Glutamic-Pyruvic Transaminase 1 Facilitates Alternative Fuels for Hepatocellular Carcinoma Growth—A Small Molecule Inhibitor, Berberine



Abstract
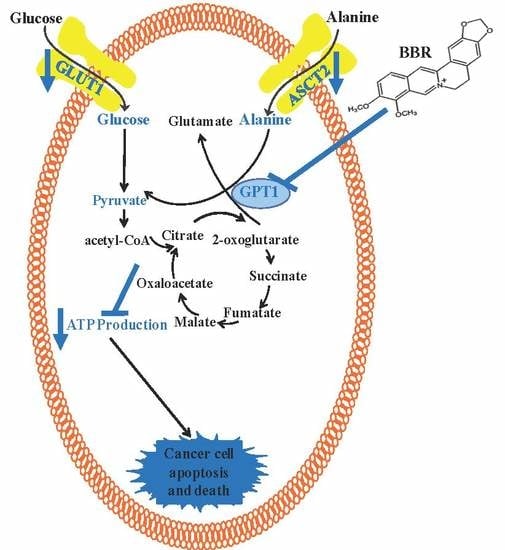
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Alanine as an Alternative Energy Source Supports HCC Growth in a Nutrient-Poor Environment
2.2. Overexpression of GPT1 in the Activated Glucose–Alanine Cycle Promotes HCC Growth
2.3. Inhibition of GPT1 Expression Reverses Alanine-Mediated HCC Growth
2.4. Targeting GPT1 by a Small Molecule Berberine Suppresses HCC Growth
2.5. Berberine Mediates Metabolic Fluctuations Primarily by Regulating the Glucose–Alanine Cycle in HCC
2.6. Berberine-Suppressed GPT1 Reverses the Dysregulated Energy Homeostasis in HCC
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Line and Cell Culture
4.3. Plasmid and Transfection
4.4. Cell Viability Assay
4.5. BrdU Incorporation Assay
4.6. Annexin V/7-AAD Staining and Flow Cytometry
4.7. ATP Content Assay
4.8. Orthotopic HCC Implantation Murine Model
4.9. Histology Examination
4.10. GC/MS-Based Metabolomics Analysis
4.11. Determination of Enzymatic Activity Inhibition
4.12. Intracellular ALT Activity Assay
4.13. Real-Time PCR
4.14. Immunoblotting
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schafer, D.F.; Sorrell, M.F. Hepatocellular carcinoma. Lancet 1999, 354, 1253–1257. [Google Scholar] [CrossRef]
- Best, J.; Schotten, C.; Theysohn, J.M.; Wetter, A.; Müller, S.; Radünz, S.; Schulze, M.; Canbay, A.; Dechêne, A.; Gerken, G. Novel implications in the treatment of hepatocellular carcinoma. Ann. Gastroenterol. 2016, 30, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Qi, F.; Cui, Y.; Zhao, L.; Sun, X.; Tang, W.; Cai, P. An update on Chinese herbal medicines as adjuvant treatment of anticancer therapeutics. Biosci. Trends 2018, 12, 220–239. [Google Scholar] [CrossRef]
- Lou, J.; Yao, P.; Tsim, K.W.; Tism, K.W.K. Cancer Treatment by Using Traditional Chinese Medicine: Probing Active Compounds in Anti-multidrug Resistance during Drug Therapy. Curr. Med. Chem. 2019, 25, 5128–5141. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Pavlova, N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Liberti, M.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2015, 35, 3619–3625. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Heiden, M.G.V.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef]
- Qian, K.; Zhong, S.; Xie, K.; Yu, D.; Yang, R.; Gong, D.-W. Hepatic ALT isoenzymes are elevated in gluconeogenic conditions including diabetes and suppressed by insulin at the protein level. Diabetes/Metab. Res. Rev. 2015, 31, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E. Control of glycaemia. Baillieres Clin. Endocrinol. Metab. 1993, 7, 551–586. [Google Scholar] [CrossRef]
- Schindhelm, R.; Diamant, M.; Dekker, J.M.; Tushuizen, M.E.; Teerlink, T.; Heine, R.J. Alanine aminotransferase as a marker of non-alcoholic fatty liver disease in relation to type 2 diabetes mellitus and cardiovascular disease. Diabetes/Metab. Res. Rev. 2006, 22, 437–443. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Lin, S.-H.; Wang, Y.; Chin, Y.E.; Kang, L.; Mi, J. Glutamic Pyruvate Transaminase GPT2 Promotes Tumorigenesis of Breast Cancer Cells by Activating Sonic Hedgehog Signaling. Theranostics 2017, 7, 3021–3033. [Google Scholar] [CrossRef]
- Smith, B.; Schafer, X.L.; Ambeskovic, A.; Spencer, C.M.; Land, H.; Munger, J. Addiction to Coupling of the Warburg Effect with Glutamine Catabolism in Cancer Cells. Cell Rep. 2016, 17, 821–836. [Google Scholar] [CrossRef]
- Hao, Y.; Samuels, Y.; Li, Q.; Krokowski, D.; Guan, B.-J.; Wang, C.; Jin, Z.; Dong, B.; Cao, B.; Feng, X.; et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat. Commun. 2016, 7, 11971. [Google Scholar] [CrossRef]
- Coloff, J.L.; Murphy, J.P.; Braun, C.R.; Harris, I.; Shelton, L.M.; Kami, K.; Gygi, S.P.; Selfors, L.M.; Brugge, J.S. Differential Glutamate Metabolism in Proliferating and Quiescent Mammary Epithelial Cells. Cell Metab. 2016, 23, 867–880. [Google Scholar] [CrossRef]
- Korangath, P.; Teo, W.W.; Sadik, H.; Han, L.; Mori, N.; Huijts, C.M.; Wildes, F.; Bharti, S.; Zhang, Z.; Santa-Maria, C.A.; et al. Targeting Glutamine Metabolism in Breast Cancer with Aminooxyacetate. Clin. Cancer Res. 2015, 21, 3263–3273. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.E.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Glinghammar, B.; Rafter, I.; Lindström, A.-K.; Hedberg, J.J.; Andersson, H.B.; Lindblom, P.; Berg, A.-L.; Cotgreave, I. Detection of the mitochondrial and catalytically active alanine aminotransferase in human tissues and plasma. Int. J. Mol. Med. 2009, 23, 621–631. [Google Scholar] [CrossRef]
- Yang, R.-Z.; Park, S.; Reagan, W.J.; Goldstein, R.; Zhong, S.; Lawton, M.; Rajamohan, F.; Qian, K.; Liu, L.; Gong, D.-W. Alanine aminotransferase isoenzymes: Molecular cloning and quantitative analysis of tissue expression in rats and serum elevation in liver toxicity. Hepatology 2008, 49, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Felig, P. The glucose-alanine cycle. Metabolism 1973, 22, 179–207. [Google Scholar] [CrossRef]
- Ye, J.; Gu, Y.; Zhang, F.; Zhao, Y.; Yuan, Y.; Hao, Z.; Sheng, Y.; Li, W.Y.; Wakeham, A.; Cairns, R.A.; et al. IDH1 deficiency attenuates gluconeogenesis in mouse liver by impairing amino acid utilization. Proc. Natl. Acad. Sci USA 2016, 114, 292–297. [Google Scholar] [CrossRef]
- Muhammad, S.A.; Fatima, N. In silico analysis and molecular docking studies of potential angiotensin-converting enzyme inhibitor using quercetin glycosides. Pharmacogn. Mag. 2015, 11, S123–S126. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.Y.; Wang, N.; Tsao, S.-W.; Zhang, Z.; Feng, Y. Suppression of Vascular Endothelial Growth Factor via Inactivation of Eukaryotic Elongation Factor 2 by Alkaloids in Coptidis rhizome in Hepatocellular Carcinoma. Integr. Cancer Ther. 2013, 13, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Tsang, C.M.; Cheung, K.C.; Cheung, Y.C.; Man, K.; Lui, V.W.Y.; Tsao, S.W.; Feng, Y. Berberine suppresses Id-1 expression and inhibits the growth and development of lung metastases in hepatocellular carcinoma. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2015, 1852, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Catapano, A.L. Berberine, a plant alkaloid with lipid- and glucose-lowering properties: From in vitro evidence to clinical studies. Atherosclerosis 2015, 243, 449–461. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouysségur, J. Tumor Cell Metabolism: Cancer’s Achilles’ Heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Sheen, J.-H.; Zoncu, R.; Kim, D.; Sabatini, D.M. Defective Regulation of Autophagy upon Leucine Deprivation Reveals a Targetable Liability of Human Melanoma Cells In Vitro and In Vivo. Cancer Cell 2011, 19, 613–628. [Google Scholar] [CrossRef]
- Kamphorst, J.J.; Gottlieb, E. Cancer metabolism: Friendly neighbours feed tumour cells. Nature 2016, 536, 401–402. [Google Scholar] [CrossRef][Green Version]
- Dickson, I. Pancreatic cancer: Stromal-cancer cell crosstalk supports tumour metabolism. Nat. Rev. Gastroentero. Hepato. 2016, 13, 558–559. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Q.; Nakayama, T.; Baytaş, O.; Davidson, S.M.; Yang, C.; Schmidt, M.; Lizarraga, S.B.; Mishra, S.; Ei-Quessny, M.; Niaz, S.; et al. Mutations in mitochondrial enzyme GPT2 cause metabolic dysfunction and neurological disease with developmental and progressive features. Proc. Natl. Acad. Sci. USA 2016, 113, E5598–E5607. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-D.; Wu, H.; Fu, G.-B.; Zhang, H.-L.; Zhou, X.; Tang, L.; Dong, L.-W.; Qin, C.-J.; Huang, S.; Zhao, L.-H.; et al. Acetyl-coenzyme A carboxylase alpha promotion of glucose-mediated fatty acid synthesis enhances survival of hepatocellular carcinoma in mice and patients. Hepatology 2016, 63, 1272–1286. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wang, X.; Tan, H.-Y.; Li, S.; Tsang, C.M.; Tsao, S.-W.; Feng, Y. Berberine Suppresses Cyclin D1 Expression through Proteasomal Degradation in Human Hepatoma Cells. Int. J. Mol. Sci. 2016, 17, 1899. [Google Scholar] [CrossRef]
- Wang, N.; Feng, Y.; Zhu, M.; Tsang, C.-M.; Man, K.; Tong, Y.; Tsao, S.-W. Berberine induces autophagic cell death and mitochondrial apoptosis in liver cancer cells: The cellular mechanism. J. Cell. Biochem. 2010, 111, 1426–1436. [Google Scholar] [CrossRef]
- Wang, N.; Feng, Y.; Lau, E.P.W.; Tsang, C.; Ching, Y.P.; Man, K.; Tong, Y.; Nagamatsu, T.; Su, W.; Tsao, S. F-Actin Reorganization and Inactivation of Rho Signaling Pathway Involved in the Inhibitory Effect of Coptidis Rhizoma on Hepatoma Cell Migration. Integr. Cancer Ther. 2010, 9, 354–364. [Google Scholar] [CrossRef]
- Wang, N.; Zhu, M.; Wang, X.; Tan, H.-Y.; Tsao, S.-W.; Feng, Y. Berberine-induced tumor suppressor p53 up-regulation gets involved in the regulatory network of MIR-23a in hepatocellular carcinoma. Biochim. Biophys. Acta (BBA)—Bioenerg. 2014, 1839, 849–857. [Google Scholar] [CrossRef]
- Mao, L.; Chen, Q.; Gong, K.; Xu, X.; Xie, Y.; Zhang, W.; Cao, H.; Hu, T.; Hong, X.; Zhan, Y.-Y. Berberine decelerates glucose metabolism via suppression of mTOR-dependent HIF-1α protein synthesis in colon cancer cells. Oncol. Rep. 2018, 39, 2436–2442. [Google Scholar] [CrossRef]
- Van Geldermalsen, M. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, W.; Tan, H.-Y.; Li, S.; Wang, N.; Feng, Y. Glutamic-Pyruvic Transaminase 1 Facilitates Alternative Fuels for Hepatocellular Carcinoma Growth—A Small Molecule Inhibitor, Berberine. Cancers 2020, 12, 1854. https://doi.org/10.3390/cancers12071854
Guo W, Tan H-Y, Li S, Wang N, Feng Y. Glutamic-Pyruvic Transaminase 1 Facilitates Alternative Fuels for Hepatocellular Carcinoma Growth—A Small Molecule Inhibitor, Berberine. Cancers. 2020; 12(7):1854. https://doi.org/10.3390/cancers12071854
Chicago/Turabian StyleGuo, Wei, Hor-Yue Tan, Sha Li, Ning Wang, and Yibin Feng. 2020. "Glutamic-Pyruvic Transaminase 1 Facilitates Alternative Fuels for Hepatocellular Carcinoma Growth—A Small Molecule Inhibitor, Berberine" Cancers 12, no. 7: 1854. https://doi.org/10.3390/cancers12071854
APA StyleGuo, W., Tan, H.-Y., Li, S., Wang, N., & Feng, Y. (2020). Glutamic-Pyruvic Transaminase 1 Facilitates Alternative Fuels for Hepatocellular Carcinoma Growth—A Small Molecule Inhibitor, Berberine. Cancers, 12(7), 1854. https://doi.org/10.3390/cancers12071854