.; Berman, B.P.; et al. Before and After: Comparison of Legacy and Harmonized TCGA Genomic Data Commons’ Data. Cell Syst. 2019, 9, 24–34.e10. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef]
- Aran, D.; Sirota, M.; Butte, A.J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015, 6, 8971. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Hieronymus, H.; Murali, R.; Tin, A.; Yadav, K.; Abida, W.; Moller, H.; Berney, D.; Scher, H.; Carver, B.; Scardino, P.; et al. Tumor copy number alteration burden is a pan-cancer prognostic factor associated with recurrence and death. eLife 2018, 7, e37294. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Cancer Type | Estimated New Cases in 2020 in the US | Estimated Deaths in 2020 in the US | 5-Year Survival (2009–2015) |
|---|---|---|---|---|
| BRCA | Breast Carcinoma | 279,100 | 42,690 | 91% |
| CESC | Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma | 13,800 | 4290 | 69% |
| COAD/READ | Colon Adenocarcinoma/Rectum Adenocarcinoma | 147,950 | 53,200 | 66% |
| ESCA | Esophageal Carcinoma | 18,440 | 16,700 | 21% |
| LUSC/LUAD | Lung Squamous Cell Carcinoma/Lung Adenocarcinoma | 228,820 | 135,720 | 21% |
| OV | Ovarian Serous Cystadenocarcinoma | 21,750 | 13,940 | 48% |
| PAAD | Pancreatic Adenocarcinoma | 57,600 | 47,050 | 10% |
| PRAD | Prostate Adenocarcinoma | 191,930 | 33,330 | 99% |
| SKCM | Skin Cutaneous Melanoma | 100,350 | 6850 | 94% |
| STAD | Stomach Adenocarcinoma | 27,600 | 11,010 | 32% |
| UCS/UCEC | Uterine Carcinosarcoma/Uterine Corpus Endometrial Carcinoma | 65,620 | 12,590 | 83% |
| Total | 1,152,960 | 377,370 |
| Abbreviation | Cancer Type | # Of Patients in TCGA | # Of Patients with Matrisome CNAs | # Of Patients with Matrisome Mutations |
|---|---|---|---|---|
| BRCA | Breast Carcinoma | 1236 | 773 (63%) | 749 (61%) |
| CESC | Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma | 312 | 276 (88%) | 278 (89%) |
| COAD/READ | Colon Adenocarcinoma/Rectum Adenocarcinoma | 545/183 | 270 (50%)/87 (48%) | 288 (53%)/87 (48%) |
| ESCA | Esophageal Carcinoma | 204 | 181 (89%) | 183 (90%) |
| LUAD/LUSC | Lung Adenocarcinoma/Lung Squamous Cell Carcinoma | 641/623 | 504 (79%)/473 (76%) | 506 (80%)/475 (76%) |
| OV | Ovarian Serous Cystadenocarcinoma | 604 | 59 (10%) | 58 (10%) |
| PAAD | Pancreatic Adenocarcinoma | 196 | 156 (80%) | 155 (79%) |
| PRAD | Prostate Adenocarcinoma | 566 | 447 (79%) | 437 (77%) |
| SKCM | Skin Cutaneous Melanoma | 479 | 359 (75%) | 356 (74%) |
| STAD | Stomach Adenocarcinoma | 511 | 424 (83%) | 429 (84%) |
| UCEC/UCS | Uterine Corpus Endometrial Carcinoma/Uterine Carcinosarcoma | 583/57 | 368 (63%)/56 (98%) | 440 (75%)/56 (98%) |
| Total | 6740 | 4433 (66%) | 4497 (67%) |
| A. Mutated core matrisome genes with a negative impact on overall survival based on univariate and multivariate analyses | ||||||
| Tumor | Gene | Matrisome Category | OS Difference | p-Value, Univariate | p-Value, Multivariate | # Cases with Mutations |
| COAD | OTOL1 | ECM Glycoproteins | −1.811 | 0.021 | 0.010 | 10 |
| COAD | MATN2 | ECM Glycoproteins | −1.675 | 0.040 | 0.048 | 11 |
| COAD | NELL2 | ECM Glycoproteins | −1.271 | 0.018 | 0.012 | 16 |
| COAD | LTBP4 | ECM Glycoproteins | −1.040 | 0.010 | 0.011 | 23 |
| LUAD | MMRN2 | ECM Glycoproteins | −1.910 | 0.000 | 0.000 | 11 |
| LUAD | LAMC2 | ECM Glycoproteins | −1.841 | 0.017 | 0.016 | 11 |
| LUAD | COL22A1 | Collagens | −1.595 | 0.012 | 0.009 | 63 |
| LUAD | LAMB3 | ECM Glycoproteins | −1.106 | 0.008 | 0.011 | 22 |
| LUSC | CILP2 | ECM Glycoproteins | −2.269 | 0.036 | 0.026 | 14 |
| LUSC | COL2A1 | Collagens | −1.099 | 0.010 | 0.026 | 22 |
| PRAD | MXRA5 | ECM Glycoproteins | −1.146 | 0.007 | 0.038 | 10 |
| SKCM | COL6A1 | Collagens | −1.478 | 0.014 | 0.028 | 10 |
| B. Mutated core matrisome genes with a positive impact on overall survival based on univariate and multivariate analyses | ||||||
| Tumor | Gene | Matrisome Category | OS Difference | p-Value, Univariate | p-Value, Multivariate | # Cases with Mutations |
| COAD | COL6A1 | Collagens | 1.026 | 0.014 | 0.036 | 17 |
| LUAD | TNC | ECM Glycoproteins | 1.382 | 0.014 | 0.040 | 18 |
| LUAD | MMRN1 | ECM Glycoproteins | 1.414 | 0.017 | 0.029 | 43 |
| LUSC | COL25A1 | Collagens | 1.860 | 0.015 | 0.038 | 16 |
| SKCM | ACAN | Proteoglycans | 1.405 | 0.025 | 0.006 | 64 |
| SKCM | COL4A6 | Collagens | 1.579 | 0.010 | 0.007 | 48 |
| SKCM | HSPG2 | Proteoglycans | 1.650 | 0.029 | 0.021 | 38 |
| SKCM | COL4A3 | Collagens | 1.755 | 0.005 | 0.004 | 49 |
| STAD | COL15A1 | Collagens | 1.201 | 0.015 | 0.012 | 31 |
| STAD | VWF | ECM Glycoproteins | 1.215 | 0.015 | 0.012 | 32 |
| STAD | TECTA | ECM Glycoproteins | 1.242 | 0.009 | 0.014 | 41 |
| STAD | NELL2 | ECM Glycoproteins | 1.398 | 0.004 | 0.023 | 19 |
| STAD | COL4A1 | Collagens | 1.495 | 0.049 | 0.026 | 31 |
| STAD | LAMB3 | ECM Glycoproteins | 1.568 | 0.026 | 0.030 | 22 |
| STAD | COL5A2 | Collagens | 1.720 | 0.012 | 0.009 | 22 |
| UCEC | FBN2 | ECM Glycoproteins | 1.059 | 0.005 | 0.036 | 79 |
| UCEC | RELN | ECM Glycoproteins | 1.163 | 0.010 | 0.044 | 61 |
| UCEC | FRAS1 | ECM Glycoproteins | 1.259 | 0.015 | 0.037 | 66 |
| A. Mutated genes encoding ECM regulators or ECM-affiliated proteins with a negative impact on overall survival based on univariate and multivariate analyses | ||||||
| Tumor | Gene | Matrisome Category | OS Difference | p-Value, Univariate | p-Value, Multivariate | # Cases with Mutations |
| BRCA | SULF2 | ECM Regulators | −1.443 | 0.037 | 0.047 | 10 |
| COAD | ADAMTS15 | ECM Regulators | −1.238 | 0.002 | 0.003 | 12 |
| COAD | GPC6 | ECM-affiliated | −1.100 | 0.031 | 0.043 | 15 |
| COAD | GPC5 | ECM-affiliated | −1.009 | 0.039 | 0.046 | 11 |
| LUAD | FCN2 | ECM-affiliated | −1.473 | 0.047 | 0.036 | 13 |
| LUAD | ADAM19 | ECM Regulators | −1.340 | 0.012 | 0.012 | 31 |
| LUAD | PLXNA4 | ECM-affiliated | −1.065 | 0.041 | 0.040 | 52 |
| LUAD | MMP16 | ECM Regulators | −1.037 | 0.015 | 0.029 | 54 |
| LUSC | PLG | ECM Regulators | −1.770 | 0.042 | 0.008 | 14 |
| LUSC | ITIH6 | ECM Regulators | −1.565 | 0.028 | 0.016 | 25 |
| SKCM | PZP | ECM Regulators | −1.565 | 0.022 | 0.031 | 33 |
| SKCM | CLEC6A | ECM-affiliated | −1.460 | 0.028 | 0.013 | 18 |
| SKCM | SEMA5A | ECM-affiliated | −1.252 | 0.022 | 0.035 | 26 |
| B. Mutated genes encoding ECM regulators or ECM-affiliated proteins with a positive impact on overall survival based on univariate and multivariate analyses | ||||||
| Tumor | Gene | Matrisome Category | OS Difference | p-Value, Univariate | p-Value, Multivariate | # Cases with Mutations |
| BRCA | PLXNA2 | ECM-affiliated | 1.011 | 0.013 | 0.030 | 15 |
| LUAD | MUC5B | ECM-affiliated | 1.296 | 0.012 | 0.014 | 53 |
| LUAD | ADAMTS5 | ECM Regulators | 1.342 | 0.015 | 0.016 | 32 |
| LUSC | ADAMTSL1 | ECM Regulators | 1.624 | 0.008 | 0.025 | 16 |
| LUSC | TGM7 | ECM Regulators | 2.123 | 0.009 | 0.043 | 10 |
| SKCM | FREM2 | ECM-affiliated | 1.371 | 0.036 | 0.004 | 51 |
| SKCM | COLEC12 | ECM-affiliated | 1.703 | 0.047 | 0.031 | 20 |
| SKCM | MUC16 | ECM-affiliated | 2.100 | 0.000 | 0.000 | 251 |
| STAD | MUC16 | ECM-affiliated | 1.077 | 0.040 | 0.027 | 145 |
| STAD | ADAMTSL3 | ECM Regulators | 1.186 | 0.025 | 0.029 | 25 |
| STAD | SULF1 | ECM Regulators | 1.246 | 0.025 | 0.035 | 30 |
| STAD | MUC4 | ECM-affiliated | 1.301 | 0.027 | 0.024 | 29 |
| STAD | CSPG4 | ECM-affiliated | 1.551 | 0.026 | 0.020 | 26 |
| STAD | ADAM12 | ECM Regulators | 1.913 | 0.046 | 0.042 | 12 |
| STAD | MMP3 | ECM Regulators | 1.916 | 0.031 | 0.026 | 13 |
| STAD | SERPINB8 | ECM Regulators | 2.546 | 0.014 | 0.027 | 10 |
| UCEC | MUC5B | ECM-affiliated | 1.128 | 0.019 | 0.043 | 102 |
| UCEC | PLXNB3 | ECM-affiliated | 1.154 | 0.026 | 0.050 | 60 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izzi, V.; Davis, M.N.; Naba, A. Pan-Cancer Analysis of the Genomic Alterations and Mutations of the Matrisome. Cancers 2020, 12, 2046. https://doi.org/10.3390/cancers12082046
Izzi V, Davis MN, Naba A. Pan-Cancer Analysis of the Genomic Alterations and Mutations of the Matrisome. Cancers. 2020; 12(8):2046. https://doi.org/10.3390/cancers12082046
Chicago/Turabian StyleIzzi, Valerio, Martin N. Davis, and Alexandra Naba. 2020. "Pan-Cancer Analysis of the Genomic Alterations and Mutations of the Matrisome" Cancers 12, no. 8: 2046. https://doi.org/10.3390/cancers12082046
APA StyleIzzi, V., Davis, M. N., & Naba, A. (2020). Pan-Cancer Analysis of the Genomic Alterations and Mutations of the Matrisome. Cancers, 12(8), 2046. https://doi.org/10.3390/cancers12082046