Targeting STAT3 and STAT5 in Cancer



{kind=link}
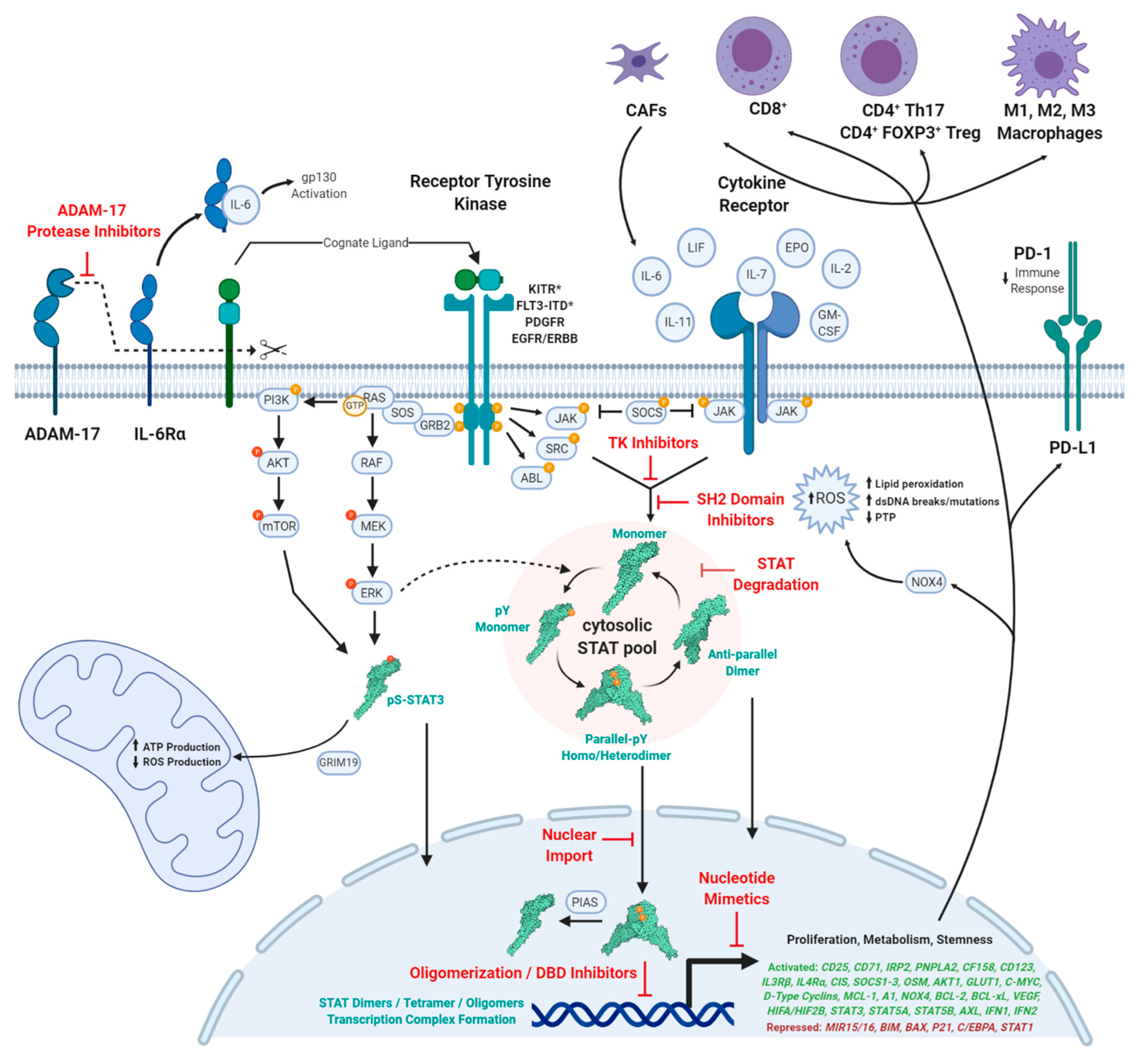
1. Introduction
2. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Abate, F.; Lasorsa, E.; Tabbo’, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccarotella, E.; Barbarossa, L.; Ercole, E.; et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 2015, 27, 516–532. [Google Scholar] [CrossRef] [PubMed]
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Müller, M.; Moriggl, R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Wang, S.Q.; Tan, D.; Gao, Y.; Lin, G.; Xi, R. EGFR, Wingless and JAK/STAT signaling cooperatively maintain Drosophila intestinal stem cells. Dev. Biol. 2011, 354, 31–43. [Google Scholar] [CrossRef]
- Zou, W.Y.; Blutt, S.E.; Zeng, X.L.; Chen, M.S.; Lo, Y.H.; Castillo-Azofeifa, D.; Klein, O.D.; Shroyer, N.F.; Donowitz, M.; Estes, M.K. Epithelial WNT Ligands Are Essential Drivers of Intestinal Stem Cell Activation. Cell Rep. 2018, 22, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 2017. [Google Scholar] [CrossRef]
- Jaiswal, S.; Libby, P. Clonal haematopoiesis: Connecting ageing and inflammation in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 137–144. [Google Scholar] [CrossRef]
- Libby, P.; Sidlow, R.; Lin, A.E.; Gupta, D.; Jones, L.W.; Moslehi, J.; Zeiher, A.; Jaiswal, S.; Schulz, C.; Blankstein, R.; et al. Clonal Hematopoiesis: Crossroads of Aging, Cardiovascular Disease, and Cancer: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 567–577. [Google Scholar] [CrossRef]
- de Araujo, E.D.; Erdogan, F.; Neubauer, H.A.; Meneksedag-Erol, D.; Manaswiyoungkul, P.; Eram, M.S.; Seo, H.S.; Qadree, A.K.; Israelian, J.; Orlova, A.; et al. Structural and functional consequences of the STAT5BN642H driver mutation. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT Pathway: Impact on Human Disease and Therapeutic Intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef]
- Kiu, H.; Nicholson, S.E. Biology and significance of the JAK/STAT signalling pathways. Growth Factors 2012, 30, 88–106. [Google Scholar] [CrossRef] [PubMed]
- Timofeeva, O.A.; Chasovskikh, S.; Lonskaya, I.; Tarasova, N.I.; Khavrutskii, L.; Tarasov, S.G.; Zhang, X.; Korostyshevskiy, V.R.; Cheema, A.; Zhang, L.; et al. Mechanisms of unphosphorylated STAT3 transcription factor binding to DNA. J. Biol. Chem. 2012, 287, 14192–14200. [Google Scholar] [CrossRef] [PubMed]
- Rincon, M.; Pereira, F.V. A new perspective: Mitochondrial stat3 as a regulator for lymphocyte function. Int. J. Mol. Sci. 2018, 19, 1656. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Vargas, H.; Ouzounova, M.; le Calvez-Kelm, F.; Lambert, M.P.; McKay-Chopin, S.; Tavtigian, S.V.; Puisieux, A.; Matar, C.; Herceg, Z. Methylome analysis reveals Jak-STAT pathway deregulation in putative breast cancer stem cells. Epigenetics 2011, 6, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017, 15. [Google Scholar] [CrossRef]
- Kim, M.; Morales, L.D.; Jang, I.S.; Cho, Y.Y.; Kim, D.J. Protein Tyrosine Phosphatases as Potential Regulators of STAT3 Signaling. Int. J. Mol. Sci. 2018, 19, 2708. [Google Scholar] [CrossRef]
- Abdeldayem, A.; Raouf, Y.S.; Constantinescu, S.N.; Moriggl, R.; Gunning, P.T. Advances in covalent kinase inhibitors. Chem. Soc. Rev. 2020, 49, 2617–2687. [Google Scholar] [CrossRef]
- Ribas, A.; Lo, R.S. Trying for a BRAF Slam Dunk. Cancer Discov. 2020, 10, 640–642. [Google Scholar] [CrossRef]
- Brachet-Botineau, M.; Polomski, M.; Neubauer, H.A.; Juen, L.; Hédou, D.; Viaud-Massuard, M.C.; Prié, G.; Gouilleux, F. Pharmacological inhibition of oncogenic STAT3 and STAT5 signaling in hematopoietic cancers. Cancers 2020, 12, 240. [Google Scholar] [CrossRef]
- Orlova, A.; Wagner, C.; De Araujo, E.D.; Bajusz, D.; Neubauer, H.A.; Herling, M.; Gunning, P.T.; Keserü, G.M.; Moriggl, R. Direct targeting options for STAT3 and STAT5 in cancer. Cancers 2019, 11, 1930. [Google Scholar] [CrossRef]
- Verdeil, G.; Lawrence, T.; Schmitt-Verhulst, A.M.; Auphan-Anezin, N. Targeting stat3 and stat5 in tumor-associated immune cells to improve immunotherapy. Cancers 2019, 11, 1832. [Google Scholar] [CrossRef] [PubMed]
- Rébé, C.; Ghiringhelli, F. STAT3, a master regulator of anti-tumor immune response. Cancers 2019, 11, 1280. [Google Scholar] [CrossRef] [PubMed]
- de Araujo, E.D.; Orlova, A.; Neubauer, H.A.; Bajusz, D.; Seo, H.S.; Dhe-Paganon, S.; Keserű, G.M.; Moriggl, R.; Gunning, P.T. Structural implications of stat3 and stat5 sh2 domain mutations. Cancers 2019, 11, 1757. [Google Scholar] [CrossRef]
- Hadzijusufovic, E.; Keller, A.; Berger, D.; Greiner, G.; Wingelhofer, B.; Witzeneder, N.; Ivanov, D.; Pecnard, E.; Nivarthi, H.; Schur, F.K.M.; et al. STAT5 is expressed in CD34+/CD38− stem cells and serves as a potential molecular target in ph-negative myeloproliferative neoplasms. Cancers 2020, 12, 1021. [Google Scholar] [CrossRef]
- Wahnschaffe, L.; Braun, T.; Timonen, S.; Giri, A.K.; Schrader, A.; Wagle, P.; Almusa, H.; Johansson, P.; Bellanger, D.; López, C.; et al. Jak/stat-activating genomic alterations are a hallmark of t-pll. Cancers 2019, 11, 1833. [Google Scholar] [CrossRef] [PubMed]
- Andersson, E.I.; Brück, O.; Braun, T.; Mannisto, S.; Saikko, L.; Lagström, S.; Ellonen, P.; Leppä, S.; Herling, M.; Kovanen, P.E.; et al. STAT3 mutation is associated with STAT3 activation in CD30+ ALK− ALCL. Cancers 2020, 12, 702. [Google Scholar] [CrossRef] [PubMed]
- Brachet-Botineau, M.; Deynoux, M.; Vallet, N.; Polomski, M.; Juen, L.; Hérault, O.; Mazurier, F.; Viaud-Massuard, M.C.; Prié, G.; Gouilleux, F. A novel inhibitor of stat5 signaling overcomes chemotherapy resistance in myeloid leukemia cells. Cancers 2019, 11, 2043. [Google Scholar] [CrossRef]
- Igelmann, S.; Neubauer, H.A.; Ferbeyre, G. STAT3 and STAT5 activation in solid cancers. Cancers 2019, 11, 1428. [Google Scholar] [CrossRef]
- Polak, K.L.; Chernosky, N.M.; Smigiel, J.M.; Tamagno, I.; Wjackson, M. Balancing STAT activity as a therapeutic strategy. Cancers 2019, 11, 1716. [Google Scholar] [CrossRef]
- Logotheti, S.; Pützer, B.M. STAT3 and STAT5 targeting for simultaneous management of melanoma and autoimmune diseases. Cancers 2019, 11, 1448. [Google Scholar] [CrossRef]
- Schumacher, N.; Rose-John, S. Adam17 activity and il-6 trans-signaling in inflammation and cancer. Cancers 2019, 11, 1736. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.J.; Sundararajan, V.; Sheu, B.C.; Huang, R.Y.J.; Wei, L.H. Activation of STAT3 and STAT5 signaling in epithelial ovarian cancer progression: Mechanism and therapeutic opportunity. Cancers 2020, 12, 24. [Google Scholar] [CrossRef] [PubMed]
- Boutillon, F.; Pigat, N.; Sala, L.S.; Reyes-Gomez, E.; Moriggl, R.; Guidotti, J.E.; Goffin, V. STAT5a/b deficiency delays, but does not prevent, prolactin-driven prostate tumorigenesis in mice. Cancers 2019, 11, 929. [Google Scholar] [CrossRef] [PubMed]
- Zerdes, I.; Wallerius, M.; Sifakis, E.G.; Wallmann, T.; Betts, S.; Bartish, M.; Tsesmetzis, N.; Tobin, N.P.; Coucoravas, C.; Bergh, J.; et al. STAT3 activity promotes programmed-death ligand 1 expression and suppresses immune responses in breast cancer. Cancers 2019, 11, 1479. [Google Scholar] [CrossRef] [PubMed]
- Linder, B.; Weirauch, U.; Ewe, A.; Uhmann, A.; Seifert, V.; Mittelbronn, M.; Harter, P.N.; Aigner, A.; Kögel, D. Therapeutic targeting of stat3 using lipopolyplex nanoparticle-formulated sirna in a syngeneic orthotopic mouse glioma model. Cancers 2019, 11, 333. [Google Scholar] [CrossRef]
- Aydin, Y.; Kurt, R.; Song, K.; Lin, D.; Osman, H.; Youngquist, B.; Scott, J.W.; Shores, N.J.; Thevenot, P.; Cohen, A.; et al. Hepatic stress response in HCV infection promotes STAT3-mediated inhibition of HNF4A-miR-122 feedback loop in liver fibrosis and cancer progression. Cancers 2019, 11, 1407. [Google Scholar] [CrossRef]
- Huang, Y.H.; Vakili, M.R.; Molavi, O.; Morrissey, Y.; Wu, C.; Paiva, I.; Soleimani, A.H.; Sanaee, F.; Lavasanifar, A.; Lai, R. Decoration of anti-CD38 on nanoparticles carrying a STAT3 inhibitor can improve the therapeutic efficacy against myeloma. Cancers 2019, 11, 248. [Google Scholar] [CrossRef]
- Kieslinger, M.; Swoboda, A.; Kramer, N.; Pratscher, B.; Wolfesberger, B.; Burgener, I.A. Companion animals as models for inhibition of STAT3 and STAT5. Cancers 2019, 11, 2035. [Google Scholar] [CrossRef]
- Jego, G.; Hermetet, F.; Girodon, F.; Garrido, C. Chaperoning STAT3/5 by heat shock proteins: Interest of their targeting in cancer therapy. Cancers 2020, 12, 21. [Google Scholar] [CrossRef]
- Lau, Y.T.K.; Ramaiyer, M.; Johnson, D.E.; Grandis, J.R. Targeting STAT3 in cancer with nucleotide therapeutics. Cancers 2019, 11, 1681. [Google Scholar] [CrossRef]
- Ernst, S.; Müller-Newen, G. Nucleocytoplasmic shuttling of stats. A target for intervention? Cancers 2019, 11, 1815. [Google Scholar] [CrossRef] [PubMed]
- Wöss, K.; Simonović, N.; Strobl, B.; Macho-Maschler, S.; Müller, M. Tyk2: An upstream kinase of stats in cancer. Cancers 2019, 11, 1728. [Google Scholar] [CrossRef] [PubMed]
- Vella, A.; D’aversa, E.; Api, M.; Breveglieri, G.; Allegri, M.; Giacomazzi, A.; Busilacchi, E.M.; Fabrizzi, B.; Cestari, T.; Sorio, C.; et al. MTOR and STAT3 pathway hyper-activation is associated with elevated interleukin-6 levels in patients with shwachman-diamond syndrome: Further evidence of lymphoid lineage impairment. Cancers 2020, 12, 597. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhong, Y.; Dong, H.; Zheng, Q.; Shi, S.; Zhu, K.; Qu, X.; Hu, W.; Zhang, X.; Wang, Y. Revisiting signal transducer and activator of transcription 3 (STAT3) as an anticancer target and its inhibitor discovery: Where are we and where should we go? Eur. J. Med. Chem. 2020, 187. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Bai, L.; Xu, R.; Zhao, Y.; Chen, J.; McEachern, D.; Chinnaswamy, K.; Wen, B.; Dai, L.; Kumar, P.; et al. Structure-Based Discovery of SD-36 as a Potent, Selective, and Efficacious PROTAC Degrader of STAT3 Protein. J. Med. Chem. 2019, 62, 11280–11300. [Google Scholar] [CrossRef] [PubMed]
- Genini, D.; Brambilla, L.; Laurini, E.; Merulla, J.; Civenni, G.; Pandit, S.; D’Antuono, R.; Perez, L.; Levy, D.E.; Pricl, S.; et al. Mitochondrial dysfunction induced by a SH2 domain-Targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E4924–E4933. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, H.; Xu, R.; Zhao, Y.; Chinnaswamy, K.; McEachern, D.; Chen, J.; Yang, C.Y.; Liu, Z.; Wang, M.; et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019, 36, 498–511.e17. [Google Scholar] [CrossRef]
- de Araujo, E.D.; Manaswiyoungkul, P.; Erdogan, F.; Qadree, A.K.; Sina, D.; Tin, G.; Toutah, K.; Yuen, K.; Gunning, P.T. A functional in vitro assay for screening inhibitors of STAT5B phosphorylation. J. Pharm. Biomed. Anal. 2019, 162, 60–65. [Google Scholar] [CrossRef]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef]
- Bournazou, E.; Bromberg, J. Targeting the tumor microenvironment. Jak-Stat 2013, 2, e23828. [Google Scholar] [CrossRef]
- Mehta, V.; Goel, S.; Kabarriti, R.; Cole, D.; Goldfinger, M.; Acuna-Villaorduna, A.; Pradhan, K.; Thota, R.; Reissman, S.; Sparano, J.A.; et al. Case Fatality Rate of Cancer Patients with COVID-19 in a New York Hospital System. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- COVID-19 More Frequent, Severe in Cancer Patients. Cancer Discov. 2020. [CrossRef]
- Guaraldi, G.; Meschiari, M.; Cozzi-Lepri, A.; Milic, J.; Tonelli, R.; Menozzi, M.; Franceschini, E.; Cuomo, G.; Orlando, G.; Borghi, V.; et al. Tocilizumab in patients with severe COVID-19: A retrospective cohort study. Lancet Rheumatol. 2020. [Google Scholar] [CrossRef]
- Horby, P.; Lim, W.S.; Emberson, J.; Mafham, M.; Bell, J.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; Elmahi, E.; et al. Effect of Dexamethasone in Hospitalized Patients with COVID-19: Preliminary Report. MedRxiv 2020, 2020.06.22.20137273. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Araujo, E.D.; Keserű, G.M.; Gunning, P.T.; Moriggl, R. Targeting STAT3 and STAT5 in Cancer. Cancers 2020, 12, 2002. https://doi.org/10.3390/cancers12082002
de Araujo ED, Keserű GM, Gunning PT, Moriggl R. Targeting STAT3 and STAT5 in Cancer. Cancers. 2020; 12(8):2002. https://doi.org/10.3390/cancers12082002
Chicago/Turabian Stylede Araujo, Elvin D., György M. Keserű, Patrick T. Gunning, and Richard Moriggl. 2020. "Targeting STAT3 and STAT5 in Cancer" Cancers 12, no. 8: 2002. https://doi.org/10.3390/cancers12082002
APA Stylede Araujo, E. D., Keserű, G. M., Gunning, P. T., & Moriggl, R. (2020). Targeting STAT3 and STAT5 in Cancer. Cancers, 12(8), 2002. https://doi.org/10.3390/cancers12082002