Clinico-Biological Features and Clonal Hematopoiesis in Patients with Severe COVID-19

 ,
,  , , ,
, , ,  on behalf of the Lille Covid Research Network (LICORNE)add
Show full author list
on behalf of the Lille Covid Research Network (LICORNE)add
Show full author list
Abstract
1. Introduction
2. Methods
2.1. Patients and Samples
2.2. Molecular Analyses
2.3. Laboratory Blood Dosages and CBC
2.4. Statistical Analyses
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Data Sharing
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CBC | Complete blood count |
| CH | Clonal hematopoiesis |
| CHIP | Clonal hematopoiesis of indeterminate potential |
| CI | Confidence interval |
| COVID-19 | Coronavirus disease 2019 |
| CRP | C-reactive protein |
| HTS | High-throughput sequencing |
| ICU | Intensive care unit |
| IL-6 | Interleukin-6 |
| OR | Odds ratio |
| OTI | Orotracheal intubation |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| VAF | Variant allele frequency |
| WBC | White blood cell count |
References
- Simonnet, A.; Chetboun, M.; Poissy, J.; Raverdy, V.; Noulette, J.; Duhamel, A.; Labreuche, J.; Mathieu, D.; Pattou, F.; Jourdain, M.; et al. High Prevalence of Obesity in Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) Requiring Invasive Mechanical Ventilation. Obesity 2020, 28, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Barhdadi, A.; Bourgoin, V.; Lépine, G.; Mollica, L.; Szuber, N.; Dubé, M.-P.; Busque, L. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 2017, 130, 753–762. [Google Scholar] [CrossRef]
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 742–752. [Google Scholar] [CrossRef]
- López-Moyado, I.F.; Rao, A. DNMT3A and TET2 mutations reshape hematopoiesis in opposing ways. Nat. Genet. 2020, 52, 554–556. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Dorsheimer, L.; Assmus, B.; Rasper, T.; Ortmann, C.A.; Ecke, A.; Abou-El-Ardat, K.; Schmid, T.; Brüne, B.; Wagner, S.; Serve, H.; et al. Association of Mutations Contributing to Clonal Hematopoiesis with Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol. 2019, 4, 25–33. [Google Scholar] [CrossRef]
- Sano, S.; Oshima, K.; Wang, Y.; Katanasaka, Y.; Sano, M.; Walsh, K. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ. Res. 2018, 123, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef]
- Sano, S.; Oshima, K.; Wang, Y.; MacLauchlan, S.; Katanasaka, Y.; Sano, M.; Zuriaga, M.A.; Yoshiyama, M.; Goukassian, D.; Cooper, M.A.; et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/NLRP3 Inflammasome. J. Am. Coll. Cardiol. 2018, 71, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Cull, A.H.; Snetsinger, B.; Buckstein, R.; Wells, R.A.; Rauh, M.J. Tet2 restrains inflammatory gene expression in macrophages. Exp. Hematol. 2017, 55, 56–70.e13. [Google Scholar] [CrossRef] [PubMed]
- Cook, E.K.; Izukawa, T.; Young, S.; Rosen, G.; Jamali, M.; Zhang, L.; Johnson, D.; Bain, E.; Hilland, J.; Ferrone, C.K.; et al. Comorbid and inflammatory characteristics of genetic subtypes of clonal hematopoiesis. Blood Adv. 2019, 3, 2482–2486. [Google Scholar] [CrossRef]
- Busque, L.; Sun, M.; Buscarlet, M.; Ayachi, S.; Feroz Zada, Y.; Provost, S.; Bourgoin, V.; Mollica, L.; Meisel, M.; Hinterleitner, R.; et al. High-sensitivity C-reactive protein is associated with clonal hematopoiesis of indeterminate potential. Blood Adv. 2020, 4, 2430–2438. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Q.; Ding, Y.; Liu, Y.; Zhao, D.; Zhao, K.; Shen, Q.; Liu, X.; Zhu, X.; Li, N.; et al. Methyltransferase Dnmt3a upregulates HDAC9 to deacetylate the kinase TBK1 for activation of antiviral innate immunity. Nat. Immunol. 2016, 17, 806–815. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Poissy, J.; Goutay, J.; Caplan, M.; Parmentier, E.; Duburcq, T.; Lassalle, F.; Jeanpierre, E.; Rauch, A.; Labreuche, J.; Susen, S.; et al. Pulmonary Embolism in COVID-19 Patients: Awareness of an Increased Prevalence. Circulation 2020. [Google Scholar] [CrossRef]
- Cook, E.K.; Luo, M.; Rauh, M.J. Clonal hematopoiesis and inflammation: Partners in leukemogenesis and comorbidity. Exp. Hematol. 2020, 83, 85–94. [Google Scholar] [CrossRef]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Elalamy, I.; Kastritis, E.; Sergentanis, T.N.; Politou, M.; Psaltopoulou, T.; Gerotziafas, G.; Dimopoulos, M.A. Hematological findings and complications of COVID-19. Am. J. Hematol. 2020, 95, 834–847. [Google Scholar] [CrossRef] [PubMed]
- Abegunde, S.O.; Buckstein, R.; Wells, R.A.; Rauh, M.J. An inflammatory environment containing TNFα favors Tet2-mutant clonal hematopoiesis. Exp. Hematol. 2018, 59, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Kotzin, J.J.; Ramdas, B.; Chen, S.; Nelanuthala, S.; Palam, L.R.; Pandey, R.; Mali, R.S.; Liu, Y.; Kelley, M.R.; et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell 2018, 23, 833–849.e5. [Google Scholar] [CrossRef] [PubMed]




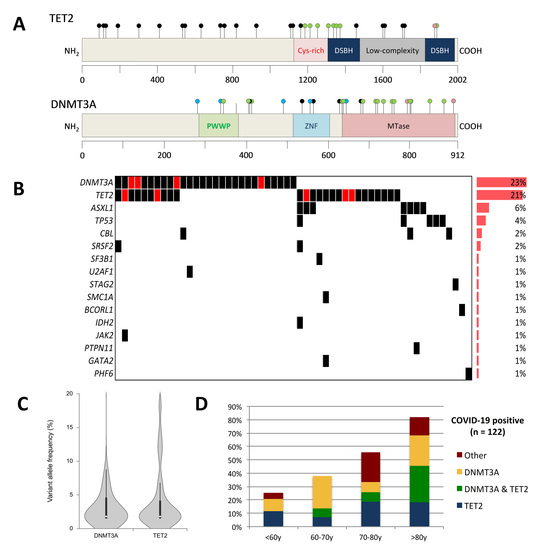
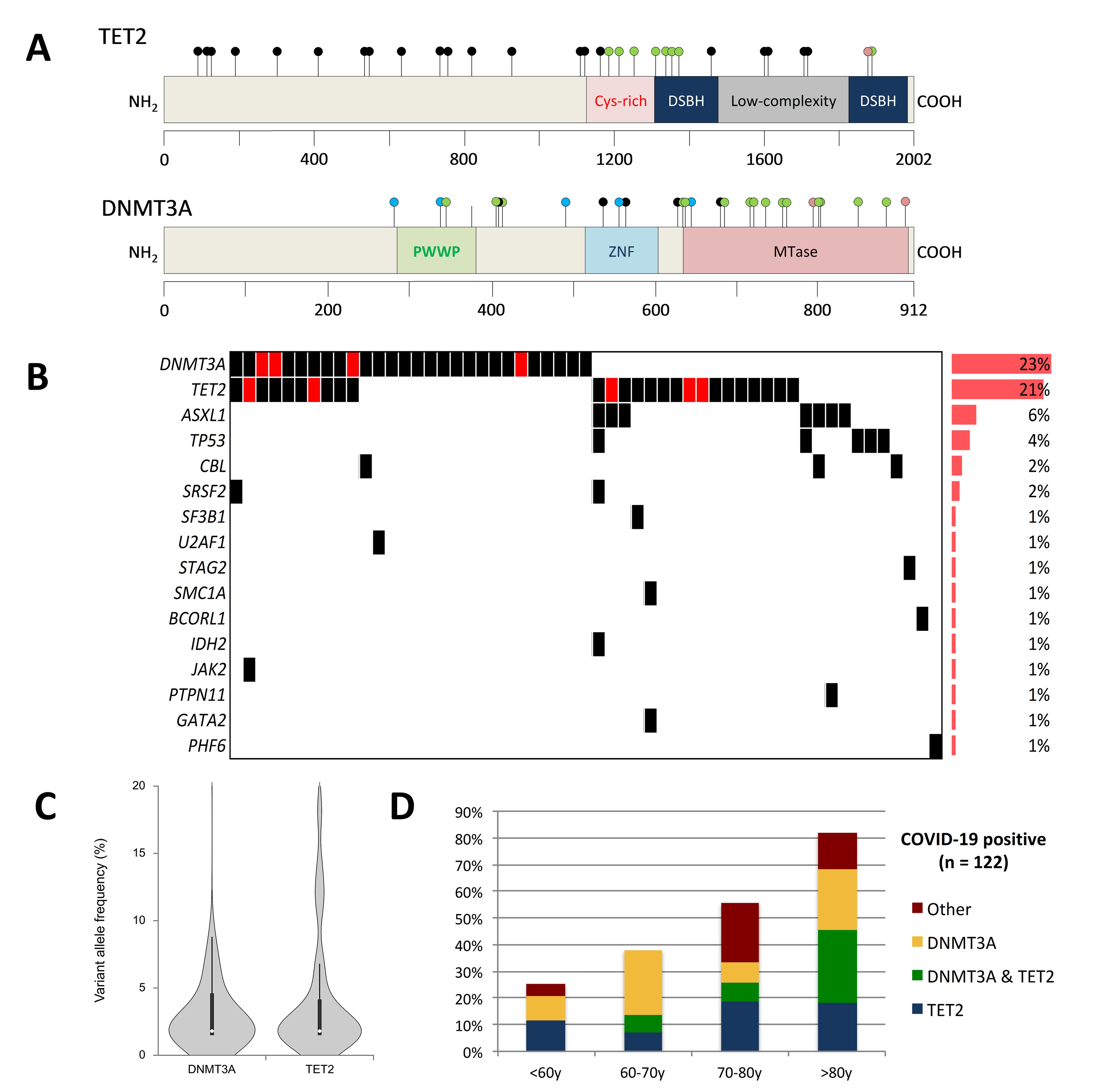{kind=link}
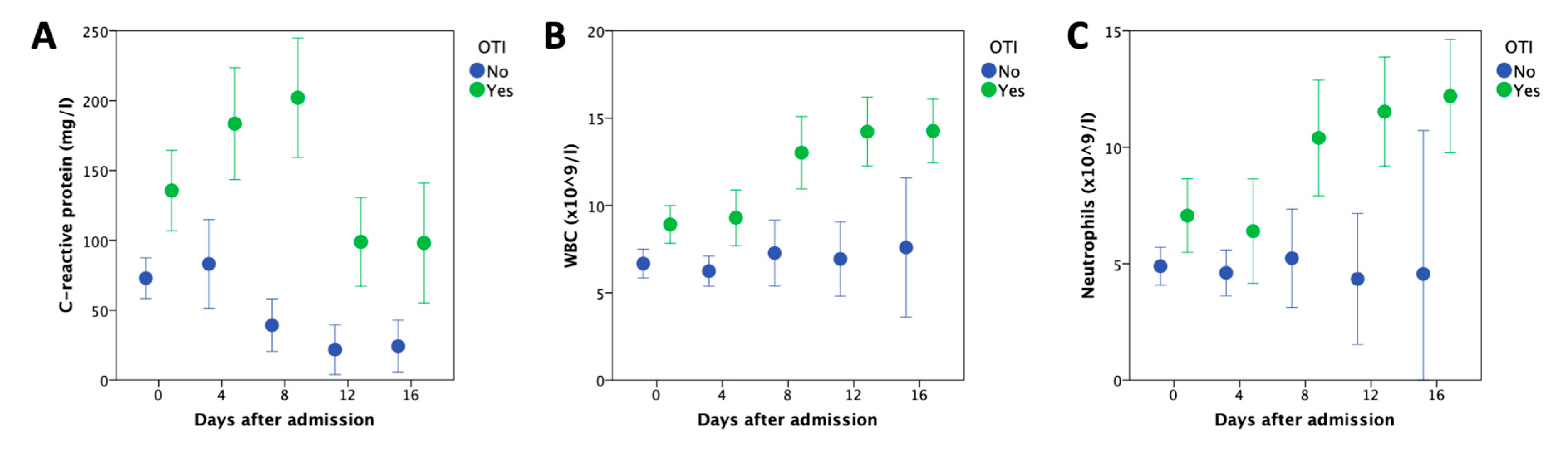{kind=link}
{kind=link}
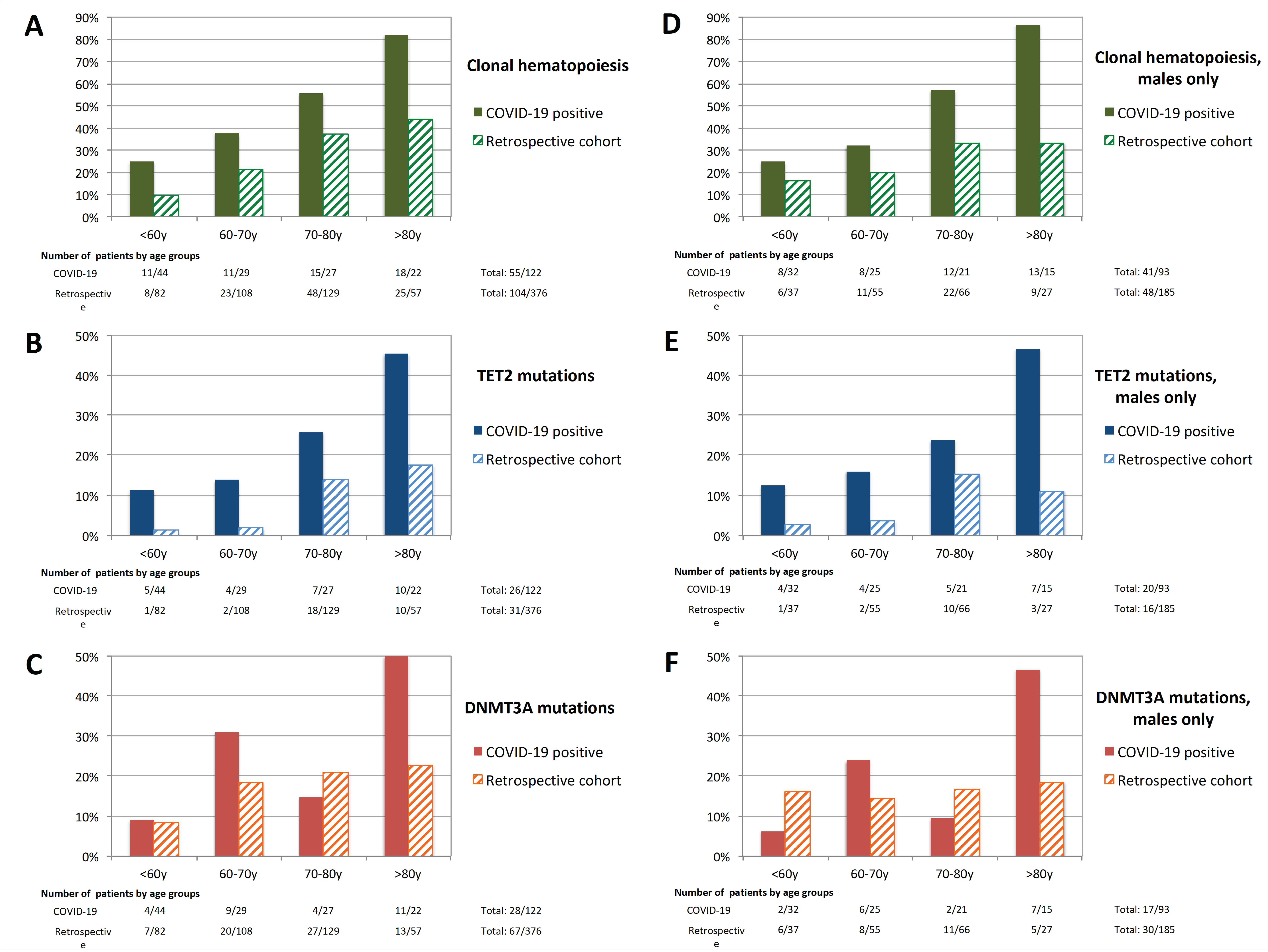{kind=link}
| Characteristics | All COVID-19 Patients | Clonal Hematopoiesis-Negative | Clonal Hematopoiesis-Positive | p-Value |
|---|---|---|---|---|
| Number of patients | 122 | 67 | 55 | |
| Male/Female, sex ratio (%) | 93/29 (76%) | 52/15 (78%) | 41/14 (75%) | 0.831 |
| Median age, years (range) | 65 (22–95) | 60.2 (22.2–87.3) | 71.7 (29.5–94.6) | <0.001 |
| <60 years | 44 (36%) | 33 (49%) | 11 (20%) | <0.001 |
| (60–70 years) | 29 (24%) | 18 (27%) | 11 (20%) | |
| (70–80 years) | 27 (22%) | 12 (18%) | 15 (27%) | |
| >80 years | 22 (18%) | 4 (6%) | 18 (33%) | |
| Body-mass index, kg/m2 (median (range)) | 28.9 (15.1–53.3) | 29.8 (16.6–53.3) | 28 (15.1–41.9) | 0.249 |
| Medical history (n (%)) | ||||
| Smoking | 21 (17%) | 10 (15%) | 11 (20%) | 0.480 |
| Diabete | 34 (28%) | 18 (27%) | 16 (29%) | 0.841 |
| Respiratory illness | 30 (25%) | 17 (25%) | 13 (24%) | 1.000 |
| Cardiac failure | 17 (14%) | 8 (12%) | 9 (16%) | 0.601 |
| Arterial hypertension | 65 (53%) | 31 (46%) | 34 (62%) | 0.102 |
| Stroke | 10 (8%) | 3 (4%) | 7 (13%) | 0.183 |
| Myocardial infarction, obliterative arterial disease | 14 (11%) | 4 (6%) | 10 (18%) | 0.046 |
| Cirrhosis | 3 (2%) | 2 (3%) | 1 (2%) | 1.000 |
| Renal insufficiency | 11 (9%) | 5 (7%) | 6 (11%) | 0.541 |
| Hematological malignancy | 6 (5%) | 4 (6%) | 2 (4%) | 0.689 |
| Solid tumor | 10 (8%) | 3 (4%) | 7 (13%) | 0.183 |
| Immunodepression | 9 (7%) | 7 (10%) | 2 (4%) | 0.183 |
| Simplified Acute Physiology Score (SAPS II) *, median (range) | 40 (12–83) | 39 (12–83) | 41 (21–81) | 0.807 |
| Symptoms at admission (n (%)) | ||||
| Fever | 101 (83%) | 57 (85%) | 44 (80%) | 0.480 |
| Cough | 82 (67%) | 43 (64%) | 39 (71%) | 0.446 |
| Expectorations | 14 (11%) | 7 (10%) | 7 (13%) | 0.779 |
| Dyspnea | 111 (91%) | 64 (96%) | 47 (85%) | 0.064 |
| Headache | 7 (6%) | 5 (8%) | 2 (4%) | 0.453 |
| Tiredness | 80 (66%) | 46 (69%) | 34 (62%) | 0.450 |
| Muscle pain | 28 (23%) | 16 (24%) | 12 (22%) | 0.832 |
| Gastrointestinal symptoms | 34 (28%) | 25 (37%) | 9 (16%) | 0.014 |
| Otolaryngeal symptoms | 16 (13%) | 7 (10%) | 9 (16%) | 0.422 |
| Measures at admission (median (range)) | ||||
| Heart rate (bpm) | 94 (50–147) | 93.5 (50–141) | 95.5 (59–147) | 0.756 |
| Systolic blood pressure (mm Hg) | 124 (58–200) | 127 (80–200) | 118 (58–160) | 0.111 |
| Diastolic blood pressure (mm Hg) | 66.5 (40–124) | 67.5 (41–124) | 66 (40–124) | 0.751 |
| Mean blood pressure (mm Hg) | 83 (51–141) | 84.5 (55–141) | 81.5 (51–133) | 0.722 |
| Body temperature (°C) | 37.7 (34.5–40.6) | 37.7 (35.5–40.6) | 37.6 (34.5–39.7) | 0.525 |
| Respiratory rate (rpm) | 24 (10–47) | 24 (13–51) | 23 (10–47) | 0.819 |
| Laboratory findings (median (range)) | ||||
| White blood cell count (number × 109/L) | 8.1 (2–28.2) | 8.8 (2–28.2) | 7.8 (2.5–26.8) | 0.762 |
| Neutrophils (number × 109/L) | 6.2 (0.9–24.3) | 6.4 (0.9–24.3) | 5.6 (1.5–22.5) | 0.931 |
| Lymphocytes (number × 109/L) | 1 (0–4.2) | 1.2 (0–4.2) | 1 (0.3–2.4) | 0.254 |
| Monocytes (number × 109/L) | 0.6 (0–1.5) | 0.6 (0–1.3) | 0.7 (0–1.5) | 0.801 |
| Red blood cell count (number × 1012/L) | 3.5 (1.9–5.1) | 3.4 (1.9–5.1) | 3.6 (2.4–5.1) | 0.175 |
| Hemoglobin concentration (g/L) | 10.3 (5.7-15.1) | 10.2 (5.7–15.1) | 10.4 (7.1–14.4) | 0.291 |
| Hematocrit (%) | 31.7 (18.2–44.5) | 31.6 (18.2–44) | 32.6 (22.4–44.5) | 0.184 |
| Mean cell volum (fl) | 90.3 (80–107.5) | 90.6 (80–107.5) | 89.6 (80.5–99) | 0.350 |
| Mean cell hemoglobin (pg) | 29.6 (22.5–37.2) | 29.8 (24.8–37.2) | 29.4 (22.5–33.6) | 0.277 |
| Mean cell hemoglobin concentration (g/L) | 32.6 (27.3–36.8) | 32.6 (29.5–36.8) | 32.6 (27.3–35.3) | 0.589 |
| Platelet count (number × 109/L) | 294 (18–1006) | 296.5 (18–763) | 281 (62–1006) | 0.220 |
| Mean platelet volume (fl) | 10.4 (8–14.5) | 10.4 (8.4–14.5) | 10.4 (8–12.4) | 0.445 |
| C-reactive protein (mg/L) | 57 (2–345) | 61 (2–322) | 55 (6–345) | 0.284 |
| Procalcitonin (µg/L) | 0.2 (0.1–188) | 0.2 (0.1–188) | 0.2 (0.1–16) | 0.194 |
| Ferritin (µg/L) | 962 (69–9900) | 1009.5 (140–9900) | 814 (69–2637) | 0.360 |
| Fibrinogen (g/L) | 6.5 (1.6–10.3) | 6.6 (2.5–10.3) | 6.1 (1.6–9.7) | 0.153 |
| Interleukine-6 (ng/L) | 38.6 (2.5–≥ 300) | 32.8 (2.5–300) | 44.2 (4.6–300) | 0.365 |
| During hospitalization (n (%)) | ||||
| Admission in reanimation intensive care unit | 89 (73%) | 56 (84%) | 33 (60%) | 0.004 |
| Orotracheal intubation | 67 (55%) | 42 (63%) | 25 (45%) | 0.069 |
| High-flow nasal cannula oxygenation | 37 (30%) | 22 (33%) | 15 (27%) | 0.557 |
| Noninvasive ventilation | 43 (35%) | 26 (39%) | 17 (31%) | 0.447 |
| ExtraCorporeal Membrane Oxygenation | 17 (14%) | 12 (18%) | 5 (9%) | 0.195 |
| Sympathomimetic amines | 59 (48%) | 37 (55%) | 22 (40%) | 0.105 |
| Extrarenal epuration | 21 (17%) | 15 (22%) | 6 (11%) | 0.147 |
| Use of corticosteroids | 54 (44%) | 32 (48%) | 22 (40%) | 0.465 |
| Outcome (n (%)) | ||||
| Venous thromboembolic disease | 22 (18%) | 12 (18%) | 10 (18%) | 1.000 |
| Arterial disease | 6 (5%) | 3 (4%) | 3 (5%) | 1.000 |
| Death † | 17 (15%) | 10 (17%) | 7 (13%) | 0.793 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duployez, N.; Demonchy, J.; Berthon, C.; Goutay, J.; Caplan, M.; Moreau, A.-S.; Bignon, A.; Marceau-Renaut, A.; Garrigue, D.; Raczkiewicz, I.; et al. Clinico-Biological Features and Clonal Hematopoiesis in Patients with Severe COVID-19. Cancers 2020, 12, 1992. https://doi.org/10.3390/cancers12071992
Duployez N, Demonchy J, Berthon C, Goutay J, Caplan M, Moreau A-S, Bignon A, Marceau-Renaut A, Garrigue D, Raczkiewicz I, et al. Clinico-Biological Features and Clonal Hematopoiesis in Patients with Severe COVID-19. Cancers. 2020; 12(7):1992. https://doi.org/10.3390/cancers12071992
Chicago/Turabian StyleDuployez, Nicolas, Jordane Demonchy, Céline Berthon, Julien Goutay, Morgan Caplan, Anne-Sophie Moreau, Anne Bignon, Alice Marceau-Renaut, Delphine Garrigue, Imelda Raczkiewicz, and et al. 2020. "Clinico-Biological Features and Clonal Hematopoiesis in Patients with Severe COVID-19" Cancers 12, no. 7: 1992. https://doi.org/10.3390/cancers12071992
APA StyleDuployez, N., Demonchy, J., Berthon, C., Goutay, J., Caplan, M., Moreau, A.-S., Bignon, A., Marceau-Renaut, A., Garrigue, D., Raczkiewicz, I., Geffroy, S., Bucci, M., Alidjinou, K., Demaret, J., Labalette, M., Brousseau, T., Dupont, A., Rauch, A., Poissy, J., ... Quesnel, B., on behalf of the Lille Covid Research Network (LICORNE). (2020). Clinico-Biological Features and Clonal Hematopoiesis in Patients with Severe COVID-19. Cancers, 12(7), 1992. https://doi.org/10.3390/cancers12071992