Depletion of Macrophages Improves Therapeutic Response to Gemcitabine in Murine Pancreas Cancer

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
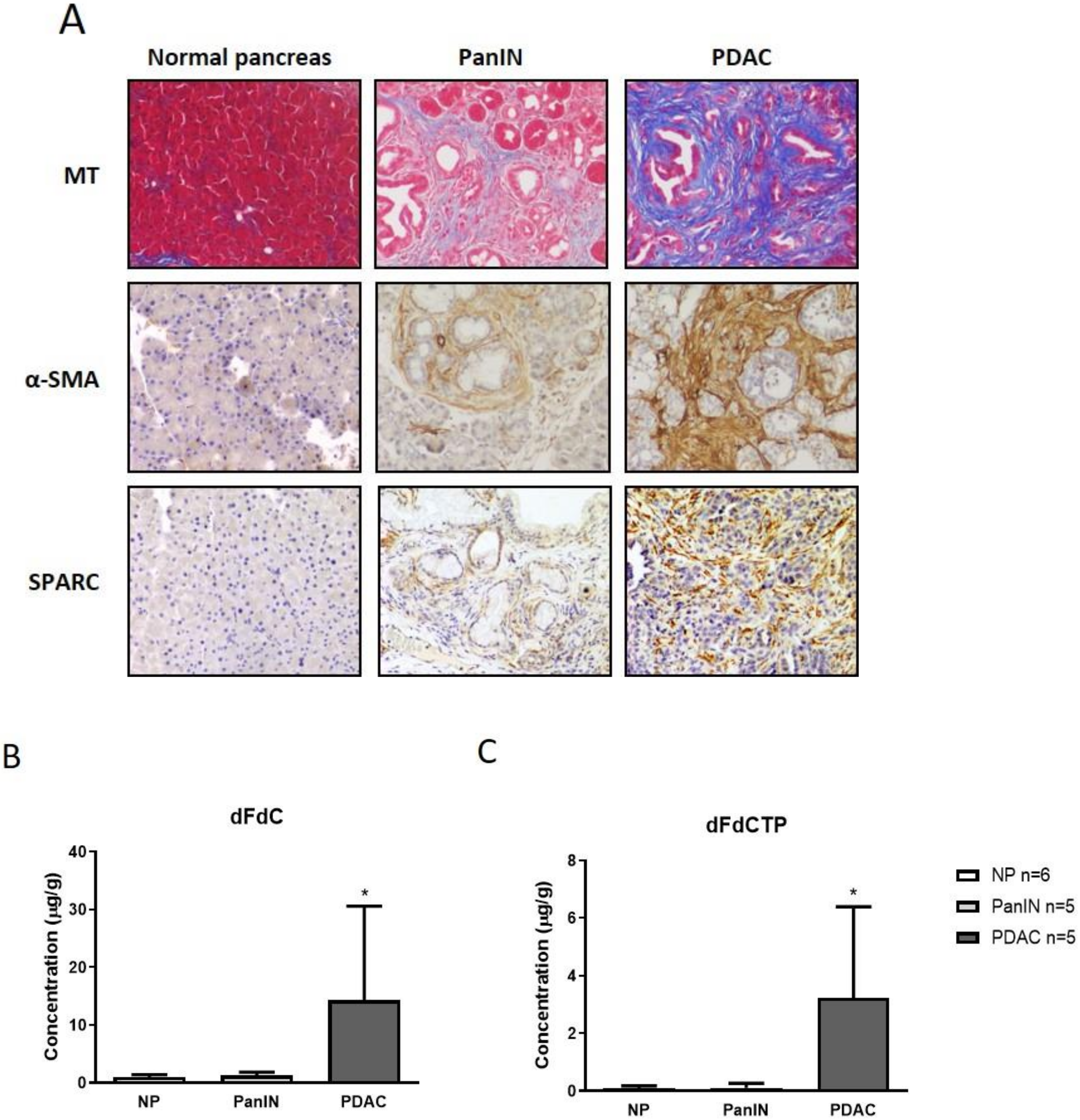
2.1. Gemcitabine Concentration is Increased in Murine Pancreatic Tumors Compared to Pancreatic Intraepithelial Neoplasia and Normal Pancreas Tissue
2.2. Murine and Human Macrophages Reduce Cytotoxicity of Gemcitabine In Vitro
2.3. Macrophages Rapidly Metabolize and Inactivate Gemcitabine
2.4. Pharmacological Depletion of TAMs Using Liposomal Clodronate Sensitizes KPC Tumors to Gemcitabine Treatment
3. Discussion
4. Materials and Methods
4.1. Genetically Engineered Mouse Models
4.2. Therapeutic Intervention
4.3. Liquid Chromatography–Mass Spectrometry/Mass Spectrometry (LC–MS/MS)
4.4. Drug Preparation
4.5. Cell Lines
4.6. Human Macrophage Polarization
4.7. Murine Macrophage Isolation
4.8. Culture with Conditioned Medium
4.9. Cell Viability Assays
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hessmann, E.; Buchholz, S.M.; Demir, I.E.; Singh, S.K.; Gress, T.M.; Ellenrieder, V.; Neesse, A. Microenvironmental determinants of pancreatic cancer. Physiol. Rev. 2020. [Google Scholar] [CrossRef]
- Mahajan, U.M.; Langhoff, E.; Goni, E.; Costello, E.; Greenhalf, W.; Halloran, C.; Ormanns, S.; Kruger, S.; Boeck, S.; Ribback, S.; et al. Immune cell and stromal signature associated with progression-free survival of patients with resected pancreatic ductal adenocarcinoma. Gastroenterology 2018, 155, 1625–1639. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Ramu, I.; Buchholz, S.M.; Patzak, M.S.; Goetze, R.G.; Singh, S.K.; Richards, F.M.; Jodrell, D.I.; Sipos, B.; Strobel, P.; Ellenrieder, V.; et al. SPARC dependent collagen deposition and gemcitabine delivery in a genetically engineered mouse model of pancreas cancer. EBioMedicine 2019, 48, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef]
- Regel, I.; Mayerle, J.; Mahajan, U.M. Current strategies and future perspectives for precision medicine in pancreatic cancer. Cancers 2020, 12, 1024. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Duluc, C.; Moatassim-Billah, S.; Chalabi-Dchar, M.; Perraud, A.; Samain, R.; Breibach, F.; Gayral, M.; Cordelier, P.; Delisle, M.B.; Bousquet-Dubouch, M.P.; et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol. Med. 2015, 7, 735–753. [Google Scholar] [CrossRef]
- Hessmann, E.; Patzak, M.S.; Klein, L.; Chen, N.; Kari, V.; Ramu, I.; Bapiro, T.E.; Frese, K.K.; Gopinathan, A.; Richards, F.M.; et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2018, 67, 497–507. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019. [Google Scholar] [CrossRef] [PubMed]
- Griesmann, H.; Drexel, C.; Milosevic, N.; Sipos, B.; Rosendahl, J.; Gress, T.M.; Michl, P. Pharmacological macrophage inhibition decreases metastasis formation in a genetic model of pancreatic cancer. Gut 2017, 66, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Winograd, R.; Evans, R.A.; Long, K.B.; Luque, S.L.; Lee, J.W.; Clendenin, C.; Gladney, W.L.; Knoblock, D.M.; Guirnalda, P.D.; et al. Exclusion of T cells from pancreatic carcinomas in mice is regulated by Ly6C(low) F4/80(+) extratumoral macrophages. Gastroenterology 2015, 149, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef]
- Zhang, Y.; Velez-Delgado, A.; Mathew, E.; Li, D.; Mendez, F.M.; Flannagan, K.; Rhim, A.D.; Simeone, D.M.; Beatty, G.L.; Pasca di Magliano, M. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 2017, 66, 124–136. [Google Scholar] [CrossRef]
- Candido, J.B.; Morton, J.P.; Bailey, P.; Campbell, A.D.; Karim, S.A.; Jamieson, T.; Lapienyte, L.; Gopinathan, A.; Clark, W.; McGhee, E.J.; et al. CSF1R(+) macrophages sustain pancreatic tumor growth through T cell suppression and maintenance of key gene programs that define the squamous subtype. Cell Rep. 2018, 23, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Abate-Daga, D.; Lagisetty, K.H.; Tran, E.; Zheng, Z.; Gattinoni, L.; Yu, Z.; Burns, W.R.; Miermont, A.M.; Teper, Y.; Rudloff, U.; et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum. Gene Ther. 2014, 25, 1003–1012. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab. 2019, 29, 1390.e6–1399.e6. [Google Scholar] [CrossRef]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef]
- Bapiro, T.E.; Richards, F.M.; Goldgraben, M.A.; Olive, K.P.; Madhu, B.; Frese, K.K.; Cook, N.; Jacobetz, M.A.; Smith, D.M.; Tuveson, D.A.; et al. A novel method for quantification of gemcitabine and its metabolites 2′,2′-difluorodeoxyuridine and gemcitabine triphosphate in tumour tissue by LC-MS/MS: Comparison with (19)F NMR spectroscopy. Cancer Chemother. Pharmacol. 2011, 68, 1243–1253. [Google Scholar] [CrossRef]
- Neesse, A.; Frese, K.K.; Bapiro, T.E.; Nakagawa, T.; Sternlicht, M.D.; Seeley, T.W.; Pilarsky, C.; Jodrell, D.I.; Spong, S.M.; Tuveson, D.A. CTGF antagonism with mAb FG-3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12325–12330. [Google Scholar] [CrossRef] [PubMed]
- Bapiro, T.E.; Frese, K.K.; Courtin, A.; Bramhall, J.L.; Madhu, B.; Cook, N.; Neesse, A.; Griffiths, J.R.; Tuveson, D.A.; Jodrell, D.I.; et al. Gemcitabine diphosphate choline is a major metabolite linked to the Kennedy pathway in pancreatic cancer models in vivo. Br. J. Cancer 2014, 111, 318–325. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Neesse, A.; Frese, K.K.; Chan, D.S.; Bapiro, T.E.; Howat, W.J.; Richards, F.M.; Ellenrieder, V.; Jodrell, D.I.; Tuveson, D.A. SPARC independent drug delivery and antitumour effects of nab-paclitaxel in genetically engineered mice. Gut 2014, 63, 974–983. [Google Scholar] [CrossRef]
- Aiello, N.M.; Bajor, D.L.; Norgard, R.J.; Sahmoud, A.; Bhagwat, N.; Pham, M.N.; Cornish, T.C.; Iacobuzio-Donahue, C.A.; Vonderheide, R.H.; Stanger, B.Z. Metastatic progression is associated with dynamic changes in the local microenvironment. Nat. Commun. 2016, 7, 12819. [Google Scholar] [CrossRef]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef]
- Neesse, A.; Bauer, C.A.; Ohlund, D.; Lauth, M.; Buchholz, M.; Michl, P.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: Ready for clinical translation? Gut 2019, 68, 159–171. [Google Scholar] [CrossRef]
- Clappaert, E.J.; Murgaski, A.; Van Damme, H.; Kiss, M.; Laoui, D. Diamonds in the Rough: Harnessing Tumor-Associated Myeloid Cells for Cancer Therapy. Front. Immunol. 2018, 9, 2250. [Google Scholar] [CrossRef]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Goetze, R.G.; Buchholz, S.M.; Patil, S.; Petzold, G.; Ellenrieder, V.; Hessmann, E.; Neesse, A. Utilizing High resolution ultrasound to monitor tumor onset and growth in genetically engineered pancreatic cancer models. Jove J. Vis. Exp. 2018. [Google Scholar] [CrossRef]
- Van Rooijen, N.; Sanders, A. Liposome mediated depletion of macrophages: Mechanism of action, preparation of liposomes and applications. J. Immunol. Methods 1994, 174, 83–93. [Google Scholar] [CrossRef]
- Tsuchiya, S.; Yamabe, M.; Yamaguchi, Y.; Kobayashi, Y.; Konno, T.; Tada, K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 1980, 26, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Jesnowski, R.; Furst, D.; Ringel, J.; Chen, Y.; Schrodel, A.; Kleeff, J.; Kolb, A.; Schareck, W.D.; Lohr, M. Immortalization of pancreatic stellate cells as an in vitro model of pancreatic fibrosis: Deactivation is induced by matrigel and N-acetylcysteine. Lab Investig. 2005, 85, 1276–1291. [Google Scholar] [CrossRef]
- Li, C.M.; Levin, M.; Kaplan, D.L. Bioelectric modulation of macrophage polarization. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Kuhnemuth, B.; Muhlberg, L.; Schipper, M.; Griesmann, H.; Neesse, A.; Milosevic, N.; Wissniowski, T.; Buchholz, M.; Gress, T.M.; Michl, P. CUX1 modulates polarization of tumor-associated macrophages by antagonizing NF-kappaB signaling. Oncogene 2015, 34, 177–187. [Google Scholar] [CrossRef]
- Muhlberg, L.; Kuhnemuth, B.; Costello, E.; Shaw, V.; Sipos, B.; Huber, M.; Griesmann, H.; Krug, S.; Schober, M.; Gress, T.M.; et al. miRNA dynamics in tumor-infiltrating myeloid cells modulating tumor progression in pancreatic cancer. Oncoimmunology 2016, 5, e1160181. [Google Scholar] [CrossRef]
- Francke, A.; Herold, J.; Weinert, S.; Strasser, R.H.; Braun-Dullaeus, R.C. Generation of mature murine monocytes from heterogeneous bone marrow and description of their properties. J. Histochem. Cytochem. 2011, 59, 813–825. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buchholz, S.M.; Goetze, R.G.; Singh, S.K.; Ammer-Herrmenau, C.; Richards, F.M.; Jodrell, D.I.; Buchholz, M.; Michl, P.; Ellenrieder, V.; Hessmann, E.; et al. Depletion of Macrophages Improves Therapeutic Response to Gemcitabine in Murine Pancreas Cancer. Cancers 2020, 12, 1978. https://doi.org/10.3390/cancers12071978
Buchholz SM, Goetze RG, Singh SK, Ammer-Herrmenau C, Richards FM, Jodrell DI, Buchholz M, Michl P, Ellenrieder V, Hessmann E, et al. Depletion of Macrophages Improves Therapeutic Response to Gemcitabine in Murine Pancreas Cancer. Cancers. 2020; 12(7):1978. https://doi.org/10.3390/cancers12071978
Chicago/Turabian StyleBuchholz, Soeren M., Robert G. Goetze, Shiv K. Singh, Christoph Ammer-Herrmenau, Frances M. Richards, Duncan I. Jodrell, Malte Buchholz, Patrick Michl, Volker Ellenrieder, Elisabeth Hessmann, and et al. 2020. "Depletion of Macrophages Improves Therapeutic Response to Gemcitabine in Murine Pancreas Cancer" Cancers 12, no. 7: 1978. https://doi.org/10.3390/cancers12071978
APA StyleBuchholz, S. M., Goetze, R. G., Singh, S. K., Ammer-Herrmenau, C., Richards, F. M., Jodrell, D. I., Buchholz, M., Michl, P., Ellenrieder, V., Hessmann, E., & Neesse, A. (2020). Depletion of Macrophages Improves Therapeutic Response to Gemcitabine in Murine Pancreas Cancer. Cancers, 12(7), 1978. https://doi.org/10.3390/cancers12071978