Tumor-Associated Neutrophils Dampen Adaptive Immunity and Promote Cutaneous Squamous Cell Carcinoma Development

 ,
,  , , , ,
, , , ,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
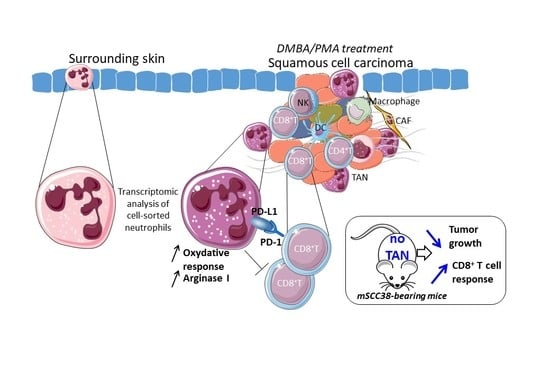
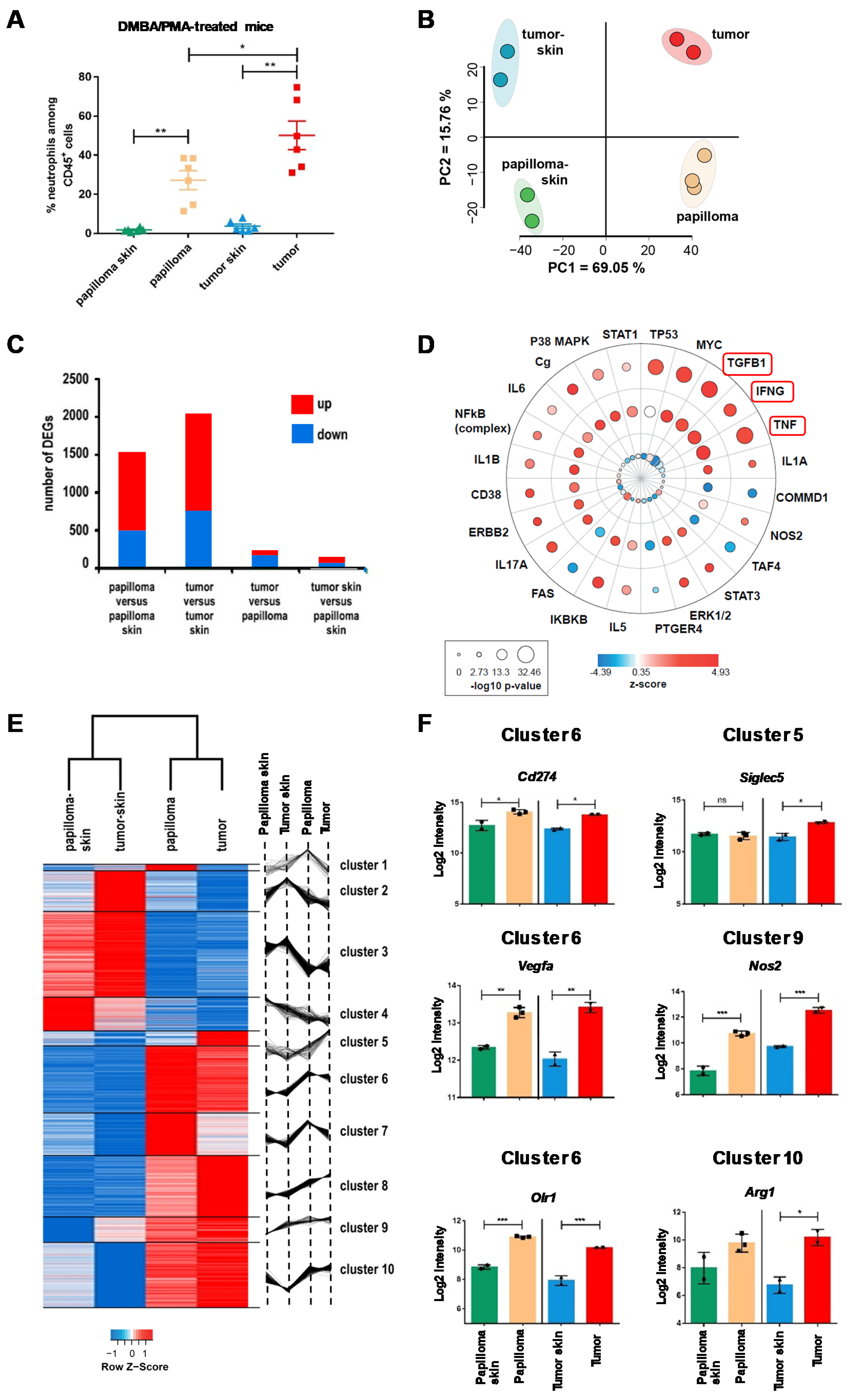
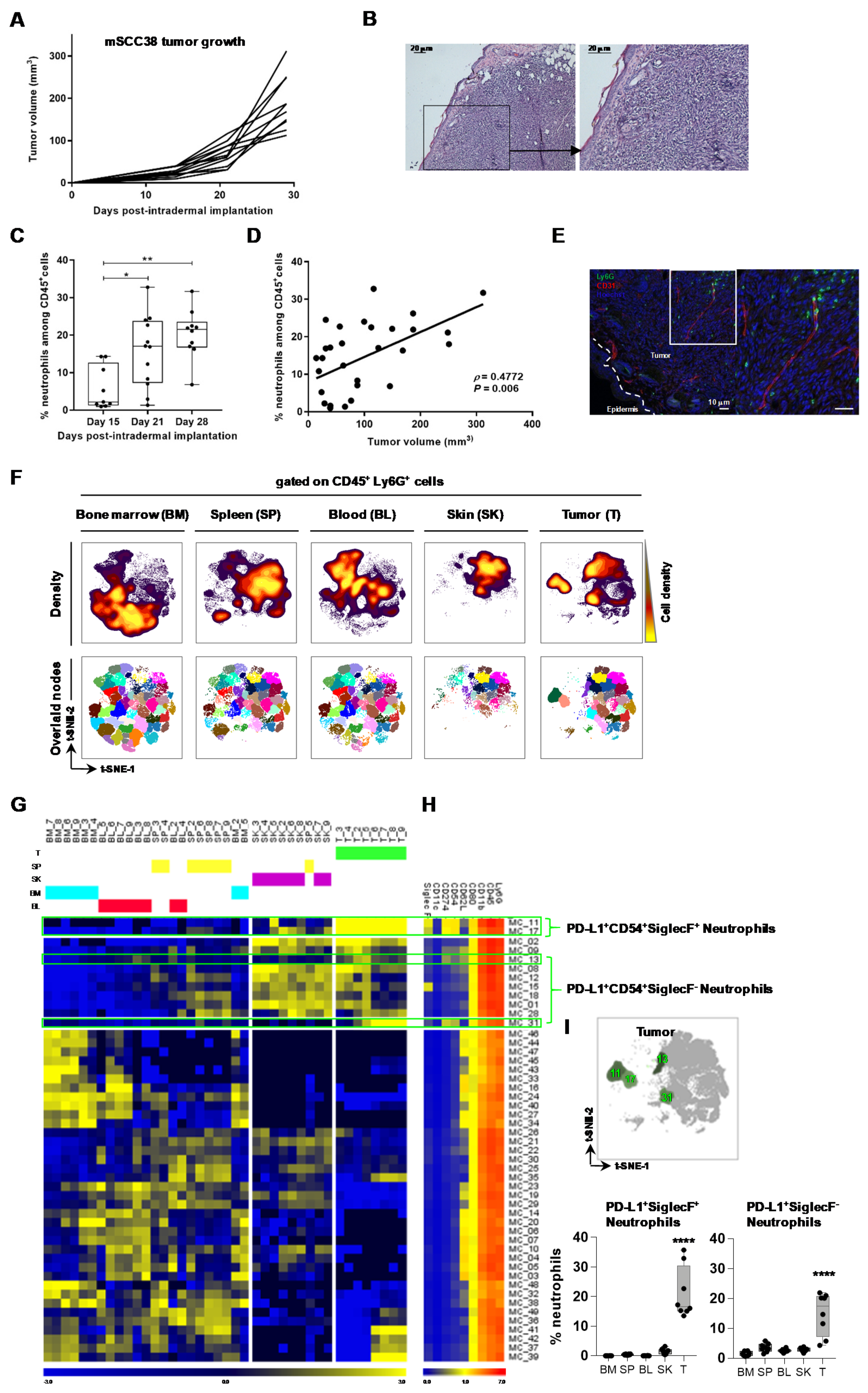
2.1. The Local Microenvironment of Cutaneous Squamous Cell Carcinoma Imprints Tumor-Associated Neutrophils Towards a Protumor Phenotype
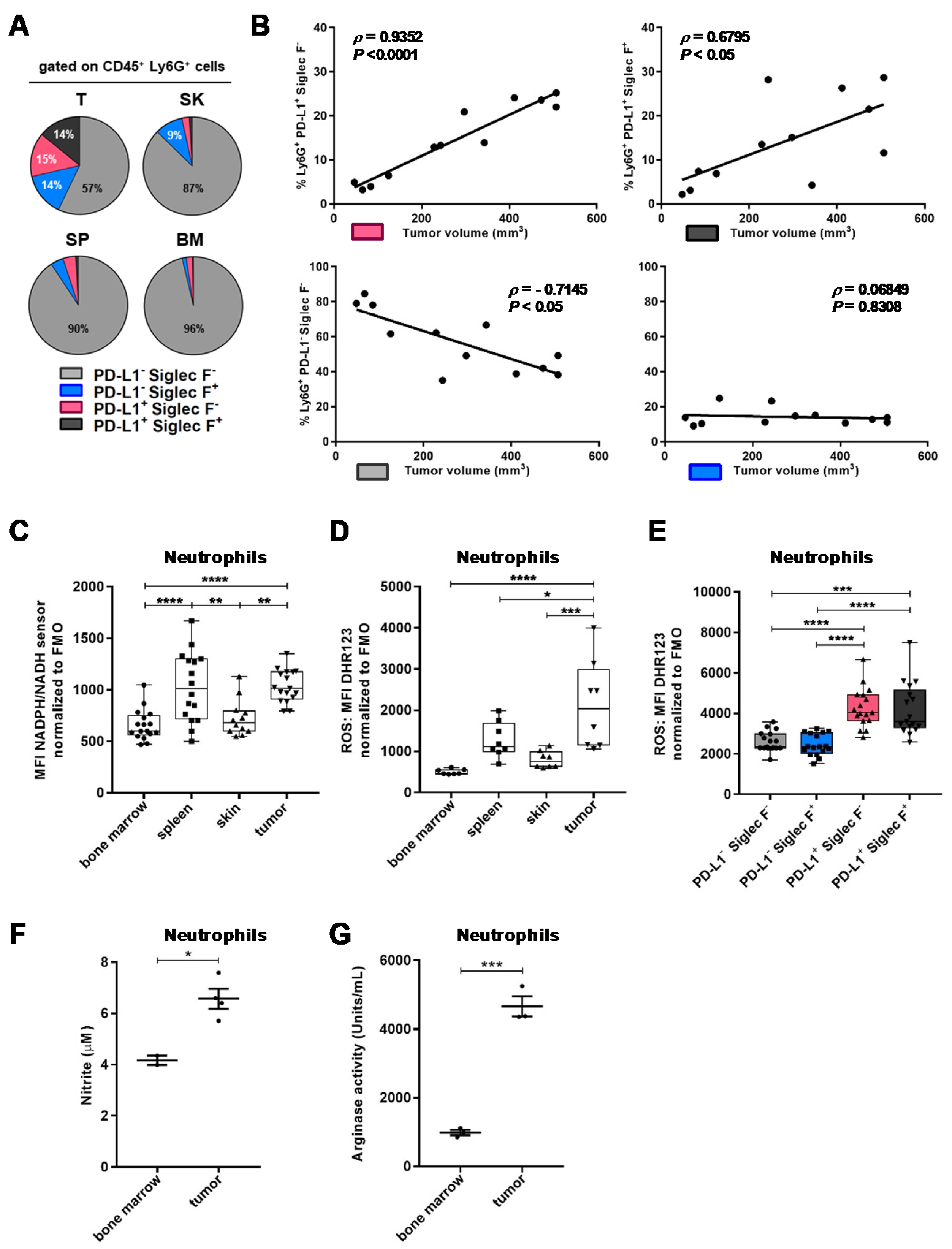
2.2. TAN Exhibit Heterogeneity in Their Phenotype and Exert Protumor Functions
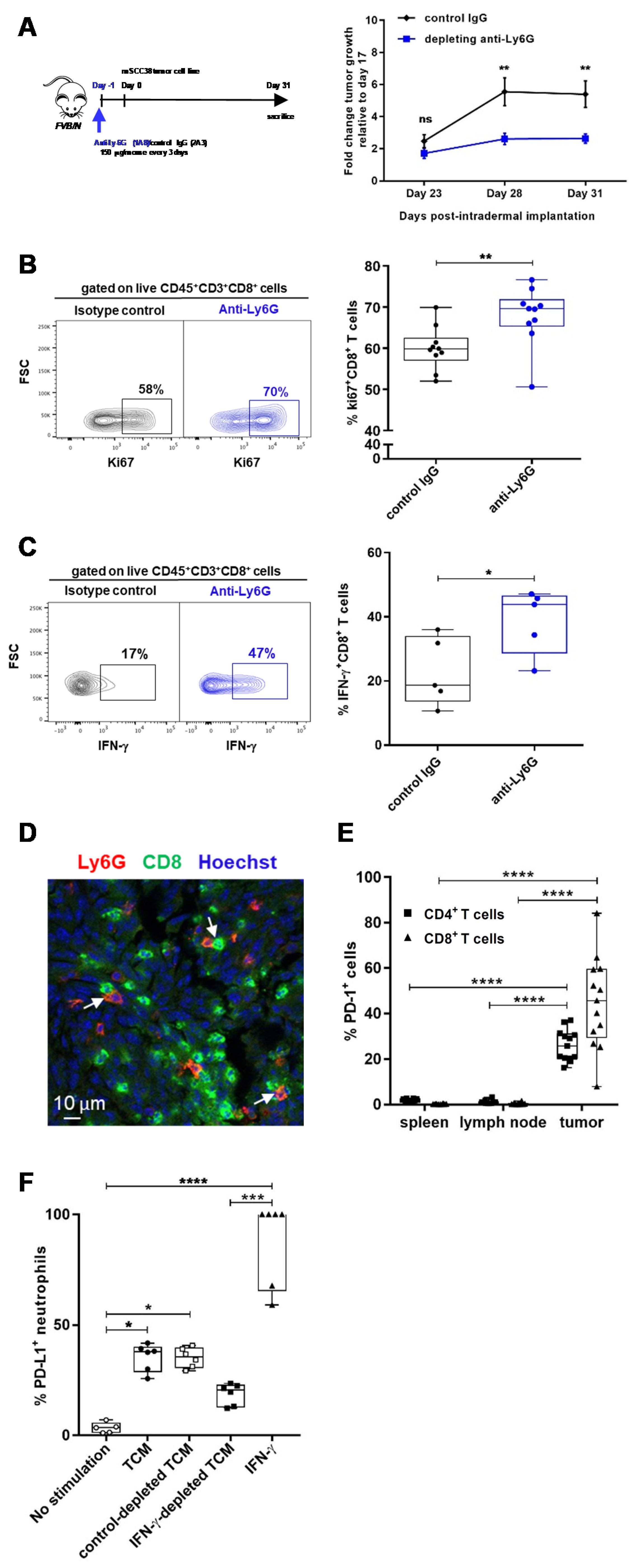
2.3. TANs Limit Antitumor CD8+ T Cell Responses and Concomitant Upregulation of PD-L1 on TANs and PD-1 on CD8+ T Cells Participates in This Process
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. In Vivo Tumor Growth
4.3. In Vivo Neutrophil Depletion
4.4. Tissue Preparation and Cell Purification
4.5. RNA Isolation and Microarray Analysis
4.6. Flow Cytometry and Computational Analysis
4.7. Immunofluorescence
4.8. Oxidative Burst Assay
4.9. NADPH/NADP
4.10. NO (Nitrite) Quantification
4.11. Arginase Activity Assay
4.12. TGF-β ELISA
4.13. Tumor Conditioned Medium and In Vitro Stimulation
4.14. Statistical Analysis
4.15. Data Deposition
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Didona, D.; Paolino, G.; Bottoni, U.; Cantisani, C. Non Melanoma Skin Cancer Pathogenesis Overview. Biomedicines 2018, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Euvrard, S.; Kanitakis, J.; Claudy, A. Skin cancers after organ transplantation. N. Engl. J. Med. 2003, 348, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; Huang, S.J.; Murphy, G.F.; Mollet, I.G.; Hijnen, D.; Muthukuru, M.; Schanbacher, C.F.; Edwards, V.; Miller, D.M.; Kim, J.E.; et al. Human squamous cell carcinomas evade the immune response by down-regulation of vascular E-selectin and recruitment of regulatory T cells. J. Exp. Med. 2008, 205, 2221–2234. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, D.E.; Roberts, S.J.; Clarke, S.L.; Filler, R.; Lewis, J.M.; Tigelaar, R.E.; Girardi, M.; Hayday, A.C. Sustained localized expression of ligand for the activating NKG2D receptor impairs natural cytotoxicity in vivo and reduces tumor immunosurveillance. Nat. Immunol. 2005, 6, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, T.; Clarke, M.; Steele, C.W.; Samuel, M.S.; Neumann, J.; Jung, A.; Huels, D.; Olson, M.F.; Das, S.; Nibbs, R.J.; et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J. Clin. Invest. 2012, 122, 3127–3144. [Google Scholar] [CrossRef]
- Seddon, A.; Hock, B.; Miller, A.; Frei, L.; Pearson, J.; McKenzie, J.; Simcock, J.; Currie, M. Cutaneous squamous cell carcinomas with markers of increased metastatic risk are associated with elevated numbers of neutrophils and/or granulocytic myeloid derived suppressor cells. J. Dermatol. Sci. 2016, 83, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; O’Leary, C.E.; Wang, L.S.; Bhatti, T.R.; Dai, N.; Kapoor, V.; Liu, P.; Mei, J.; Guo, L.; Oliver, P.M.; et al. CD11b+Ly6G+ cells inhibit tumor growth by suppressing IL-17 production at early stages of tumorigenesis. Oncoimmunology 2016, 5, e1061175. [Google Scholar] [CrossRef] [PubMed]
- Ponzetta, A.; Carriero, R.; Carnevale, S.; Barbagallo, M.; Molgora, M.; Perucchini, C.; Magrini, E.; Gianni, F.; Kunderfranco, P.; Polentarutti, N.; et al. Neutrophils Driving Unconventional T Cells Mediate Resistance against Murine Sarcomas and Selected Human Tumors. Cell 2019, 178, 346–360. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Zhou, J.; Nefedova, Y.; Lei, A.; Gabrilovich, D. Neutrophils and PMN-MDSC: Their biological role and interaction with stromal cells. Semin. Immunol. 2018, 35, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Brandau, S. The dichotomy of neutrophil granulocytes in cancer. Semin. Cancer Biol. 2013, 23, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Andzinski, L.; Kasnitz, N.; Stahnke, S.; Wu, C.F.; Gereke, M.; von Kockritz-Blickwede, M.; Schilling, B.; Brandau, S.; Weiss, S.; Jablonska, J. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int. J. Cancer 2016, 138, 1982–1993. [Google Scholar] [CrossRef] [PubMed]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Templeton, A.J.; McNamara, M.G.; Seruga, B.; Vera-Badillo, F.E.; Aneja, P.; Ocana, A.; Leibowitz-Amit, R.; Sonpavde, G.; Knox, J.J.; Tran, B.; et al. Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: A systematic review and meta-analysis. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Brandau, S.; Dumitru, C.A.; Lang, S. Protumor and antitumor functions of neutrophil granulocytes. Semin. Immunopathol. 2013, 35, 163–176. [Google Scholar] [CrossRef]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 2019, 50, 1317–1334.e1310. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Mishalian, I.; Singhal, S.; Cheng, G.; Kapoor, V.; Horng, W.; Fridlender, G.; Bayuh, R.; Worthen, G.S.; et al. Transcriptomic analysis comparing tumor-associated neutrophils with granulocytic myeloid-derived suppressor cells and normal neutrophils. PLos ONE 2012, 7, e31524. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.I.; Collazo, M.; Shalova, I.N.; Biswas, S.K.; Gabrilovich, D.I. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J. Leukoc. Biol. 2012, 91, 167–181. [Google Scholar] [CrossRef]
- Lakschevitz, F.S.; Visser, M.B.; Sun, C.; Glogauer, M. Neutrophil transcriptional profile changes during transit from bone marrow to sites of inflammation. Cell Mol. Immunol. 2015, 12, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.M.; Wei, S.; Smart, R.C. A multihit, multistage model of chemical carcinogenesis. Carcinogenesis 1999, 20, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Gastaldi, C.; Bertero, T.; Xu, N.; Bourget-Ponzio, I.; Lebrigand, K.; Fourre, S.; Popa, A.; Cardot-Leccia, N.; Meneguzzi, G.; Sonkoly, E.; et al. miR-193b/365a cluster controls progression of epidermal squamous cell carcinoma. Carcinogenesis 2014, 35, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.L.; Angel, J.M.; Kiguchi, K.; DiGiovanni, J. Multi-stage chemical carcinogenesis in mouse skin: Fundamentals and applications. Nat. Protoc. 2009, 4, 1350–1362. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Thomay, A.A.; Connolly, M.D.; Reichner, J.S.; Albina, J.E. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J. Leukoc. Biol. 2008, 83, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.P.; Rothenberg, M.E. Eosinophils and cancer. Cancer Immunol. Res. 2014, 2, 1–8. [Google Scholar] [CrossRef]
- Glodde, N.; Bald, T.; van den Boorn-Konijnenberg, D.; Nakamura, K.; O’Donnell, J.S.; Szczepanski, S.; Brandes, M.; Eickhoff, S.; Das, I.; Shridhar, N.; et al. Reactive Neutrophil Responses Dependent on the Receptor Tyrosine Kinase c-MET Limit Cancer Immunotherapy. Immunity 2017, 47, 789–802.e789. [Google Scholar] [CrossRef] [PubMed]
- De Kleijn, S.; Langereis, J.D.; Leentjens, J.; Kox, M.; Netea, M.G.; Koenderman, L.; Ferwerda, G.; Pickkers, P.; Hermans, P.W. IFN-gamma-stimulated neutrophils suppress lymphocyte proliferation through expression of PD-L1. PLos ONE 2013, 8, e72249. [Google Scholar] [CrossRef]
- Goncalves-Maia, M.; Gache, Y.; Basante, M.; Cosson, E.; Salavagione, E.; Muller, M.; Bernerd, F.; Avril, M.F.; Schaub, S.; Sarasin, A.; et al. NK Cell and Fibroblast-Mediated Regulation of Skin Squamous Cell Carcinoma Invasion by CLEC2A Is Compromised in Xeroderma Pigmentosum. J. Invest. Dermatol. 2020. [Google Scholar] [CrossRef]
- Moses, K.; Brandau, S. Human neutrophils: Their role in cancer and relation to myeloid-derived suppressor cells. Semin. Immunol. 2016, 28, 187–196. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Engblom, C.; Pfirschke, C.; Zilionis, R.; Da Silva Martins, J.; Bos, S.A.; Courties, G.; Rickelt, S.; Severe, N.; Baryawno, N.; Faget, J.; et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecF(high) neutrophils. Science 2017, 358. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Dominguez, G.A.; Youn, J.I.; Kossenkov, A.V.; Mony, S.; Alicea-Torres, K.; Tcyganov, E.; Hashimoto, A.; Nefedova, Y.; Lin, C.; et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 2016, 1. [Google Scholar] [CrossRef]
- He, G.; Zhang, H.; Zhou, J.; Wang, B.; Chen, Y.; Kong, Y.; Xie, X.; Wang, X.; Fei, R.; Wei, L.; et al. Peritumoural neutrophils negatively regulate adaptive immunity via the PD-L1/PD-1 signalling pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2015, 34, 141. [Google Scholar] [CrossRef]
- Mollaoglu, G.; Jones, A.; Wait, S.J.; Mukhopadhyay, A.; Jeong, S.; Arya, R.; Camolotto, S.A.; Mosbruger, T.L.; Stubben, C.J.; Conley, C.J.; et al. The Lineage-Defining Transcription Factors SOX2 and NKX2-1 Determine Lung Cancer Cell Fate and Shape the Tumor Immune Microenvironment. Immunity 2018, 49, 764–779. [Google Scholar] [CrossRef]
- Gregory, A.D.; Houghton, A.M. Tumor-associated neutrophils: New targets for cancer therapy. Cancer Res. 2011, 71, 2411–2416. [Google Scholar] [CrossRef]
- Raber, P.L.; Thevenot, P.; Sierra, R.; Wyczechowska, D.; Halle, D.; Ramirez, M.E.; Ochoa, A.C.; Fletcher, M.; Velasco, C.; Wilk, A.; et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int. J. Cancer 2014, 134, 2853–2864. [Google Scholar] [CrossRef]
- Negorev, D.; Beier, U.H.; Zhang, T.; Quatromoni, J.G.; Bhojnagarwala, P.; Albelda, S.M.; Singhal, S.; Eruslanov, E.; Lohoff, F.W.; Levine, M.H.; et al. Human neutrophils can mimic myeloid-derived suppressor cells (PMN-MDSC) and suppress microbead or lectin-induced T cell proliferation through artefactual mechanisms. Sci. Rep. 2018, 8, 3135. [Google Scholar] [CrossRef]
- Wang, T.T.; Zhao, Y.L.; Peng, L.S.; Chen, N.; Chen, W.; Lv, Y.P.; Mao, F.Y.; Zhang, J.Y.; Cheng, P.; Teng, Y.S.; et al. Tumour-activated neutrophils in gastric cancer foster immune suppression and disease progression through GM-CSF-PD-L1 pathway. Gut 2017, 66, 1900–1911. [Google Scholar] [CrossRef]
- Eruslanov, E.B.; Bhojnagarwala, P.S.; Quatromoni, J.G.; Stephen, T.L.; Ranganathan, A.; Deshpande, C.; Akimova, T.; Vachani, A.; Litzky, L.; Hancock, W.W.; et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J. Clin. Invest. 2014, 124, 5466–5480. [Google Scholar] [CrossRef]
- Kargl, J.; Busch, S.E.; Yang, G.H.; Kim, K.H.; Hanke, M.L.; Metz, H.E.; Hubbard, J.J.; Lee, S.M.; Madtes, D.K.; McIntosh, M.W.; et al. Neutrophils dominate the immune cell composition in non-small cell lung cancer. Nat. Commun. 2017, 8, 14381. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Elpek, K.G.; Cremasco, V.; Shen, H.; Harvey, C.J.; Wucherpfennig, K.W.; Goldstein, D.R.; Monach, P.A.; Turley, S.J. The tumor microenvironment shapes lineage, transcriptional, and functional diversity of infiltrating myeloid cells. Cancer Immunol. Res. 2014, 2, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Sagiv, J.Y.; Michaeli, J.; Assi, S.; Mishalian, I.; Kisos, H.; Levy, L.; Damti, P.; Lumbroso, D.; Polyansky, L.; Sionov, R.V.; et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015, 10, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Bhojnagarwala, P.S.; O’Brien, S.; Moon, E.K.; Garfall, A.L.; Rao, A.S.; Quatromoni, J.G.; Stephen, T.L.; Litzky, L.; Deshpande, C.; et al. Origin and Role of a Subset of Tumor-Associated Neutrophils with Antigen-Presenting Cell Features in Early-Stage Human Lung Cancer. Cancer Cell 2016, 30, 120–135. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Seignez, C.; Racoeur, C.; Isambert, N.; Mabrouk, N.; Scagliarini, A.; Reveneau, S.; Arnould, L.; Bettaieb, A.; Jeannin, J.F.; et al. Tumor-derived granzyme B-expressing neutrophils acquire antitumor potential after lipid A treatment. Oncotarget 2018, 9, 28364–28378. [Google Scholar] [CrossRef]
- Finisguerra, V.; Di Conza, G.; Di Matteo, M.; Serneels, J.; Costa, S.; Thompson, A.A.; Wauters, E.; Walmsley, S.; Prenen, H.; Granot, Z.; et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature 2015, 522, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef]
- Fortunati, E.; Kazemier, K.M.; Grutters, J.C.; Koenderman, L.; Van den Bosch v, J. Human neutrophils switch to an activated phenotype after homing to the lung irrespective of inflammatory disease. Clin. Exp. Immunol. 2009, 155, 559–566. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, J.; Cai, Y.; Fu, S.; Zhang, N.; Fu, X.; Li, L. IFN-gamma-mediated inhibition of lung cancer correlates with PD-L1 expression and is regulated by PI3K-AKT signaling. Int. J. Cancer 2018, 143, 931–943. [Google Scholar] [CrossRef]
- Minn, A.J.; Wherry, E.J. Combination Cancer Therapies with Immune Checkpoint Blockade: Convergence on Interferon Signaling. Cell 2016, 165, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [PubMed]
- Evrard, M.; Kwok, I.W.H.; Chong, S.Z.; Teng, K.W.W.; Becht, E.; Chen, J.; Sieow, J.L.; Penny, H.L.; Ching, G.C.; Devi, S.; et al. Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity 2018, 48, 364–379.e368. [Google Scholar] [CrossRef] [PubMed]
- Rice, C.M.; Davies, L.C.; Subleski, J.J.; Maio, N.; Gonzalez-Cotto, M.; Andrews, C.; Patel, N.L.; Palmieri, E.M.; Weiss, J.M.; Lee, J.M.; et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat. Commun. 2018, 9, 5099. [Google Scholar] [CrossRef] [PubMed]
- Kleinovink, J.W.; van Hall, T.; Ossendorp, F.; Fransen, M.F. PD-L1 immune suppression in cancer: Tumor cells or host cells? Oncoimmunology 2017, 6, e1325982. [Google Scholar] [CrossRef] [PubMed]
- Migden, M.R.; Khushalani, N.I.; Chang, A.L.S.; Lewis, K.D.; Schmults, C.D.; Hernandez-Aya, L.; Meier, F.; Schadendorf, D.; Guminski, A.; Hauschild, A.; et al. Cemiplimab in locally advanced cutaneous squamous cell carcinoma: Results from an open-label, phase 2, single-arm trial. Lancet Oncol. 2020, 21, 294–305. [Google Scholar] [CrossRef]
- Tachinami, H.; Nishii, N.; Xia, Y.; Kashima, Y.; Ohno, T.; Nagai, S.; Li, L.; Lau, W.; Tomihara, K.; Noguchi, M.; et al. Differences of tumor-recruiting myeloid cells in murine squamous cell carcinoma influence the efficacy of immunotherapy combined with a TLR7 agonist and PD-L1 blockade. Oral Oncol. 2019, 91, 21–28. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khou, S.; Popa, A.; Luci, C.; Bihl, F.; Meghraoui-Kheddar, A.; Bourdely, P.; Salavagione, E.; Cosson, E.; Rubod, A.; Cazareth, J.; et al. Tumor-Associated Neutrophils Dampen Adaptive Immunity and Promote Cutaneous Squamous Cell Carcinoma Development. Cancers 2020, 12, 1860. https://doi.org/10.3390/cancers12071860
Khou S, Popa A, Luci C, Bihl F, Meghraoui-Kheddar A, Bourdely P, Salavagione E, Cosson E, Rubod A, Cazareth J, et al. Tumor-Associated Neutrophils Dampen Adaptive Immunity and Promote Cutaneous Squamous Cell Carcinoma Development. Cancers. 2020; 12(7):1860. https://doi.org/10.3390/cancers12071860
Chicago/Turabian StyleKhou, Sokchea, Alexandra Popa, Carmelo Luci, Franck Bihl, Aida Meghraoui-Kheddar, Pierre Bourdely, Emie Salavagione, Estelle Cosson, Alain Rubod, Julie Cazareth, and et al. 2020. "Tumor-Associated Neutrophils Dampen Adaptive Immunity and Promote Cutaneous Squamous Cell Carcinoma Development" Cancers 12, no. 7: 1860. https://doi.org/10.3390/cancers12071860
APA StyleKhou, S., Popa, A., Luci, C., Bihl, F., Meghraoui-Kheddar, A., Bourdely, P., Salavagione, E., Cosson, E., Rubod, A., Cazareth, J., Barbry, P., Mari, B., Rezzonico, R., Anjuère, F., & Braud, V. M. (2020). Tumor-Associated Neutrophils Dampen Adaptive Immunity and Promote Cutaneous Squamous Cell Carcinoma Development. Cancers, 12(7), 1860. https://doi.org/10.3390/cancers12071860