High-Throughput Sequencing of Gastric Cancer Patients: Unravelling Genetic Predispositions Towards an Early-Onset Subtype

 , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Group
DNA Extraction
2.2. Library Preparation for Next-Generation Sequencing
2.3. Data Processing
2.4. In Silico Estimation of the Detected Variants
3. Age-Dependent Genotypic and Phenotypic Characteristics of Gastric Cancer Subtypes
In Silico Estimation
4. High-Throughput Mutation Profiling Identifies the Most Frequent Mutations in EOGC and CGC Samples
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CGC | conventional gastric carcinoma |
| EOGC | early-onset gastric cancer |
| FA | Fanconi anemia |
| FFPE | formalin-fixed paraffin-embedded |
| GC | gastric cancer |
| GIST | gastrointestinal stromal tumor |
| MEN1 | multiple endocrine neoplasia type 1 |
| NGS | next-generation sequencing |
| SNPs | single nucleotide polymorphisms |
References
- Gullo, I.; Carneiro, F.; Oliveira, C.; Almeida, G.M. Heterogeneity in Gastric Cancer: From Pure Morphology to Molecular Classifications. Pathobiology 2018, 85, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.P.; Xu, W.; Liu, W.T.; Yan, M.; Zhu, Z.G. Tumor heterogeneity of gastric cancer: From the perspective of tumor-initiating cell. World J. Gastroenterol. 2018, 24, 2567–2581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Zhang, P.Y. Gastric cancer: Somatic genetics as a guide to therapy. J. Med. Genet. 2017, 54, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Kokkola, A.; Sipponen, P. Gastric carcinoma in young adults. Hepato Gastroenterol. 2001, 48, 1552–1555. [Google Scholar]
- Milne, A.N.; Carvalho, R.; Morsink, F.M.; Musler, A.R.; de Leng, W.W.; Ristimäki, A.; Offerhaus, G.J. Early-onset gastric cancers have a different molecular expression profile than conventional gastric cancers. Mod. Pathol. 2006, 19, 564–572. [Google Scholar] [CrossRef]
- Pucułek, M.; Machlowska, J.; Wierzbicki, R.; Baj, J.; Maciejewski, R.; Sitarz, R. Helicobacter pylori associated factors in the development of gastric cancer with special reference to the early-onset subtype. Oncotarget 2018, 9, 31146–31162. [Google Scholar] [CrossRef]
- Rocco, A.; Nardone, G. Diet, H pylori infection and gastric cancer: Evidence and controversies. World J. Gastroenterol. 2007, 13, 2901–2912. [Google Scholar] [CrossRef]
- Baj, J.; Brzozowska, K.; Forma, A.; Maani, A.; Sitarz, E.; Portincasa, P. Immunological Aspects of the Tumor Microenvironment and Epithelial-Mesenchymal Transition in Gastric Carcinogenesis. Int. J. Mol. Sci. 2020, 21, 2544. [Google Scholar] [CrossRef]
- Baj, J.; Korona-Głowniak, I.; Forma, A.; Maani, A.; Sitarz, E.; Rahnama-Hezavah, M.; Radzikowska, E.; Portincasa, P. Mechanisms of the Epithelial–Mesenchymal Transition and Tumor Microenvironment in Helicobacter pylori-Induced Gastric Cancer. Cells 2020, 9, 1055. [Google Scholar] [CrossRef] [PubMed]
- Machlowska, J.; Maciejewski, R.; Sitarz, R. The Pattern of Signatures in Gastric Cancer Prognosis. Int. J. Mol. Sci. 2018, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Machlowska, J.; Baj, J.; Sitarz, M.; Maciejewski, R.; Sitarz, R. Gastric Cancer: Epidemiology, Risk Factors, Classification, Genomic Characteristics and Treatment Strategies. Int. J. Mol. Sci. 2020, 21, 4012. [Google Scholar] [CrossRef] [PubMed]
- Rimini, M.; Casadei-Gardini, A.; Ravaioli, A.; Rovesti, G.; Conti, F.; Borghi, A.; Dall’Aglio, A.C.; Bedogni, G.; Domenicali, M.; Giacomoni, P.; et al. Could Inflammatory Indices and Metabolic Syndrome Predict the Risk of Cancer Development? Analysis from the Bagnacavallo Population Study. J. Clin. Med. 2020, 9, 1177. [Google Scholar] [CrossRef] [PubMed]
- Salati, M.; Pipitone, S.; Rimini, M.; Gelsomino, F.; Gardini, A.C.; Andrikou, K.; Schipilliti, F.; Cortesi, G.; Cassanelli, L.; Caffari, E.; et al. Immune-inflammatory and clinicopathologic prognostic factors in a Western cohort of resected gastric cancers (GCs). Ann. Oncol. 2019, 30, 75. [Google Scholar] [CrossRef]
- Laurén, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar]
- Bosman, F.T.; Carneiro, F.; Hruban, R.H.; Theise, N.D. WHO Classification of Tumours of the Digestive System, 4th ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2010. [Google Scholar]
- Liang, H.; Kim, Y.H. Identifying molecular drivers of gastric cancer through next-generation sequencing. Cancer Lett. 2013, 340, 241–246. [Google Scholar] [CrossRef][Green Version]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef]
- Riquelme, I.; Saavedra, K.; Espinoza, J.A.; Weber, H.; García, P.; Nervi, B.; Garrido, M.; Corvalán, A.H.; Roa, J.C.; Bizma, C. Molecular classification of gastric cancer: Towards a pathway-driven targeted therapy. Oncotarget 2015, 6, 24750–24779. [Google Scholar] [CrossRef]
- Sitarz, R.; Leguit, R.J.; de Leng, W.W.; Polak, M.; Morsink, F.M.; Bakker, O.; Maciejewski, R.; Offerhaus, G.J.; Milne, A.N. The COX-2 promoter polymorphism -765 G>C is associated with early-onset, conventional and stump gastric cancers. Mod. Pathol. 2008, 21, 685–690. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Narzisi, G.; O’Rawe, J.A.; Iossifov, I.; Fang, H.; Lee, Y.H.; Wang, Z.; Wu, Y.; Lyon, G.J.; Wigler, M.; Schatz, M.C. Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nat. Methods 2014, 11, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Patru, C.L.; Surlin, V.; Georgescu, I.; Patru, E. Current issues in gastric cancer epidemiology. Rev. Med. Chir. Soc. Med. Nat. Iasi 2013, 117, 199–204. [Google Scholar] [PubMed]
- Kamps, R.; Brandão, R.D.; Bosch, B.J.; Paulussen, A.D.; Xanthoulea, S.; Blok, M.J.; Romano, A. Next-Generation Sequencing in Oncology: Genetic Diagnosis, Risk Prediction and Cancer Classification. Int. J. Mol. Sci. 2017, 18, 308. [Google Scholar] [CrossRef] [PubMed]
- Shyr, D.; Liu, Q. Next generation sequencing in cancer research and clinical application. Biol. Proced. Online 2013, 15, 4. [Google Scholar] [CrossRef]
- Marini, F.; Falchetti, A.; Luzi, E.; Tonelli, F.; Maria Luisa, B. Multiple Endocrine Neoplasia Type 1 (MEN1) Syndrome. In Cancer Syndromes; Riegert-Johnson, D.L., Boardman, L.A., Hefferon, T., Roberts, M., Eds.; National Center for Biotechnology Information: Bethesda, MD, USA, 2009. [Google Scholar]
- Debelenko, L.V.; Emmert-Buck, M.R.; Zhuang, Z.; Epshteyn, E.; Moskaluk, C.A.; Jensen, R.T.; Liotta, L.A.; Lubensky, I.A. The multiple endocrine neoplasia type I gene locus is involved in the pathogenesis of type II gastric carcinoids. Gastroenterology 1997, 113, 773–781. [Google Scholar] [CrossRef]
- Capelli, L.; Petracci, E.; Quagliuolo, V.; Saragoni, L.; Colombo, P.; Morgagni, P.; Calistri, D.; Tomezzoli, A.; Di Cosmo, M.; Roviello, F.; et al. GISTs: Analysis of c-Kit, PDGFRA and BRAF mutations in relation to prognosis and clinical pathological characteristics of patients—A GIRCG study. Eur. J. Surg. Oncol. 2016, 42, 1206–1214. [Google Scholar] [CrossRef]
- Swift, M. Fanconi’s anaemia in the genetics of neoplasia. Nature 1971, 230, 370–373. [Google Scholar] [CrossRef]
- Goto, M.; Miller, R.W.; Ishikawa, Y.; Sugano, H. Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol. Prev. Biomark. 1996, 5, 239–246. [Google Scholar]
- Kastan, M.B. DNA damage responses: Mechanisms and roles in human disease: 2007 G.H.A. Clowes Memorial Award Lecture. Mol. Cancer Res. 2008, 6, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell. 2007, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Wada, R.; Ishino, K.; Kudo, M.; Uchida, E.; Naito, Z. Expression of DNA damage response proteins in gastric cancer: Comprehensive protein profiling and histological analysis. Int. J. Oncol. 2018, 52, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, L.; Melucci, E.; De Nicola, F.; Goeman, F.; Casini, B.; Sperati, F.; Pallocca, M.; Terrenato, I.; Pizzuti, L.; Vici, P.; et al. DNA damage repair and survival outcomes in advanced gastric cancer patients treated with first-line chemotherapy. Int. J. Cancer. 2017, 140, 2587–2595. [Google Scholar] [CrossRef]
- Lee, H.E.; Han, N.; Kim, M.A.; Lee, H.S.; Yang, H.K.; Lee, B.L.; Kim, W.H. DNA damage response-related proteins in gastric cancer: ATM, Chk2 and p53 expression and their prognostic value. Pathobiology 2014, 81, 25–35. [Google Scholar] [CrossRef]
- Wang, W. A major switch for the Fanconianemia DNA damage-response pathway. Nat. Struct. Mol. Biol. 2008, 15, 1128–1130. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Lasota, J. Succinate dehydrogenase deficient gastrointestinal stromal tumors (GISTs)—A review. Int. J. Biochem. Cell Biol. 2014, 53, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.C.; Luo, Y.H.; Yang, S.M.; Li, X.A.; Ling, X.L.; Fang, L. Mutation analysis of APC gene in gastric cancer with microsatellite instability. World J. Gastroenterol. 2002, 8, 787–791. [Google Scholar] [CrossRef]
- Wong, H.; Yau, T. Molecular targeted therapies in advanced gastric cancer: Does tumor histologymatter. Therap. Adv. Gastroenterol. 2013, 6, 15–31. [Google Scholar] [CrossRef]
- Ishimoto, T.; Miyake, K.; Nandi, T.; Yashiro, M.; Onishi, N.; Huang, K.K.; Lin, S.J.; Kalpana, R.; Tay, S.T.; Suzuki, Y.; et al. Activation of Transforming Growth Factor Beta 1 Signaling in Gastric Cancer-associated Fibroblasts Increases Their Motility, via Expression of Rhomboid 5 Homolog 2, and Ability to Induce Invasiveness of Gastric Cancer Cells. Gastroenterology 2017, 153, 191–204. [Google Scholar] [CrossRef]
- Fenoglio-Preiser, C.M.; Wang, J.; Stemmermann, G.N.; Noffsinger, A. TP53 and gastric carcinoma: A review. Hum. Mutat. 2003, 21, 258–270. [Google Scholar] [CrossRef] [PubMed]



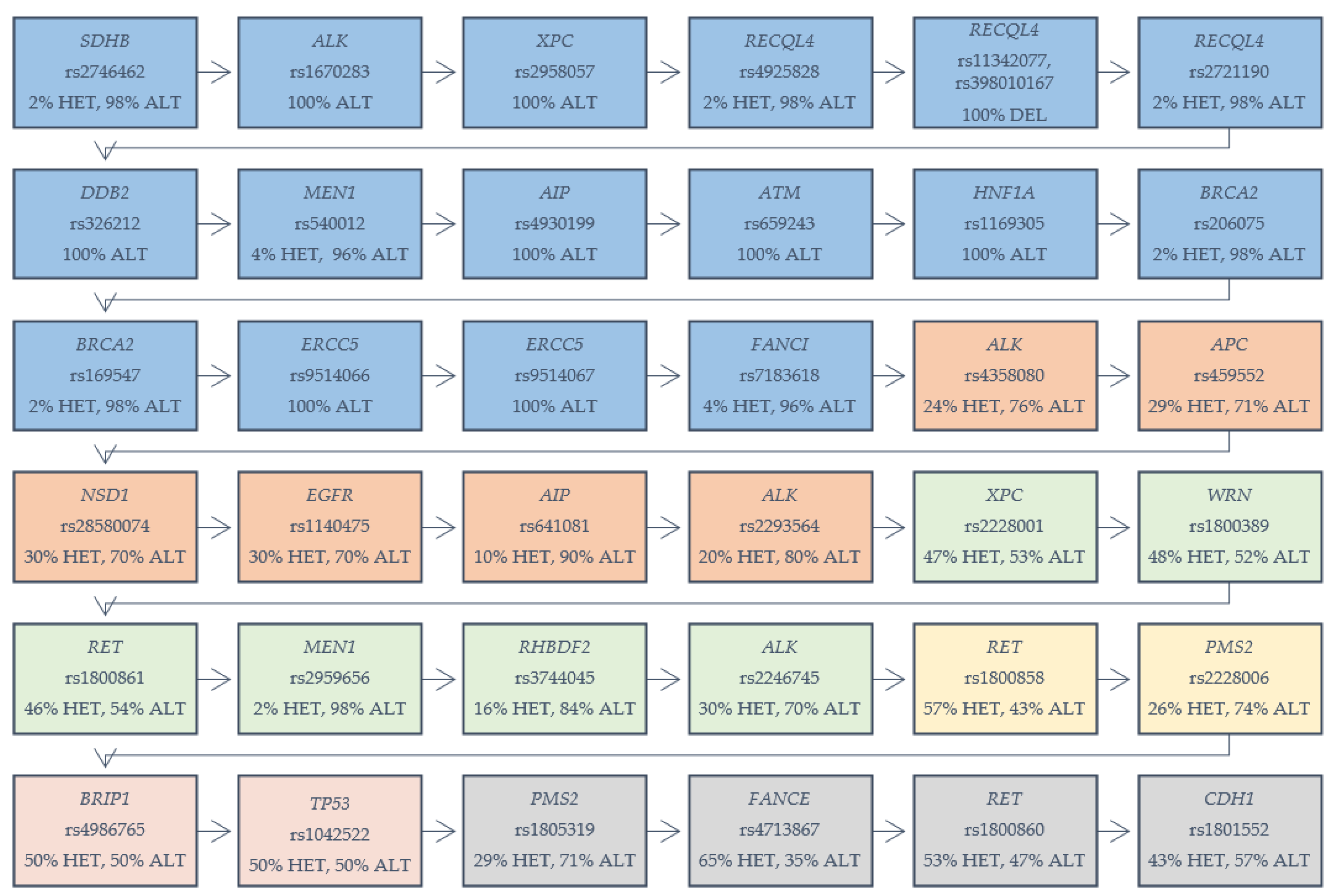{kind=link}
{kind=link}
| Phenotype | Genotype | Reference | Frequency | p Value | Chr | Gene | Variant Type | dbSNP |
|---|---|---|---|---|---|---|---|---|
| CGC | a * | b * | 0.471 | 0.004 | 19 | STK11 | Splice_acceptor | NA |
| EOGC | a * | b * | 0.067 | |||||
| CGC | C/T | C | 0.313 | 0.006 | 10 | RET | Synonymous | rs1800862 |
| CGC | T/T | C | 0.063 | |||||
| EOGC | C/T | C | 0.031 | |||||
| EOGC | T/T | C | 0.000 | |||||
| CGC | C/T | C | 0.389 | 0.009 | 14 | FANCM | Missense | rs10138997 |
| EOGC | C/T | C | 0.059 | |||||
| CGC | G/A | G | 0.059 | 0.010 | 10 | RET | Missense | rs1799939 |
| CGC | A/A | G | 0.000 | |||||
| EOGC | G/A | G | 0.462 | |||||
| EOGC | A/A | G | 0.038 | |||||
| CGC | A/G | A | 0.353 | 0.018 | 16 | SLX4 | Synonymous | rs3810812 |
| CGC | G/G | A | 0.588 | |||||
| EOGC | A/G | A | 0.324 | |||||
| EOGC | G/G | A | 0.265 | |||||
| CGC | G/A | G | 0.278 | 0.024 | 8 | WRN | Missense | rs2230009 |
| EOGC | G/A | G | 0.029 | |||||
| CGC | T/C | T | 0.059 | 0.041 | 11 | MEN1 | Missense | rs2959656 |
| CGC | C/C | T | 0.824 | |||||
| EOGC | T/C | T | 0.000 | |||||
| EOGC | C/C | T | 1.000 | |||||
| CGC | A/G | A | 0.000 | 0.052 | 4 | KIT | Synonymous | rs55986963 |
| CGC | G/G | A | 0.056 | |||||
| EOGC | A/G | A | 0.206 | |||||
| EOGC | G/G | A | 0.000 | |||||
| CGC | G/A | G | 0.222 | 0.054 | 16 | SLX4 | Synonymous | rs28516461 |
| CGC | A/A | G | 0.000 | |||||
| EOGC | G/A | G | 0.029 | |||||
| EOGC | A/A | G | 0.059 |
| dbSNP | CADD Score | DANN Score | FATHMM-XF Prediction | SIFT Prediction | PROVEAN Prediction | gnomAD MAF (European Non-Finnish) |
|---|---|---|---|---|---|---|
| rs1800862 | 10.26 | 0.6808 | Benign (high conf.) | Tolerated | Neutral | 0.04899 |
| rs10138997 | 14.99 | 0.9662 | Benign | Tolerated | Neutral | 0.05851 |
| rs1799939 | 8.26 | 0.8595 | Benign | Tolerated | Neutral | 0.1847 |
| rs3810812 | 1.21 | 0.3514 | Benign (high conf.) | Tolerated | Neutral | 0.5139 |
| rs2230009 | 13.29 | 0.3339 | Benign (high conf.) | Tolerated | Neutral | 0.05857 |
| rs2959656 | 11.00 | 0.5414 | Benign (high conf.) | Tolerated | Neutral | 0.9960 |
| rs55986963 | 7.36 | 0.4165 | Benign (high conf.) | Tolerated | Neutral | 0.03027 |
| rs28516461 | 0.14 | 0.8924 | Benign (high conf.) | Tolerated | Neutral | 0.01392 |
| dbSNP | Variant Type | Chr | Gene | CADD Score (Scaled) | DANN Score | FATHMM-XF Prediction | SIFT Prediction | PROVEAN Prediction | gnomAD MAF (European Non-Finnish) |
|---|---|---|---|---|---|---|---|---|---|
| rs2746462 | Synonymous | 1 | SDHB | 6.608 | 0.6939 | Benign (high conf.) | Tolerated | Neutral | 0.9724 |
| rs1670283 | Missense | 2 | ALK | 0.648 | 0.5289 | Benign (high conf.) | Tolerated | Neutral | 0.9998 |
| rs2958057 | Synonymous | 3 | XPC | 6.712 | 0.7056 | Benign (high conf.) | Tolerated | Neutral | 1.000 |
| rs4925828 | Synonymous | 8 | RECQL4 | 5.112 | 0.6599 | Benign (high conf.) | Tolerated | NA | 0.9994 |
| rs11342077, rs398010167 | Frameshift | 8 | RECQL4 | NA | NA | NA | NA | NA | NA |
| rs2721190 | Missense | 8 | RECQL4 | 5.653 | 0.4532 | Benign | Tolerated | NA | 0.9993 |
| rs326212 | Synonymous | 11 | DDB2 | 9.276 | 0.5475 | Benign (high conf.) | Tolerated | Neutral | 1.000 |
| rs540012 | Synonymous | 11 | MEN1 | 8.171 | 0.5409 | Benign (high conf.) | Tolerated | Neutral | 0.9999 |
| rs4930199 | Missense | 11 | AIP | 15.65 | 0.7273 | Benign | Tolerated | Neutral | 1.000 |
| rs659243 | Missense | 11 | ATM | 7.875 | 0.7855 | Benign (high conf.) | Tolerated | Neutral | 1.000 |
| rs1169305 | Missense | 12 | HNF1A | 9.746 | 0.7163 | Benign | Tolerated | Neutral | 0.9998 |
| rs206075 | Synonymous | 13 | BRCA2 | 2.651 | 0.4723 | Benign (high conf.) | Tolerated | Neutral | 0.9996 |
| rs169547 | Missense | 13 | BRCA2 | 11.56 | 0.1694 | Benign (high conf.) | Tolerated | Neutral | 0.9997 |
| rs9514066 | Missense | 13 | ERCC5 | 21.0 | 0.9962 | Benign | Tolerated, Damaging | Neutral | 1.000 |
| rs9514067 | Missense | 13 | ERCC5 | 1.290 | 0.6273 | Benign | Tolerated | Neutral | 0.9999 |
| rs7183618 | Synonymous | 15 | FANCI | 3.241 | 0.4015 | Benign (high conf.) | Tolerated | Neutral | 0.9469 |
| rs4358080 | Synonymous | 2 | ALK | 8.136 | 0.5627 | Benign (high conf.) | Tolerated | Neutral | 0.9086 |
| rs459552 | Missense | 5 | APC | 18.00 | 0.8086 | Benign (high conf.) | Tolerated | Neutral | 0.7677 |
| rs28580074 | Synonymous | 5 | NSD1 | 5.986 | 0.7262 | Benign (high conf.) | Tolerated | Neutral | 0.8768 |
| rs1140475 | Synonymous | 7 | EGFR | 6.939 | 0.4885 | Benign (high conf.) | Tolerated | Neutral | 0.8741 |
| rs641081 | Missense | 11 | AIP | 4.743 | 0.6594 | Benign | Tolerated | Neutral | 0.9979 |
| rs2293564 | Synonymous | 2 | ALK | 1.005 | 0.3863 | Benign (high conf.) | Tolerated | Neutral | 0.9187 |
| rs2228001 | Missense | 3 | XPC | 17.09 | 0.9017 | Benign | Tolerated | Neutral | 0.5939 |
| rs1800389 | Synonymous | 8 | WRN | 4.911 | 0.4329 | Benign (high conf.) | Tolerated | Neutral | 0.7031 |
| rs1800861 | Synonymous | 10 | RET | 10.76 | 0.7308 | Benign (high conf.) | Tolerated | Neutral | 0.7668 |
| rs2959656 | Missense | 11 | MEN1 | 11.00 | 0.5414 | Benign (high conf.) | Tolerated | Neutral | 0.9960 |
| rs3744045 | Missense | 17 | RHBDF2 | 21.1 | 0.9951 | Pathogenic | Tolerated, Damaging | Neutral | 0.9366 |
| rs2246745 | Synonymous | 2 | ALK | 4.814 | 0.5041 | Benign (high conf.) | Tolerated | Neutral | 0.8143 |
| rs1800858 | Synonymous | 10 | RET | 1.167 | 0.3128 | Benign (high conf.) | Tolerated | Neutral | 0.7384 |
| rs2228006 | Missense | 7 | PMS2 | 10.11 | 0.2203 | Benign (high conf.) | Tolerated | Neutral | 0.8501 |
| rs4986765 | Synonymous | 17 | BRIP1 | 9.957 | 0.6153 | Benign (high conf.) | Tolerated | Neutral | 0.6603 |
| rs1042522 | Missense | 17 | TP53 | 9.176 | 0.5704 | Benign | Tolerated | Neutral | 0.7366 |
| rs1805319 | Synonymous | 7 | PMS2 | 0.168 | 0.7105 | Benign (high conf.) | Tolerated | Neutral | 0.8160 |
| rs4713867 | Synonymous | 6 | FANCE | 1.716 | 0.3909 | Benign (high conf.) | Tolerated | Neutral | 0.6747 |
| rs1800860 | Synonymous | 10 | RET | 6.248 | 0.3745 | Benign (high conf.) | Tolerated | Neutral | 0.6900 |
| rs1801552 | Synonymous | 16 | CDH1 | 5.661 | 0.4494 | Benign (high conf.) | Tolerated | Neutral | 0.6252 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machlowska, J.; Kapusta, P.; Baj, J.; Morsink, F.H.M.; Wołkow, P.; Maciejewski, R.; Offerhaus, G.J.A.; Sitarz, R. High-Throughput Sequencing of Gastric Cancer Patients: Unravelling Genetic Predispositions Towards an Early-Onset Subtype. Cancers 2020, 12, 1981. https://doi.org/10.3390/cancers12071981
Machlowska J, Kapusta P, Baj J, Morsink FHM, Wołkow P, Maciejewski R, Offerhaus GJA, Sitarz R. High-Throughput Sequencing of Gastric Cancer Patients: Unravelling Genetic Predispositions Towards an Early-Onset Subtype. Cancers. 2020; 12(7):1981. https://doi.org/10.3390/cancers12071981
Chicago/Turabian StyleMachlowska, Julita, Przemysław Kapusta, Jacek Baj, Folkert H. M. Morsink, Paweł Wołkow, Ryszard Maciejewski, G. Johan A. Offerhaus, and Robert Sitarz. 2020. "High-Throughput Sequencing of Gastric Cancer Patients: Unravelling Genetic Predispositions Towards an Early-Onset Subtype" Cancers 12, no. 7: 1981. https://doi.org/10.3390/cancers12071981
APA StyleMachlowska, J., Kapusta, P., Baj, J., Morsink, F. H. M., Wołkow, P., Maciejewski, R., Offerhaus, G. J. A., & Sitarz, R. (2020). High-Throughput Sequencing of Gastric Cancer Patients: Unravelling Genetic Predispositions Towards an Early-Onset Subtype. Cancers, 12(7), 1981. https://doi.org/10.3390/cancers12071981