Human Papillomavirus 16 E7 Promotes EGFR/PI3K/AKT1/NRF2 Signaling Pathway Contributing to PIR/NF-κB Activation in Oral Cancer Cells

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Results
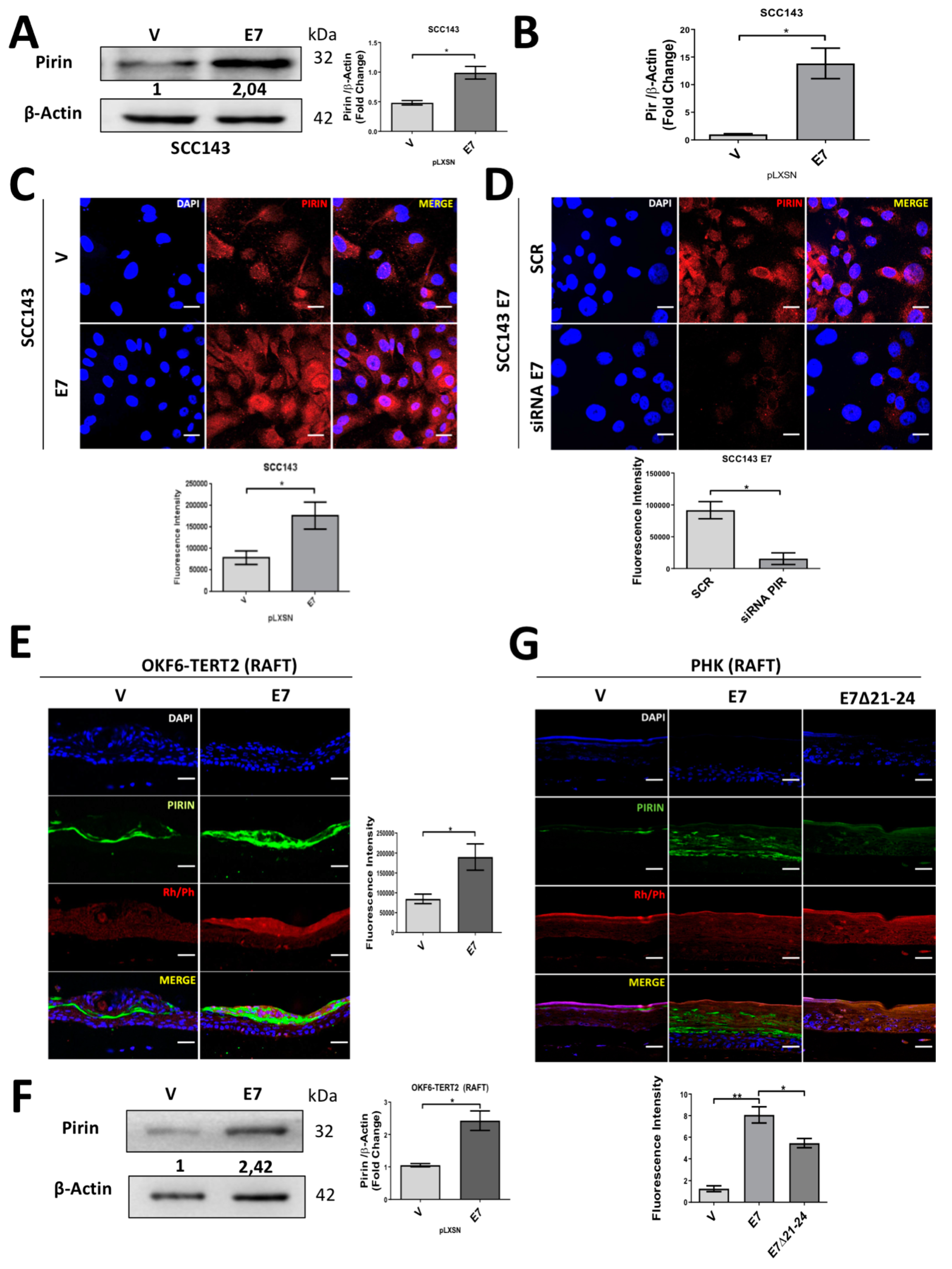
2.1. HPV16 E7 Oncoprotein Upregulates the Levels of Pirin in Oral Cells
2.2. HPV16 E7 Promotes NF-κB Activation
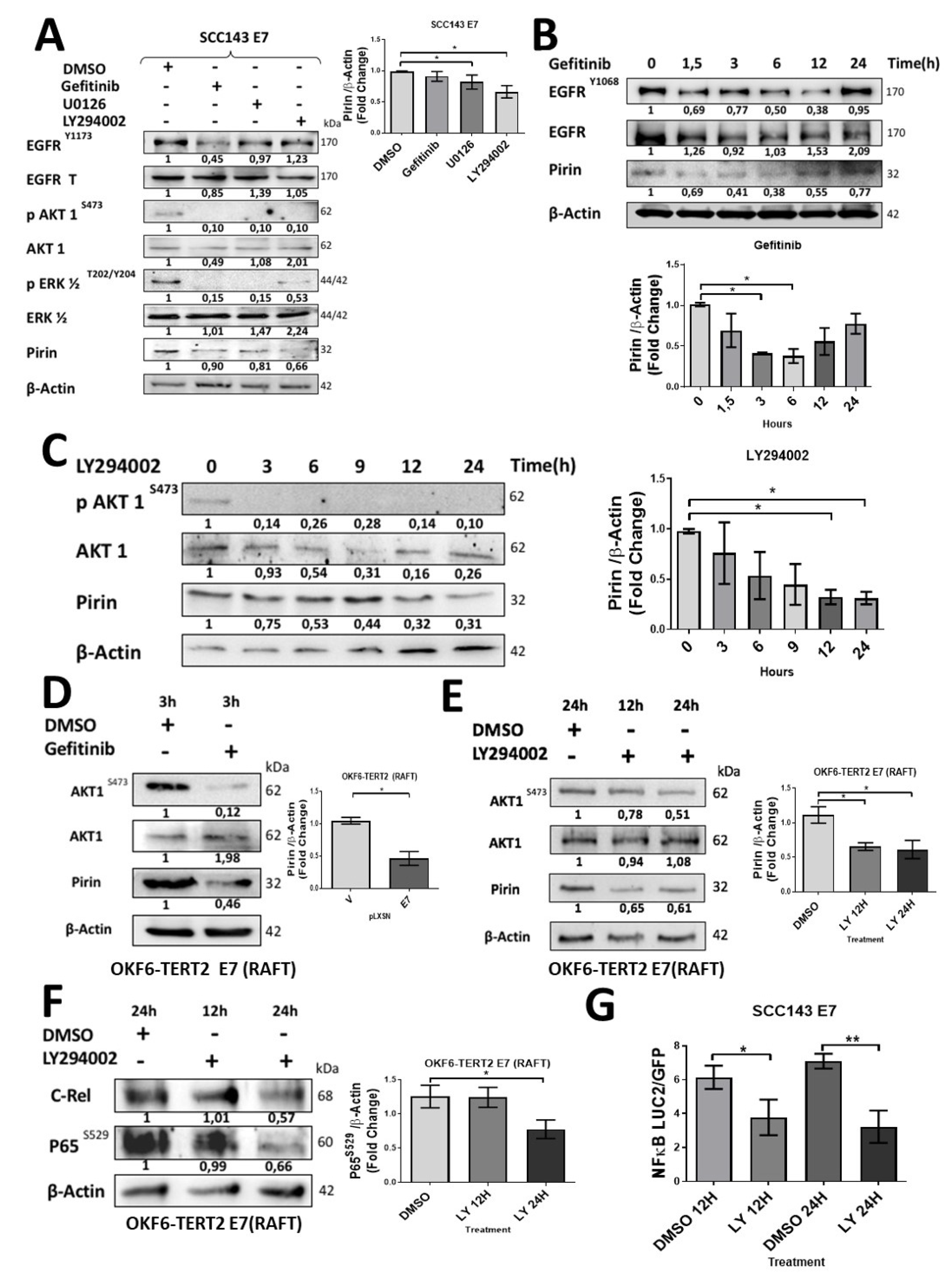
2.3. HPV16 E7 Induces EGFR/PI3K/AKT1 Signaling for PIR/NF-κB Activation
2.4. HPV16 E7 Activates PIR-Promoter through NRF2 Transcription Factor in Oral Cells
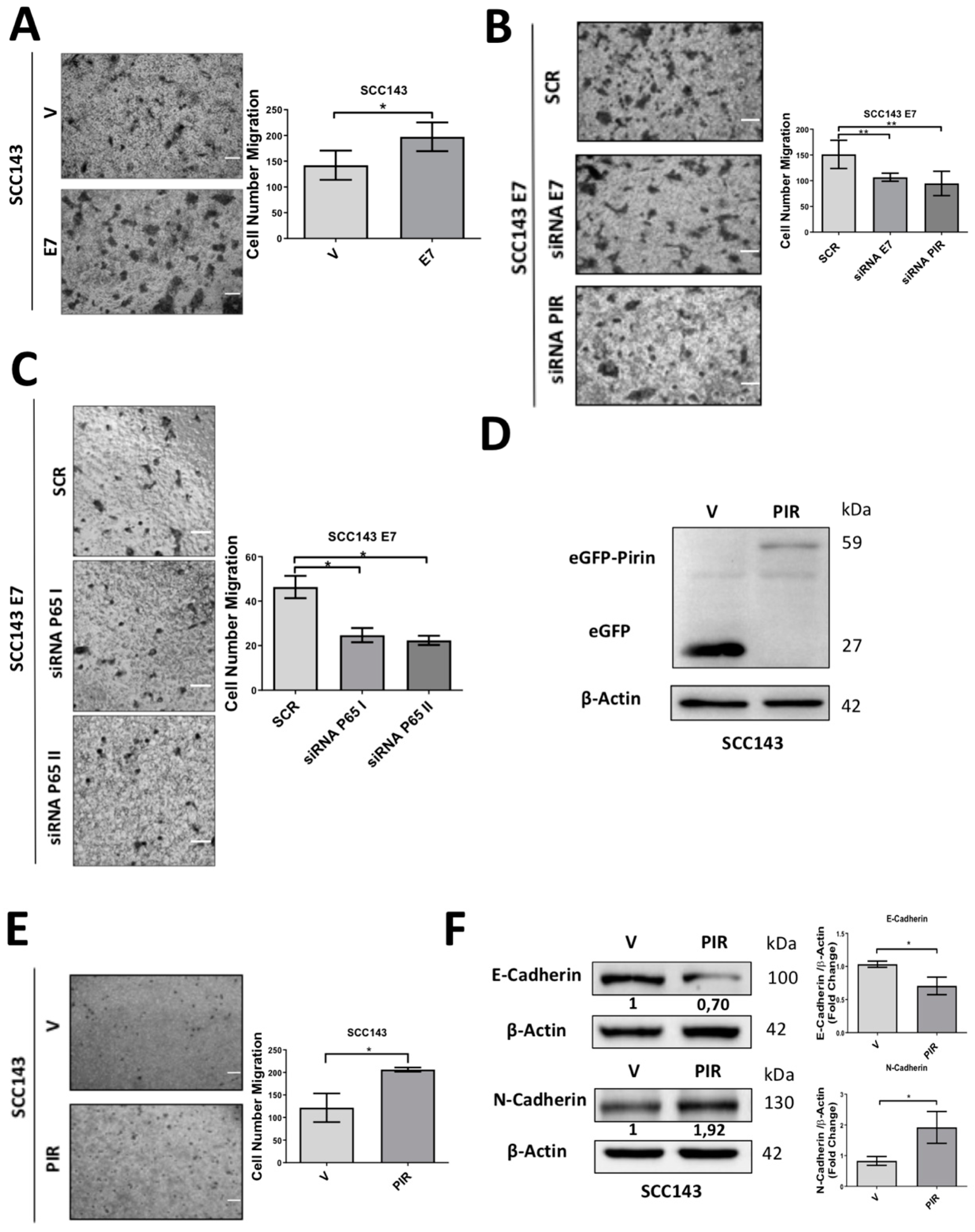
2.5. HPV16 E7-Mediated Pirin Overexpression Positively Regulates Cell Migration and Epithelial–Mesenchymal Transition (EMT)
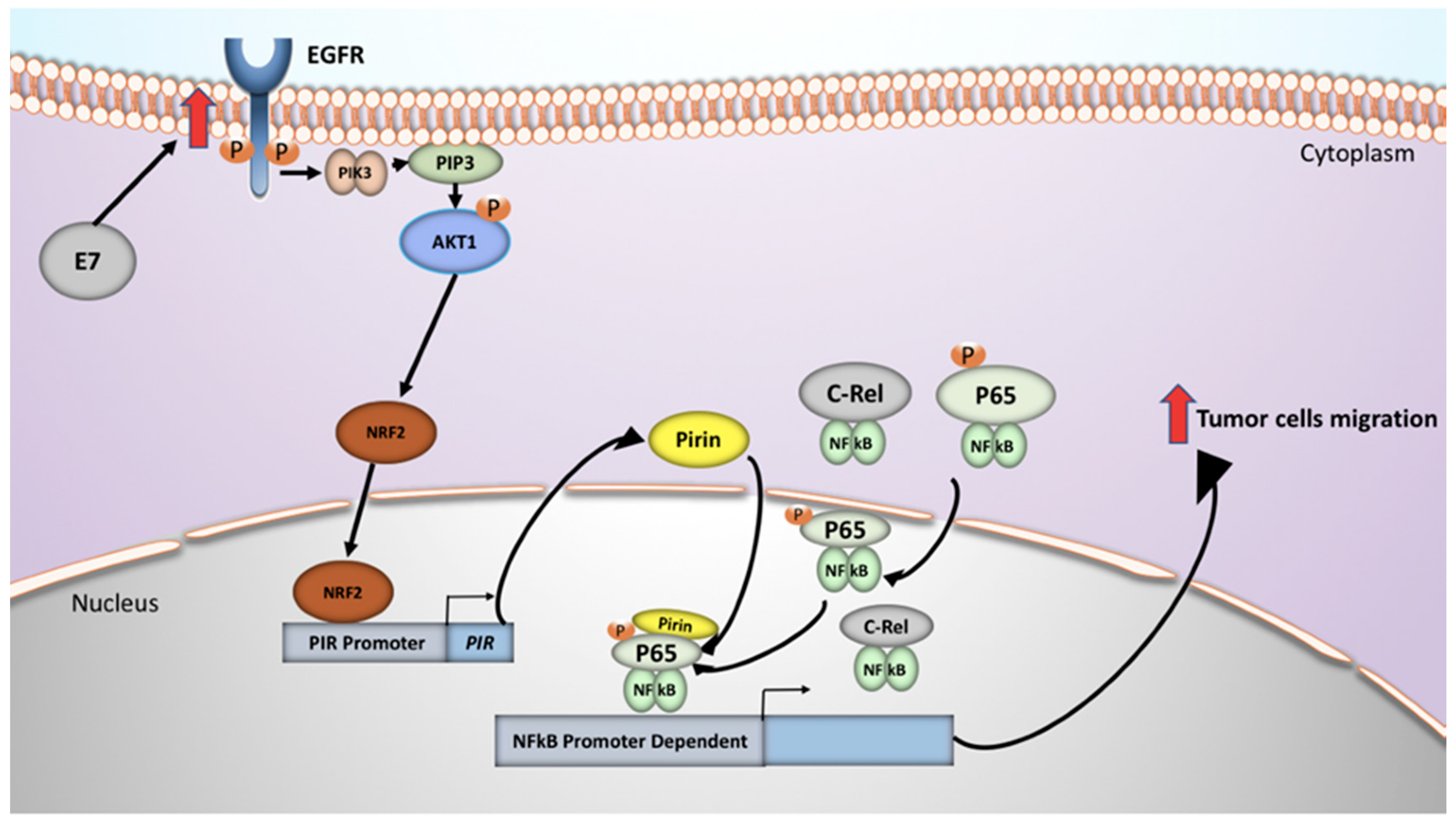
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Culture, Vectors, Transfections and Transductions
4.2. Organotypic Raft Cultures
4.3. Cell Viability Assay
4.4. Luciferase and NF-κB Activation Assays
4.5. Proteome Profiler Phospho MAPK and NF-κB Array
4.6. Western Blot
4.7. Pharmacological Inhibition Assay
4.8. Immunofluorescence and Confocal Microscopy
4.9. Reverse Transcriptase–Quantitative Polymerase Chain Reaction
4.10. Cell Migration Assays
4.11. Chromatin Immunoprecipitation
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hausen, H.Z. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Hausen, H.Z. Human papillomavirus & cervical cancer. Indian J. Med. Res. 2009, 130, 209. [Google Scholar] [PubMed]
- Herrero, R.; Castellsagué, X.; Pawlita, M.; Lissowska, J.; Kee, F.; Balaram, P.; Rajkumar, T.; Sridhar, H.; Rose, B.; Pintos, J.; et al. Human papillomavirus and oral cancer: The International Agency for Research on Cancer multicenter study. J. Natl. Cancer Inst. 2003, 95, 1772–1783. [Google Scholar] [CrossRef]
- Kreimer, A.R.; Clifford, G.M.; Boyle, P.; Franceschi, S. Human Papillomavirus Types in Head and Neck Squamous Cell Carcinomas Worldwide: A Systematic Review. Cancer Epidemiol. Biomark. Prev. 2005, 14, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Wakeham, K.; Kavanagh, K. The Burden of HPV-Associated Anogenital Cancers. Curr. Oncol. Rep. 2014, 16. [Google Scholar] [CrossRef] [PubMed]
- Kreimer, A.R.; Clifford, G.M.; Snijders, P.J.; Castellsagué, X.; Meijer, C.J.; Pawlita, M.; Viscidi, R.; Herrero, R.; Franceschi, S. HPV16 semiquantitative viral load and serologic biomarkers in oral and oropharyngeal squamous cell carcinomas. Int. J. Cancer 2005, 115, 329–332. [Google Scholar] [CrossRef]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Heck, D.V.; Yee, C.L.; Howley, P.; Munger, K. Efficiency of binding the retinoblastoma protein correlates with the transforming capacity of the E7 oncoproteins of the human papillomaviruses. Proc. Natl. Acad. Sci. USA 1992, 89, 4442–4446. [Google Scholar] [CrossRef]
- Yeo-Teh, N.S.L.; Ito, Y.; Jha, S. High-Risk Human Papillomaviral Oncogenes E6 and E7 Target Key Cellular Pathways to Achieve Oncogenesis. Int. J. Mol. Sci. 2018, 19, 1706. [Google Scholar] [CrossRef] [PubMed]
- Marullo, R.; Werner, E.; Zhang, H.; Chen, G.Z.; Shin, N.M.; Doetsch, P.W. HPV16 E6 and E7 proteins induce a chronic oxidative stress response via NOX2 that causes genomic instability and increased susceptibility to DNA damage in head and neck cancer cells. Carcinogenesis 2015, 36, 1397–1406. [Google Scholar] [CrossRef]
- Spardy, N.; Covella, K.; Cha, E.; Hoskins, E.E.; Wells, S.I.; Duensing, A.; Duensing, S. Human papillomavirus 16 E7 oncoprotein attenuates DNA damage checkpoint control by increasing the proteolytic turnover of claspin. Cancer Res. 2009, 69, 7022–7029. [Google Scholar] [CrossRef]
- Funk, J.O.; Waga, S.; Harry, J.B.; Espling, E.; Stillman, B.; Galloway, D.A. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genome Res. 1997, 11, 2090–2100. [Google Scholar] [CrossRef] [PubMed]
- Zerfass-Thome, K.; Zwerschke, W.; Mannhardt, B.; Tindle, R.; Botz, J.W.; Jansen-Dürr, P. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene 1996, 13, 2323–2330. [Google Scholar]
- Hu, T.; Ferril, S.; Snider, A.; Barbosa, M. In-vivo analysis of hpv e7 protein association with prb, p107 and p130. Int. J. Oncol. 1995, 6, 167–174. [Google Scholar] [CrossRef]
- Akerman, G.S.; Tolleson, W.H.; Brown, K.L.; Zyzak, L.L.; Mourateva, E.; Engin, T.S.; Basaraba, A.; Coker, A.L.; Creek, K.E.; Pirisi, L. Human papillomavirus type 16 E6 and E7 cooperate to increase epidermal growth factor receptor (EGFR) mRNA levels, overcoming mechanisms by which excessive EGFR signaling shortens the life span of normal human keratinocytes. Cancer Res. 2001, 61, 3837–3843. [Google Scholar]
- Menges, C.W.; Baglia, L.A.; Lapoint, R.; McCance, D.J. Human Papillomavirus Type 16 E7 Up-regulates AKT Activity through the Retinoblastoma Protein. Cancer Res. 2006, 66, 5555–5559. [Google Scholar] [CrossRef]
- Fruman, D.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef]
- Charette, S.T.; McCance, D.J. The E7 protein from human papillomavirus type 16 enhances keratinocyte migration in an Akt-dependent manner. Oncogene 2007, 26, 7386–7390. [Google Scholar] [CrossRef]
- Tankere, F.; Camproux, A.; Barry, B.; Guedon, C.; Depondt, J.; Gehanno, P.; Tankere, F. Prognostic Value of Lymph Node Involvement in Oral Cancers: A Study of 137 Cases. Laryngoscope 2000, 110, 2061–2065. [Google Scholar] [CrossRef] [PubMed]
- Sano, D.; Myers, J.N. Metastasis of squamous cell carcinoma of the oral tongue. Cancer Metastasis Rev. 2007, 26, 645–662. [Google Scholar] [CrossRef]
- Carrillo, D.; Muñoz, J.P.; Huerta, H.; Leal, G.; Corvalán, A.H.; León, O.; Calaf, G.M.; Urzúa, U.; Boccardo, E.; Tapia, J.C.; et al. Upregulation of PIR gene expression induced by human papillomavirus E6 and E7 in epithelial oral and cervical cells. Open Biol. 2017, 7, 170111. [Google Scholar] [CrossRef]
- Komai, K.; Niwa, Y.; Sasazawa, Y.; Simizu, S. Pirin regulates epithelial to mesenchymal transition independently of Bcl3-Slug signaling. FEBS Lett. 2015, 589, 738–743. [Google Scholar] [CrossRef]
- Pires, B.; Silva, R.C.M.C.; Ferreira, G.M.; Abdelhay, E. NF-kappaB: Two Sides of the Same Coin. Genes 2018, 9, 24. [Google Scholar] [CrossRef]
- Mitchell, J.P.; Carmody, R.J. NF-κB and the Transcriptional Control of Inflammation. Int. Rev. Cell Mol. Biol. 2018, 335, 41–84. [Google Scholar] [CrossRef]
- Vandermark, E.R.; DeLuca, K.A.; Gardner, C.R.; Marker, D.F.; Schreiner, C.N.; Strickland, D.; Wilton, K.M.; Mondal, S.; Woodworth, C. Human papillomavirus type 16 E6 and E 7 proteins alter NF-kB in cultured cervical epithelial cells and inhibition of NF-kB promotes cell growth and immortalization. Virology 2012, 425, 53–60. [Google Scholar] [CrossRef]
- Morgan, E.L.; Macdonald, A. Autocrine STAT3 activation in HPV positive cervical cancer through a virus-driven Rac1—NFkappaB—IL-6 signalling axis. PLoS Pathog. 2019, 15, e1007835. [Google Scholar] [CrossRef]
- Tummers, B.; Goedemans, R.; Pelascini, L.P.L.; Jordanova, E.S.; Van Esch, E.M.G.; Meyers, C.; Melief, C.J.M.; Boer, J.M.; Van Der Burg, S.H. The interferon-related developmental regulator 1 is used by human papillomavirus to suppress NFkappaB activation. Nat. Commun. 2015, 6, 6537. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Hayden, M.; Ghosh, S. Crosstalk in NF-kappaB signaling pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef]
- He, C.; Mao, D.; Hua, G.; Lv, X.; Chen, X.; Angeletti, P.C.; Dong, J.; Remmenga, S.W.; Rodabaugh, K.J.; Zhou, J.; et al. The Hippo/YAP pathway interacts with EGFR signaling and HPV oncoproteins to regulate cervical cancer progression. EMBO Mol. Med. 2015, 7, 1426–1449. [Google Scholar] [CrossRef]
- Brzóska, K.; Stępkowski, T.; Kruszewski, M. Basal PIR expression in HeLa cells is driven by NRF2 via evolutionary conserved antioxidant response element. Mol. Cell. Biochem. 2014, 389, 99–111. [Google Scholar] [CrossRef]
- Williams, V.M.; Filippova, M.; Filippov, V.; Payne, K.J.; Duerksen-Hughes, P.J. Human Papillomavirus Type 16 E6* Induces Oxidative Stress and DNA Damage. J. Virol. 2014, 88, 6751–6761. [Google Scholar] [CrossRef]
- Prati, B.; Abjaude, W.D.S.; Termini, L.; Morale, M.; Herbster, S.; Longatto-Filho, A.; Nunes, R.A.L.; Camacho, L.C.C.; Rabelo-Santos, S.H.; Zeferino, L.C.; et al. Three Prime Repair Exonuclease 1 (TREX1) expression correlates with cervical cancer cells growth in vitro and disease progression in vivo. Sci. Rep. 2019, 9, 351. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Hausen, H.Z. Papillomaviruses in anogenital cancer as a model to understand the role of viruses in human cancers. Cancer Res. 1989, 49, 4677–4681. [Google Scholar]
- Balsitis, S.J.; Sage, J.; Duensing, S.; Munger, K.; Jacks, T.; Lambert, P.F. Recapitulation of the Effects of the Human Papillomavirus Type 16 E7 Oncogene on Mouse Epithelium by Somatic Rb Deletion and Detection of pRb-Independent Effects of E7 In Vivo. Mol. Cell. Biol. 2003, 23, 9094–9103. [Google Scholar] [CrossRef]
- Shin, M.-K.; Sage, J.; Lambert, P.F. Inactivating all three rb family pocket proteins is insufficient to initiate cervical cancer. Cancer Res. 2012, 72, 5418–5427. [Google Scholar] [CrossRef][Green Version]
- Branca, M.; Giorgi, C.; Ciotti, M.; Santini, D.; Di Bonito, L.; Costa, S.; Benedetto, A.; Bonifacio, D.; Paba, P.; Accardi, L.; et al. Upregulation of nuclear factor-kappaB (NF-kappaB) is related to the grade of cervical intraepithelial neoplasia, but is not an independent predictor of high-risk human papillomavirus or disease outcome in cervical cancer. Diagn. Cytopathol. 2006, 34, 555–563. [Google Scholar] [CrossRef]
- Hayden, M.; Ghosh, S. Shared Principles in NF-kappaB Signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Sun, S.C. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef]
- Jiang, T.; Grabiner, B.; Zhu, Y.; Jiang, C.; Li, H.; You, Y.; Lang, J.; Hung, M.C.; Lin, X. CARMA3 is crucial for EGFR-Induced activation of NF-kappaB and tumor progression. Cancer Res. 2011, 71, 2183–2192. [Google Scholar] [CrossRef]
- Pan, D.; Jiang, C.; Ma, Z.; Blonska, M.; You, M.J.; Lin, X. MALT1 is required for EGFR-induced NF-kappaB activation and contributes to EGFR-driven lung cancer progression. Oncogene 2015, 35, 919–928. [Google Scholar] [CrossRef]
- Li, Z.; Liao, J.; Yang, Z.; Choi, E.Y.; Lapidus, R.G.; Liu, X.; Cullen, K.J.; Dan, H. Co-targeting EGFR and IKKβ/NF-kappaB signalling pathways in head and neck squamous cell carcinoma: A potential novel therapy for head and neck squamous cell cancer. Br. J. Cancer 2018, 120, 306–316. [Google Scholar] [CrossRef]
- Chua, H.L.; Bhat-Nakshatri, P.; Clare, S.E.; Morimiya, A.; Badve, S.; Nakshatri, H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: Potential involvement of ZEB-1 and ZEB-2. Oncogene 2006, 26, 711–724. [Google Scholar] [CrossRef]
- Kang, B.-H.; Shu, C.; Chao, J.-K.; Lee, C.-H.; Fu, T.-Y.; Liou, H.-H.; Ger, L.-P.; Liu, P. HSPD1 repressed E-cadherin expression to promote cell invasion and migration for poor prognosis in oral squamous cell carcinoma. Sci. Rep. 2019, 9, 8932. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Liu, F.; Rehmani, I.; Esaki, S.; Fu, R.; Chen, L.; De Serrano, V.; Liu, A. Pirin is an iron-Dependent redox regulator of NF-kappaB. Proc. Natl. Acad. Sci. USA 2013, 110, 9722–9727. [Google Scholar] [CrossRef]
- Miyazaki, I.; Simizu, S.; Okumura, H.; Takagi, S.; Osada, H. A small-molecule inhibitor shows that pirin regulates migration of melanoma cells. Nat. Methods 2010, 6, 667–673. [Google Scholar] [CrossRef]
- Zhou, Y.; Pan, Y.; Zhang, S.; Shi, X.; Ning, T.; Ke, Y. Increased phosphorylation of p70 S6 kinase is associated with HPV16 infection in cervical cancer and esophageal cancer. Br. J. Cancer 2007, 97, 218–222. [Google Scholar] [CrossRef]
- Bai, D.; Ueno, L.; Vogt, P.K. Akt-Mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int. J. Cancer 2009, 125, 2863–2870. [Google Scholar] [CrossRef]
- Madrid, L.V.; Mayo, M.W.; Reuther, J.Y.; Baldwin, A.S. Akt Stimulates the Transactivation Potential of the RelA/p65 Subunit of NF-kappaB through Utilization of kappaB Kinase and Activation of the Mitogen-activated Protein Kinase p38. J. Biol. Chem. 2001, 276, 18934–18940. [Google Scholar] [CrossRef]
- Gustin, J.A.; Ozes, O.N.; Akca, H.; Pincheira, R.; Mayo, L.D.; Li, Q.; Guzman, J.R.; Korgaonkar, C.K.; Donner, D.B. Cell Type-Specific Expression of the kappaB Kinases Determines the Significance of Phosphatidylinositol 3-Kinase/Akt Signaling to NF-kappaB Activation. J. Biol. Chem. 2003, 279, 1615–1620. [Google Scholar] [CrossRef] [PubMed]
- Hausen, H.Z. Papillomaviruses causing cancer: Evasion from host-cell control in early events in carcinogenesis. J. Natl. Cancer Inst. 2000, 92, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.-Q.; Tuersun, H.; Jiao, S.-J.; Zheng, J.-H.; Xiao, J.-B.; Hasim, A. Functional Role of NRF2 in Cervical Carcinogenesis. PLoS ONE 2015, 10, e0133876. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.-M.; Kim, Y.S.; Hur, D.Y. LMP1 and 2A Induce the Expression of Nrf2 Through Akt Signaling Pathway in Epstein-Barr Virus-Transformed B Cells. Transl. Oncol. 2019, 12, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, Y.; Feng, Z.; Bergan, R.; Li, B.; Qin, Y.; Zhao, L.; Zhang, Z.; Shi, M. Involvement of the PI3K/Akt/Nrf2 Signaling Pathway in Resveratrol-Mediated Reversal of Drug Resistance in HL-60/ADR Cells. Nutr. Cancer 2019, 71, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Reddy, N.M.; Potteti, H.R.; Vegiraju, S.; Chen, H.-J.; Tamatam, C.M.; Reddy, S.P. PI3K-AKT Signaling via Nrf2 Protects against Hyperoxia-Induced Acute Lung Injury, but Promotes Inflammation Post-Injury Independent of Nrf2 in Mice. PLoS ONE 2015, 10, e0129676. [Google Scholar] [CrossRef]
- Rojo, A.I.; Sagarra, M.R.; Cuadrado, A. GSK-3beta down-Regulates the transcription factor Nrf2 after oxidant damage: Relevance to exposure of neuronal cells to oxidative stress. J. Neurochem. 2008, 105, 192–202. [Google Scholar] [CrossRef]
- White, J.S.; Weissfeld, J.L.; Ragin, C.C.R.; Rossie, K.M.; Martin, C.L.; Shuster, M.; Ishwad, C.S.; Law, J.C.; Myers, E.N.; Johnson, J.T.; et al. The influence of clinical and demographic risk factors on the establishment of head and neck squamous cell carcinoma cell lines. Oral Oncol. 2006, 43, 701–712. [Google Scholar] [CrossRef]
- Valdano, M.B.; Cavatorta, A.L.; Morale, M.G.; Marziali, F.; Lino, V.D.S.; Steenbergen, R.D.M.; Boccardo, E.; Gardiol, D. Disc large 1 expression is altered by human papillomavirus E6/E7 proteins in organotypic cultures of human keratinocytes. J. Gen. Virol. 2016, 97, 453–462. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Forward 5′-3′ | Reverse 5′-3′ | Amplicon (bp) |
|---|---|---|---|
| PIR | TCAAATTGGACCCAGGAGCC | TCCAAGCACTGCTGTGTGAT | 131 |
| E7 HPV16 | CAATATTGTAATGGGCTCTGTCC | ATTTGCAACCAGAGACAACTGAT | 120 |
| ARE CHIP | GTGAGCATCTCTTCCCGGCA | TGACCGCCAGCATTCCCTCA | 347 |
| β-actin | AGCGAGCATCCCCCAAAG | GGGCACGAAGGCTCATCA | 289 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrillo-Beltrán, D.; Muñoz, J.P.; Guerrero-Vásquez, N.; Blanco, R.; León, O.; de Souza Lino, V.; Tapia, J.C.; Maldonado, E.; Dubois-Camacho, K.; Hermoso, M.A.; et al. Human Papillomavirus 16 E7 Promotes EGFR/PI3K/AKT1/NRF2 Signaling Pathway Contributing to PIR/NF-κB Activation in Oral Cancer Cells. Cancers 2020, 12, 1904. https://doi.org/10.3390/cancers12071904
Carrillo-Beltrán D, Muñoz JP, Guerrero-Vásquez N, Blanco R, León O, de Souza Lino V, Tapia JC, Maldonado E, Dubois-Camacho K, Hermoso MA, et al. Human Papillomavirus 16 E7 Promotes EGFR/PI3K/AKT1/NRF2 Signaling Pathway Contributing to PIR/NF-κB Activation in Oral Cancer Cells. Cancers. 2020; 12(7):1904. https://doi.org/10.3390/cancers12071904
Chicago/Turabian StyleCarrillo-Beltrán, Diego, Juan P. Muñoz, Nahir Guerrero-Vásquez, Rancés Blanco, Oscar León, Vanesca de Souza Lino, Julio C. Tapia, Edio Maldonado, Karen Dubois-Camacho, Marcela A. Hermoso, and et al. 2020. "Human Papillomavirus 16 E7 Promotes EGFR/PI3K/AKT1/NRF2 Signaling Pathway Contributing to PIR/NF-κB Activation in Oral Cancer Cells" Cancers 12, no. 7: 1904. https://doi.org/10.3390/cancers12071904
APA StyleCarrillo-Beltrán, D., Muñoz, J. P., Guerrero-Vásquez, N., Blanco, R., León, O., de Souza Lino, V., Tapia, J. C., Maldonado, E., Dubois-Camacho, K., Hermoso, M. A., Corvalán, A. H., Calaf, G. M., Boccardo, E., & Aguayo, F. (2020). Human Papillomavirus 16 E7 Promotes EGFR/PI3K/AKT1/NRF2 Signaling Pathway Contributing to PIR/NF-κB Activation in Oral Cancer Cells. Cancers, 12(7), 1904. https://doi.org/10.3390/cancers12071904