Mambalgin-2 Induces Cell Cycle Arrest and Apoptosis in Glioma Cells via Interaction with ASIC1a

 , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Results
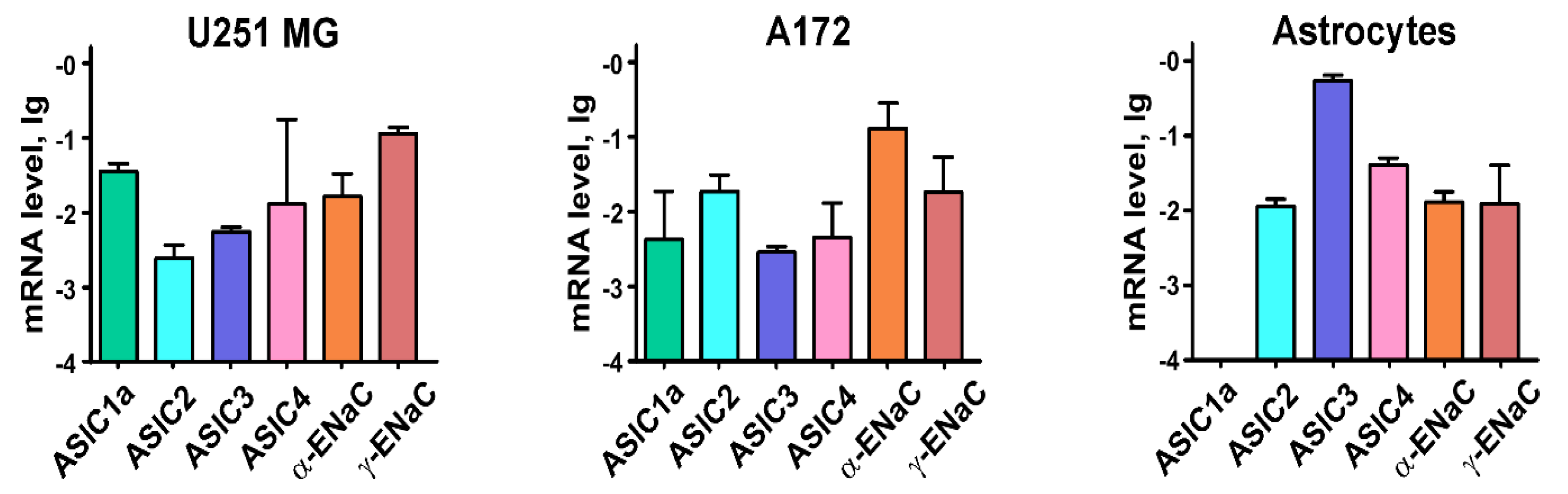
2.1. Astrocytes and Glioma Cells Express Different ASICs and ENaCs
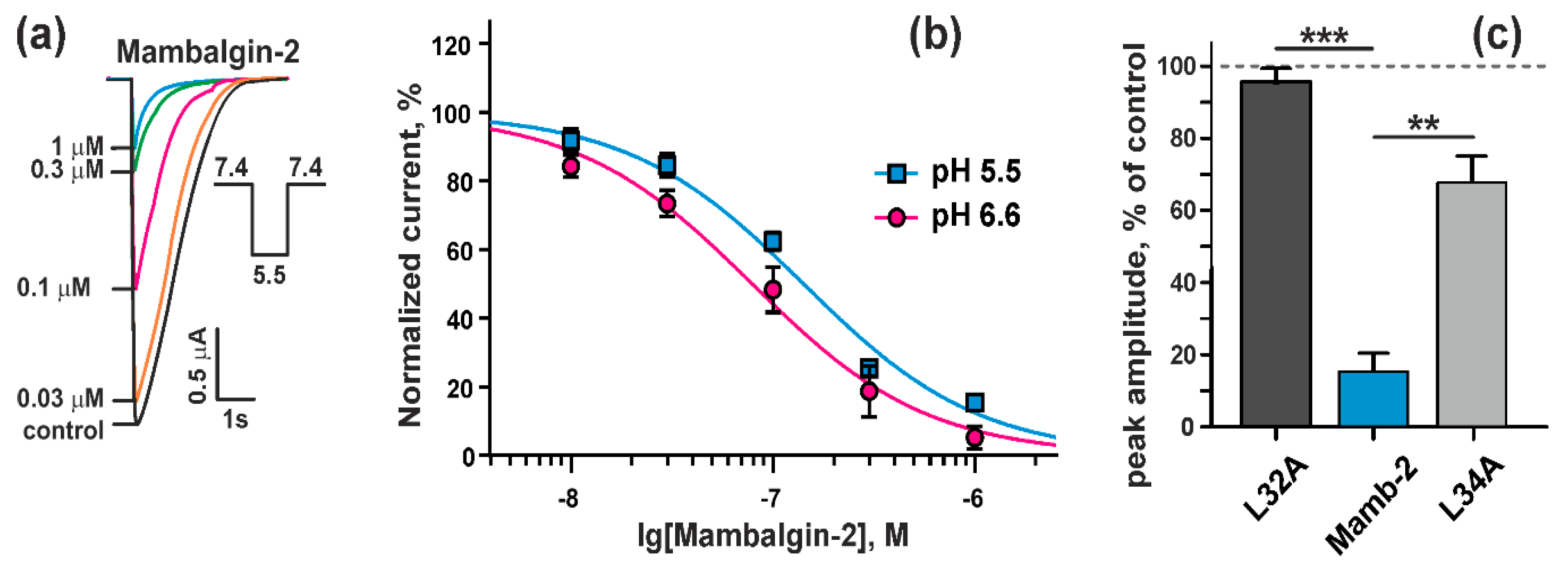
2.2. Mambalgin-2 Inhibits ASIC1a in Xenopus laevis oocytes
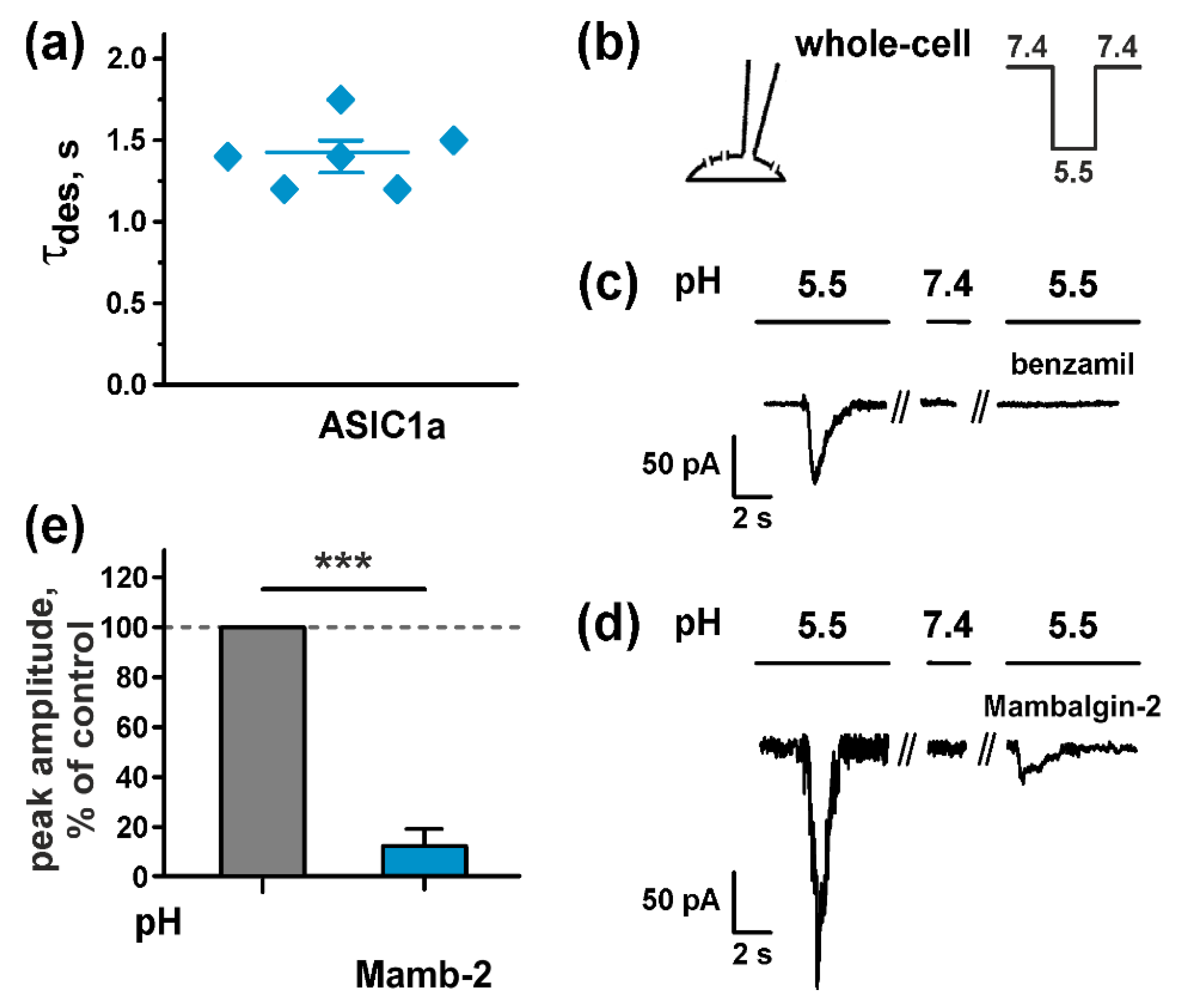
2.3. Mambalgin-2 Inhibits ASICs Activity in U251 MG Glioma Cells
2.4. Mambalgin-2 Inhibits Growth of Glioma Cells via Interaction with ASIC1a
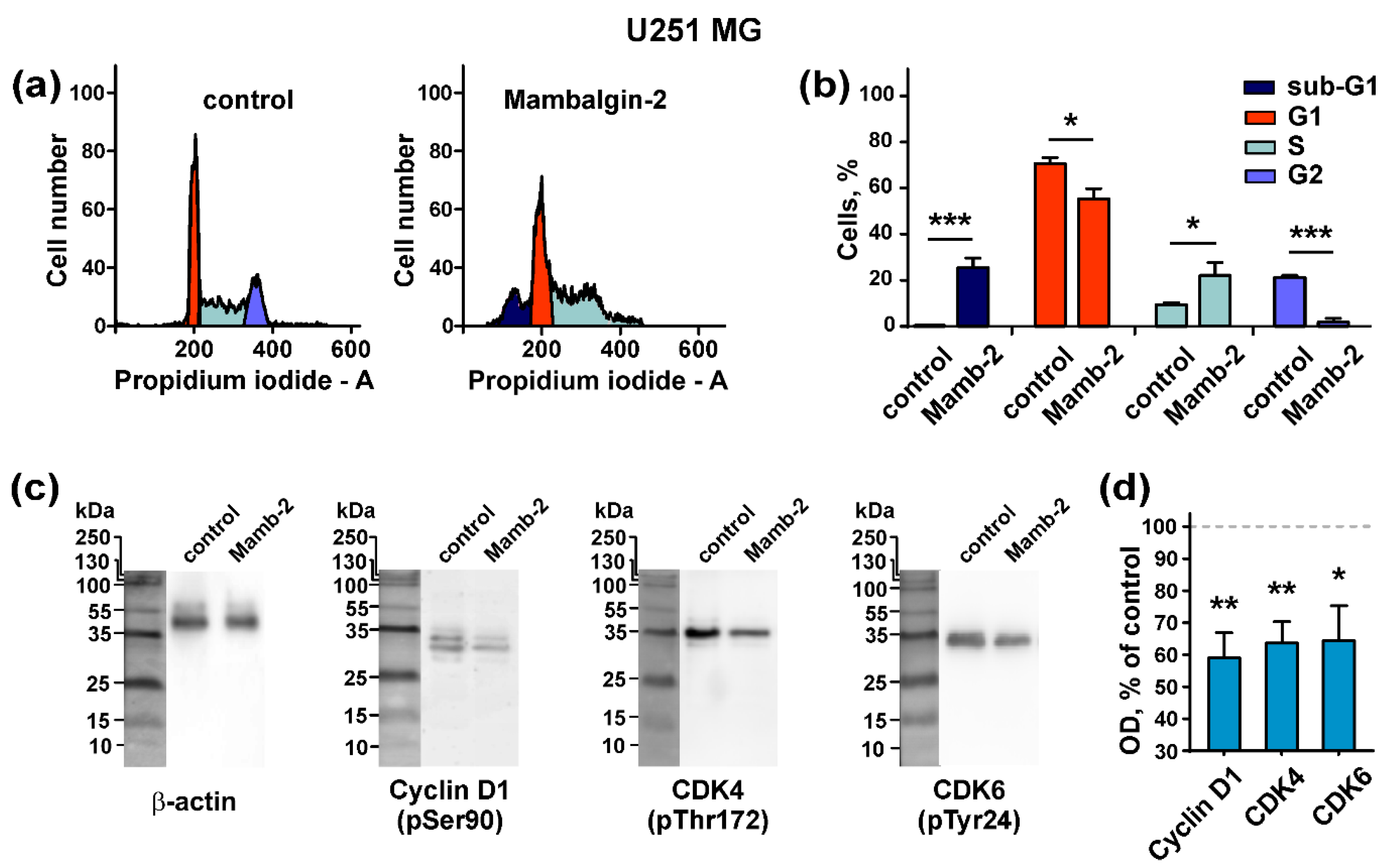
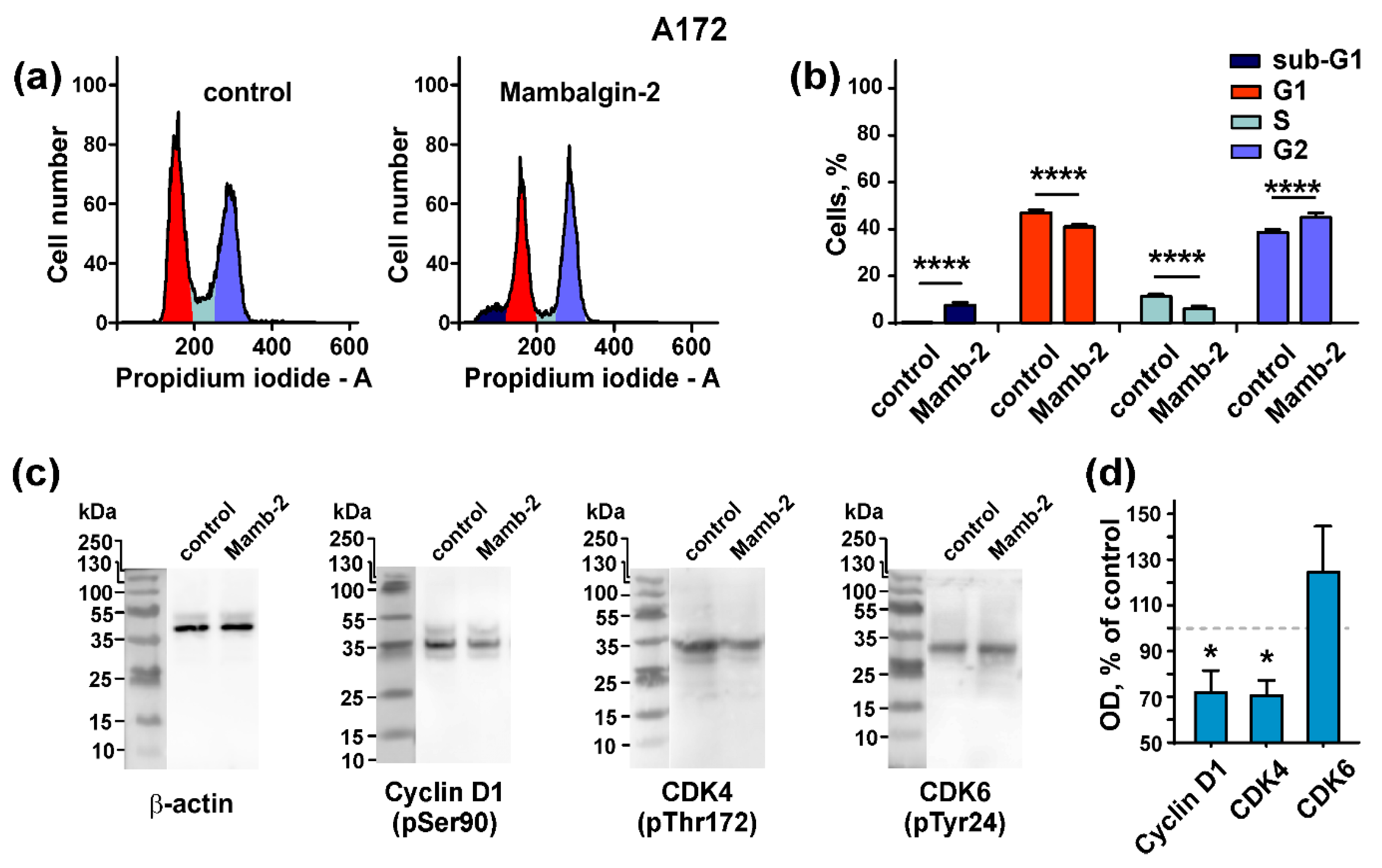
2.5. Mambalgin-2 Induces Cell Cycle Arrest in U251 MG and A172 Cells
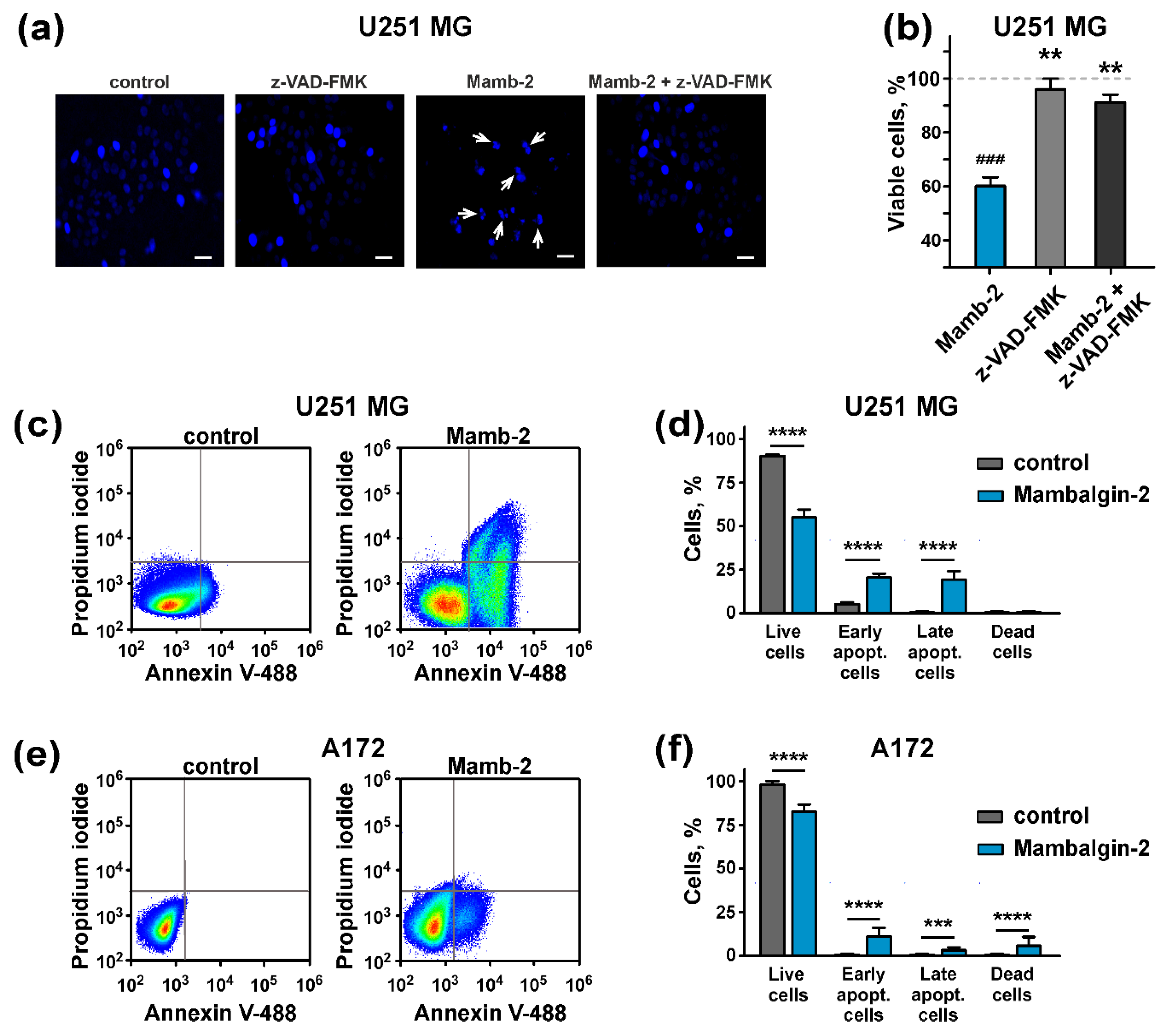
2.6. Mambalgin-2 Induces Apoptosis in U251 MG and A172 Cells
2.7. Mambalgin-2 Inhibits Growth of Primary Cells Obtained from GBM Patient
3. Discussion
4. Materials and Methods
4.1. Recombinant Mambalgin-2 Production
4.2. Real-Time PCR
4.3. Electrophysiological Recordings in Xenopus oocytes
4.4. Patch Clamp on U251 MG Cells
4.5. Cell Cultivation and Proliferation Assay
4.6. Primary Cell Culture from a GBM Patient
4.7. Cell Cycle Analysis
4.8. Western Blotting
4.9. Fluorescent Microscopy
4.10. Analysis of Phosphatidylserine Externalization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Honasoge, A.; Sontheimer, H. Involvement of tumor acidification in brain cancer pathophysiology. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar] [PubMed]
- Hjelmeland, A.B.; Wu, Q.; Heddleston, J.M.; Choudhary, G.S.; MacSwords, J.; Lathia, J.D.; McLendon, R.; Lindner, D.; Sloan, A.; Rich, J.N. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011, 18, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Harguindey, S.; Stanciu, D.; Devesa, J.; Alfarouk, K.; Cardone, R.A.; Polo Orozco, J.D.; Devesa, P.; Rauch, C.; Orive, G.; Anitua, E.; et al. Cellular acidification as a new approach to cancer treatment and to the understanding and therapeutics of neurodegenerative diseases. Semin. Cancer Biol. 2017, 43, 157–179. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, S.; Schild, L. International Union of Basic and Clinical Pharmacology. XCI. structure, function, and pharmacology of acid-sensing ion channels and the epithelial Na+ channel. Pharmacol. Rev. 2015, 67, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Qadri, Y.J.; Rooj, A.K.; Fuller, C.M. ENaCs and ASICs as therapeutic targets. Am. J. Physiol. Cell Physiol. 2012, 302, C943–C965. [Google Scholar] [CrossRef] [PubMed]
- Wemmie, J.A.; Taugher, R.J.; Kreple, C.J. Acid-sensing ion channels in pain and disease. Nat. Rev. Neurosci. 2013, 14, 461–471. [Google Scholar] [CrossRef]
- Berdiev, B.K.; Xia, J.; McLean, L.A.; Markert, J.M.; Gillespie, G.Y.; Mapstone, T.B.; Naren, A.P.; Jovov, B.; Bubien, J.K.; Ji, H.-L.; et al. Acid-sensing ion channels in malignant gliomas. J. Biol. Chem. 2003, 278, 15023–15034. [Google Scholar] [CrossRef]
- Kapoor, N.; Bartoszewski, R.; Qadri, Y.J.; Bebok, Z.; Bubien, J.K.; Fuller, C.M.; Benos, D.J. Knockdown of ASIC1 and epithelial sodium channel subunits inhibits glioblastoma whole cell current and cell migration. J. Biol. Chem. 2009, 284, 24526–24541. [Google Scholar] [CrossRef]
- Kapoor, N.; Lee, W.; Clark, E.; Bartoszewski, R.; McNicholas, C.M.; Latham, C.B.; Bebok, Z.; Parpura, V.; Fuller, C.M.; Palmer, C.A.; et al. Interaction of ASIC1 and ENaC subunits in human glioma cells and rat astrocytes. Am. J. Physiol. Cell Physiol. 2011, 300, C1246–C1259. [Google Scholar] [CrossRef][Green Version]
- Rooj, A.K.; McNicholas, C.M.; Bartoszewski, R.; Bebok, Z.; Benos, D.J.; Fuller, C.M. Glioma-specific cation conductance regulates migration and cell cycle progression. J. Biol. Chem. 2012, 287, 4053–4065. [Google Scholar] [CrossRef] [PubMed]
- Rooj, A.K.; Liu, Z.; McNicholas, C.M.; Fuller, C.M. Physical and functional interactions between a glioma cation channel and integrin-β1 require α-actinin. Am. J. Physiol. Cell Physiol. 2015, 309, C308–C319. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. bioRxiv 2019, 326470. [Google Scholar] [CrossRef]
- Wu, Y.; Gao, B.; Xiong, Q.-J.; Wang, Y.-C.; Huang, D.-K.; Wu, W.-N. Acid-sensing ion channels contribute to the effect of extracellular acidosis on proliferation and migration of A549 cells. Tumour Biol. 2017, 39, 1010428317705750. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, T.; Wu, C.; Xia, Q.; Xu, D. ASIC1a mediates the drug resistance of human hepatocellular carcinoma via the Ca2+/PI3-kinase/AKT signaling pathway. Lab. Investig. 2017, 97, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Singh, R.; Asters, M.; Liu, J.; Zhang, X.; Pabbidi, M.R.; Watabe, K.; Mo, Y.-Y. Regulation of breast tumorigenesis through acid sensors. Oncogene 2016, 35, 4102–4111. [Google Scholar] [CrossRef]
- Jin, C.; Ye, Q.-H.; Yuan, F.-L.; Gu, Y.-L.; Li, J.-P.; Shi, Y.-H.; Shen, X.-M.; Liu, B.; Lin, Z.-H. Involvement of acid-sensing ion channel 1α in hepatic carcinoma cell migration and invasion. Tumour. Biol. 2015, 36, 4309–4317. [Google Scholar] [CrossRef]
- Zhu, S.; Zhou, H.-Y.; Deng, S.-C.; Deng, S.-J.; He, C.; Li, X.; Chen, J.-Y.; Jin, Y.; Hu, Z.-L.; Wang, F.; et al. ASIC1 and ASIC3 contribute to acidity-induced EMT of pancreatic cancer through activating Ca 2+ /RhoA pathway. Cell Death Dis. 2017, 8, e2806. [Google Scholar] [CrossRef]
- Xu, S.; Liu, C.; Ma, Y.; Ji, H.-L.; Li, X. Potential Roles of Amiloride-Sensitive Sodium Channels in Cancer Development. Biomed Res. Int. 2016, 2016, 2190216. [Google Scholar] [CrossRef]
- Liu, C.; Zhu, L.-L.; Xu, S.-G.; Ji, H.-L.; Li, X.-M. ENaC/DEG in Tumor Development and Progression. J. Cancer 2016, 7, 1888–1891. [Google Scholar] [CrossRef]
- Hegde, M.; Roscoe, J.; Cala, P.; Gorin, F. Amiloride kills malignant glioma cells independent of its inhibition of the sodium-hydrogen exchanger. J. Pharmacol. Exp. Ther. 2004, 310, 67–74. [Google Scholar] [CrossRef]
- Matthews, H.; Ranson, M.; Kelso, M.J. Anti-tumour/metastasis effects of the potassium-sparing diuretic amiloride: An orally active anti-cancer drug waiting for its call-of-duty? Int. J. Cancer 2011, 129, 2051–2061. [Google Scholar] [CrossRef]
- Escoubas, P.; De Weille, J.R.; Lecoq, A.; Diochot, S.; Waldmann, R.; Champigny, G.; Moinier, D.; Ménez, A.; Lazdunski, M. Isolation of a tarantula toxin specific for a class of proton-gated Na+ channels. J. Biol. Chem. 2000, 275, 25116–25121. [Google Scholar] [CrossRef] [PubMed]
- Bubien, J.K.; Ji, H.-L.; Gillespie, G.Y.; Fuller, C.M.; Markert, J.M.; Mapstone, T.B.; Benos, D.J. Cation selectivity and inhibition of malignant glioma Na+ channels by Psalmotoxin 1. Am. J. Physiol. Cell Physiol. 2004, 287, C1282–C1291. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hoagland, E.N.; Sherwood, T.W.; Lee, K.G.; Walker, C.J.; Askwith, C.C. Identification of a Calcium Permeable Human Acid-sensing Ion Channel 1 Transcript Variant. J. Biol. Chem. 2010, 285, 41852–41862. [Google Scholar] [CrossRef] [PubMed]
- Er, S.Y.; Cristofori-Armstrong, B.; Escoubas, P.; Rash, L.D. Discovery and molecular interaction studies of a highly stable, tarantula peptide modulator of acid-sensing ion channel 1. Neuropharmacology 2017, 127, 185–195. [Google Scholar] [CrossRef]
- Diochot, S.; Baron, A.; Salinas, M.; Douguet, D.; Scarzello, S.; Dabert-Gay, A.-S.; Debayle, D.; Friend, V.; Alloui, A.; Lazdunski, M.; et al. Black mamba venom peptides target acid-sensing ion channels to abolish pain. Nature 2012, 490, 552–555. [Google Scholar] [CrossRef]
- Baron, A.; Diochot, S.; Salinas, M.; Deval, E.; Noël, J.; Lingueglia, E. Venom toxins in the exploration of molecular, physiological and pathophysiological functions of acid-sensing ion channels. Toxicon 2013, 75, 187–204. [Google Scholar] [CrossRef]
- Shulepko, M.A.; Lyukmanova, E.N.; Shenkarev, Z.O.; Dubovskii, P.V.; Astapova, M.V.; Feofanov, A.V.; Arseniev, A.S.; Utkin, Y.N.; Kirpichnikov, M.P.; Dolgikh, D.A. Towards universal approach for bacterial production of three-finger Ly6/uPAR proteins: Case study of cytotoxin I from cobra N. oxiana. Protein Expr. Purif. 2017, 130, 13–20. [Google Scholar] [CrossRef]
- Osmakov, D.I.; Koshelev, S.G.; Lyukmanova, E.N.; Shulepko, M.A.; Andreev, Y.A.; Illes, P.; Kozlov, S.A. Multiple Modulation of Acid-Sensing Ion Channel 1a by the Alkaloid Daurisoline. Biomolecules 2019, 9, 336. [Google Scholar] [CrossRef]
- Mourier, G.; Salinas, M.; Kessler, P.; Stura, E.A.; Leblanc, M.; Tepshi, L.; Besson, T.; Diochot, S.; Baron, A.; Douguet, D.; et al. Mambalgin-1 Pain-relieving Peptide, Stepwise Solid-phase Synthesis, Crystal Structure, and Functional Domain for Acid-sensing Ion Channel 1a Inhibition. J. Biol. Chem. 2016, 291, 2616–2629. [Google Scholar] [CrossRef]
- Leng, T.-D.; Si, H.-F.; Li, J.; Yang, T.; Zhu, M.; Wang, B.; Simon, R.P.; Xiong, Z.-G. Amiloride Analogs as ASIC1a Inhibitors. CNS Neurosci. Ther. 2016, 22, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Kajstura, M.; Halicka, H.D.; Pryjma, J.; Darzynkiewicz, Z. Discontinuous fragmentation of nuclear DNA during apoptosis revealed by discrete “sub-G1” peaks on DNA content histograms. Cytom. A 2007, 71, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Pavlyukov, M.S.; Yu, H.; Bastola, S.; Minata, M.; Shender, V.O.; Lee, Y.; Zhang, S.; Wang, J.; Komarova, S.; Wang, J.; et al. Apoptotic cell-derived extracellular vesicles promote malignancy of glioblastoma via intercellular transfer of splicing factors. Cancer Cell 2018, 34, 119–135.e10. [Google Scholar] [CrossRef] [PubMed]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef] [PubMed]
- Shulepko, M.A.; Bychkov, M.L.; Lyukmanova, E.N.; Kirpichnikov, M.P. Recombinant analogue of the human protein SLURP-1 inhibits the growth of U251 MG and A172 glioma cells. Dokl. Biochem. Biophys. 2020, 489, 392–395. [Google Scholar] [CrossRef]
- Bychkov, M.; Shenkarev, Z.; Shulepko, M.; Shlepova, O.; Kirpichnikov, M.; Lyukmanova, E. Water-soluble variant of human Lynx1 induces cell cycle arrest and apoptosis in lung cancer cells via modulation of α7 nicotinic acetylcholine receptors. PLoS ONE 2019, 14, e0217339. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Hu, Z.; Wu, W.-N.; Yu, D.-F.; Xiong, Q.-J.; Song, J.-R.; Shu, Q.; Fu, H.; Wang, F.; Chen, J.-G. Existence and distinction of acid-evoked currents in rat astrocytes. Glia 2010, 58, 1415–1424. [Google Scholar] [CrossRef]
- Yang, F.; Sun, X.; Ding, Y.; Ma, H.; Yang, T.O.; Ma, Y.; Wei, D.; Li, W.; Xu, T.; Jiang, W. Astrocytic Acid-Sensing Ion Channel 1a Contributes to the Development of Chronic Epileptogenesis. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Ugawa, S.; Ueda, T.; Ishida, Y.; Nishigaki, M.; Shibata, Y.; Shimada, S. Amiloride-blockable acid-sensing ion channels are leading acid sensors expressed in human nociceptors. J. Clin. Investig. 2002, 110, 1185–1190. [Google Scholar] [CrossRef]
- Tikhonov, D.B.; Magazanik, L.G.; Nagaeva, E.I. Ligands of Acid-Sensing Ion Channel 1a: Mechanisms of Action and Binding Sites. Acta Nat. 2019, 11, 4–13. [Google Scholar] [CrossRef]
- Cristofori-Armstrong, B.; Saez, N.J.; Chassagnon, I.R.; King, G.F.; Rash, L.D. The modulation of acid-sensing ion channel 1 by PcTx1 is pH-, subtype- and species-dependent: Importance of interactions at the channel subunit interface and potential for engineering selective analogues. Biochem. Pharmacol. 2019, 163, 381–390. [Google Scholar] [CrossRef]
- Sherwood, T.W.; Lee, K.G.; Gormley, M.G.; Askwith, C.C. Heteromeric acid-sensing ion channels (ASICs) composed of ASIC2b and ASIC1a display novel channel properties and contribute to acidosis-induced neuronal death. J. Neurosci. 2011, 31, 9723–9734. [Google Scholar] [CrossRef]
- Joeres, N.; Augustinowski, K.; Neuhof, A.; Assmann, M.; Gründer, S. Functional and pharmacological characterization of two different ASIC1a/2a heteromers reveals their sensitivity to the spider toxin PcTx1. Sci. Rep. 2016, 6, 27647. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kalbacher, H.; Gründer, S. Interaction of Acid-sensing Ion Channel (ASIC) 1 with the Tarantula Toxin Psalmotoxin 1 is State Dependent. J. Gen. Physiol. 2006, 127, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hagan, R.; Schoellerman, J. Dual actions of Psalmotoxin at ASIC1a and ASIC2a heteromeric channels (ASIC1a/2a). Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Bresenitz, P.; Reska, A.; El Moussaoui, L.; Beier, C.P.; Gründer, S. Glioblastoma cancer stem cell lines express functional acid sensing ion channels ASIC1a and ASIC3. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.U.; Coman, D.; Walsh, J.J.; Ali, M.M.; Huang, Y.; Hyder, F. Temozolomide arrests glioma growth and normalizes intratumoral extracellular pH. Sci. Rep. 2017, 7, 7865. [Google Scholar] [CrossRef]
- Zhang, P.; Canessa, C.M. Single channel properties of rat acid-sensitive ion channel-1alpha, -2a, and -3 expressed in Xenopus oocytes. J. Gen. Physiol. 2002, 120, 553–566. [Google Scholar] [CrossRef]
- Khleif, S.N.; DeGregori, J.; Yee, C.L.; Otterson, G.A.; Kaye, F.J.; Nevins, J.R.; Howley, P.M. Inhibition of cyclin D-CDK4/CDK6 activity is associated with an E2F-mediated induction of cyclin kinase inhibitor activity. Proc. Natl. Acad. Sci. USA 1996, 93, 4350–4354. [Google Scholar] [CrossRef]
- Gladden, A.B.; Diehl, J.A. Location, location, location: The role of cyclin D1 nuclear localization in cancer. J. Cell. Biochem. 2005, 96, 906–913. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wang, L.; Chen, L.; Zhang, Y.; Shi, P. Induction of cell cycle arrest via the p21, p27-cyclin E,A/Cdk2 pathway in SMMC-7721 hepatoma cells by clioquinol. Acta Pharm. 2015, 65, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Wolter, F.; Akoglu, B.; Clausnitzer, A.; Stein, J. Downregulation of the Cyclin D1/Cdk4 Complex Occurs during Resveratrol-Induced Cell Cycle Arrest in Colon Cancer Cell Lines. J. Nutr. 2001, 131, 2197–2203. [Google Scholar] [CrossRef]
- Rezaei, P.F.; Fouladdel, S.; Ghaffari, S.M.; Amin, G.; Azizi, E. Induction of G1 cell cycle arrest and cyclin D1 down-regulation in response to pericarp extract of Baneh in human breast cancer T47D cells. Daru 2012, 20, 101. [Google Scholar] [CrossRef]
- Dolatabadi, S.; Candia, J.; Akrap, N.; Vannas, C.; Tesan Tomic, T.; Losert, W.; Landberg, G.; Åman, P.; Ståhlberg, A. Cell Cycle and Cell Size Dependent Gene Expression Reveals Distinct Subpopulations at Single-Cell Level. Front. Genet. 2017, 8. [Google Scholar] [CrossRef]
- Shenkarev, Z.O.; Shulepko, M.A.; Bychkov, M.L.; Kulbatskii, D.S.; Shlepova, O.V.; Vasilyeva, N.A.; Andreev-Andrievsky, A.A.; Popova, A.S.; Lagereva, E.A.; Loktyushov, E.V.; et al. Water-Soluble Variant of Human Lynx1 Positively Modulates Synaptic Plasticity and Ameliorates Cognitive Impairment Associated with a7-nAChR Dysfunction. J. Neurochem. 2020. [Google Scholar] [CrossRef]
- Lin, L.-H.; Jin, J.; Nashelsky, M.B.; Talman, W.T. Acid-sensing ion channel 1 and nitric oxide synthase are in adjacent layers in the wall of rat and human cerebral arteries. J. Chem. Neuroanat. 2014, 61, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | Amiloride | Mambalgin-2 | ||
|---|---|---|---|---|
| A0, % | EC50, nM | A0, % | EC50, nM | |
| U251 MG | 44.84 ± 4.0 | 3.6 ± 0.2 | 61.78 ± 1.4 | 0.5 ± 0.2 |
| A172 | 48.06 ± 4.6 | 0.8 ± 0.02 | 60.15 ± 3.0 | 10 ± 1.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bychkov, M.; Shulepko, M.; Osmakov, D.; Andreev, Y.; Sudarikova, A.; Vasileva, V.; Pavlyukov, M.S.; Latyshev, Y.A.; Potapov, A.A.; Kirpichnikov, M.; et al. Mambalgin-2 Induces Cell Cycle Arrest and Apoptosis in Glioma Cells via Interaction with ASIC1a. Cancers 2020, 12, 1837. https://doi.org/10.3390/cancers12071837
Bychkov M, Shulepko M, Osmakov D, Andreev Y, Sudarikova A, Vasileva V, Pavlyukov MS, Latyshev YA, Potapov AA, Kirpichnikov M, et al. Mambalgin-2 Induces Cell Cycle Arrest and Apoptosis in Glioma Cells via Interaction with ASIC1a. Cancers. 2020; 12(7):1837. https://doi.org/10.3390/cancers12071837
Chicago/Turabian StyleBychkov, Maxim, Mikhail Shulepko, Dmitry Osmakov, Yaroslav Andreev, Anastasia Sudarikova, Valeria Vasileva, Marat S. Pavlyukov, Yaroslav A. Latyshev, Alexander A. Potapov, Mikhail Kirpichnikov, and et al. 2020. "Mambalgin-2 Induces Cell Cycle Arrest and Apoptosis in Glioma Cells via Interaction with ASIC1a" Cancers 12, no. 7: 1837. https://doi.org/10.3390/cancers12071837
APA StyleBychkov, M., Shulepko, M., Osmakov, D., Andreev, Y., Sudarikova, A., Vasileva, V., Pavlyukov, M. S., Latyshev, Y. A., Potapov, A. A., Kirpichnikov, M., Shenkarev, Z. O., & Lyukmanova, E. (2020). Mambalgin-2 Induces Cell Cycle Arrest and Apoptosis in Glioma Cells via Interaction with ASIC1a. Cancers, 12(7), 1837. https://doi.org/10.3390/cancers12071837