DrugSniper, a Tool to Exploit Loss-Of-Function Screens, Identifies CREBBP as a Predictive Biomarker of VOLASERTIB in Small Cell Lung Carcinoma (SCLC)

 , , , , ,
, , , , ,  and
and 
Abstract
1. Introduction
2. Results
2.1. DrugSniper: A Computational Pipeline to Predict Putative Mutation Biomarkers of Targeted Drug Sensitivity
2.2. Validation with Previously Known Associations
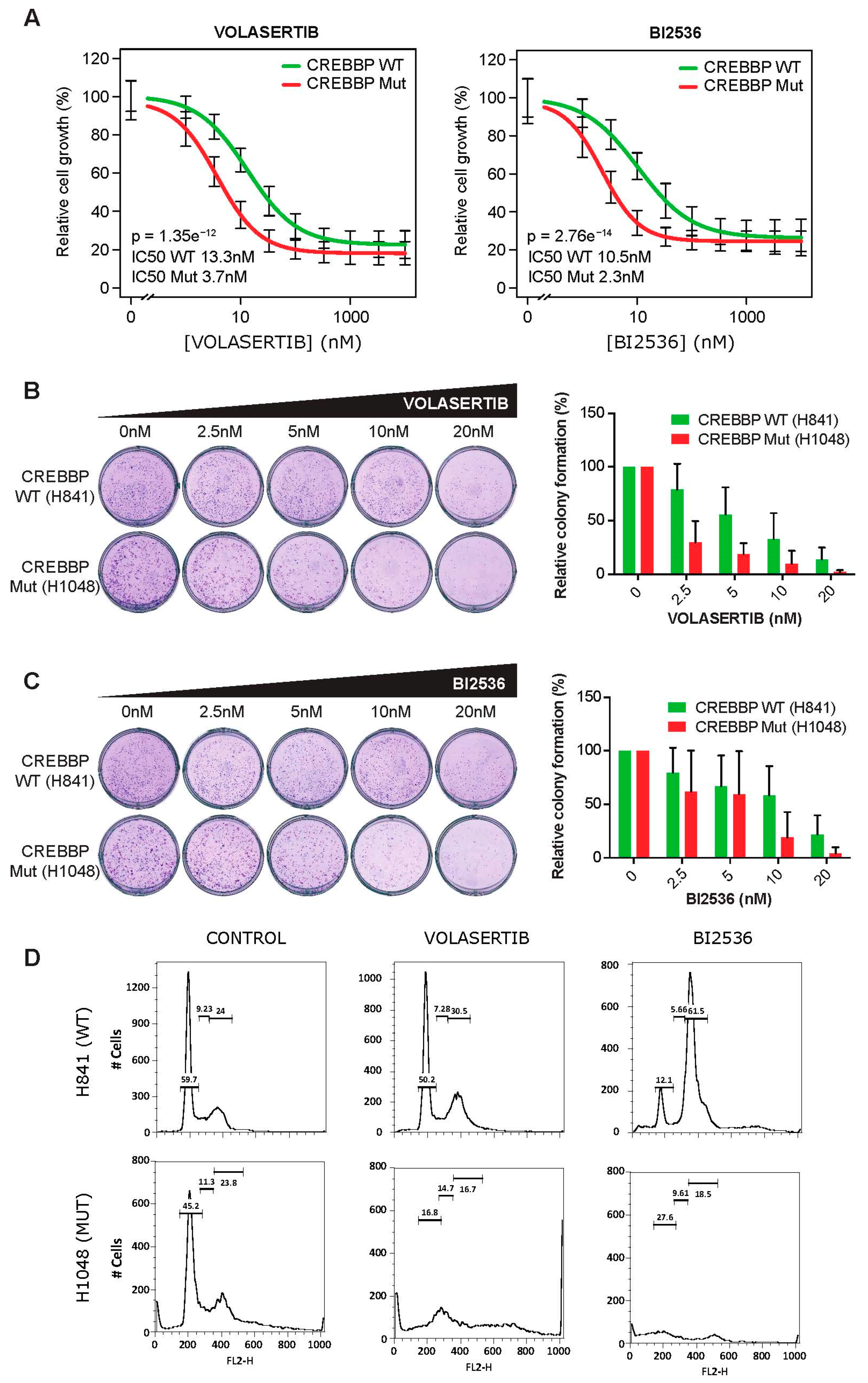
2.3. CREBBP Mutation is a Predictive Biomarker for PLK1 Inhibitors Efficacy in Small Cell Lung Cancer Cell Lines
- To be essential in >20% of SCLC cell lines, with a threshold of −2 for the DEMETER score for essentiality. Using this filter, we require that at least 20% of SCLC samples die when genes considered “essential” are knocked down.
- To be specific for SCLC cell lines with an odds ratio >1 (i.e., the percentage of cell lines in which the selected genes were essential should be larger for SCLC than for cell lines derived from any other tumor types). This specificity parameter states that at most for 20% of the other cell-lines, the selected gene is essential. Henceforth, the gene is hypothesized to be a good drug target specifically for SCLC.
- To have a minimal expression score of 1 TPM (transcripts per million) in more than 75% of the SCLC cell lines.
3. Discussion
4. Materials and Methods
4.1. Data Sources and Preprocessing
4.2. Statistical Model
4.3. Cell Culture
4.4. Cell Viability Assays
4.5. Drug Sensitivity Assay (IC50)
4.6. Colony Formation Assay
4.7. Cell Cycle Analysis
4.8. Statistical Analysis
4.9. Availability of Data and Materials
5. Conclusion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CCLE | the Cancer Cell Line Encyclopedia |
| FDR | false discovery rate |
| Lfdr | local false discovery rate |
| RNAi | RNA interference |
| SCLC | Small cell lung cancer |
References
- Hofree, M.; Shen, J.P.; Carter, H.; Gross, A.; Ideker, T. Network-based stratification of tumor mutations. Nat. Methods 2013, 10, 1108–1115. [Google Scholar] [CrossRef]
- Kawakami, M.; Mustachio, L.M.; Rodriguez-Canales, J.; Mino, B.; Roszik, J.; Tong, P.; Wang, J.; Lee, J.; Myung, J.H.; Heymach, J.V.; et al. Next-Generation CDK2/9 Inhibitors and Anaphase Catastrophe in Lung Cancer. J. Natl. Cancer Inst. 2017, 109, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Chu, Q.S.-C.; Gandhi, L.; Stephenson, J.J.; Govindan, R.; Bradford, D.S.; Bonomi, P.D.; Ellison, D.M.; Eaton, K.D.; Fritsch, H.; et al. An open-label, phase II study of the polo-like kinase-1 (Plk-1) inhibitor, BI 2536, in patients with relapsed small cell lung cancer (SCLC). Lung Cancer 2017, 104, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Wildey, G.; Chen, Y.; Lent, I.; Stetson, L.; Pink, J.; Barnholtz-Sloan, J.S.; Dowlati, A. Pharmacogenomic Approach to Identify Drug Sensitivity in Small-Cell Lung Cancer. PLoS ONE 2014, 9, e106784. [Google Scholar] [CrossRef] [PubMed]
- Sabari, J.K.; Lok, B.H.; Laird, J.H.; Poirier, J.T.; Rudin, C.M. Unravelling the biology of SCLC: implications for therapy. Nat. Rev. Clin. Oncol. 2017, 14, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.; Lok, B.H.; Schneeberger, V.E.; Desmeules, P.; Miles, L.A.; Arnold, P.; Ni, A.; Khodos, I.; De Stanchina, E.; Nguyen, T.; et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 2017, 31, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.; MacPherson, D.; et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat. Rev. Cancer 2019, 19, 289–297. [Google Scholar] [CrossRef]
- Shao, D.D.; Tsherniak, A.; Gopal, S.; Weir, B.A.; Tamayo, P.; Stransky, N.; Schumacher, S.E.; Zack, T.I.; Beroukhim, R.; Garraway, L.A.; et al. ATARiS: Computational quantification of gene suppression phenotypes from multisample RNAi screens. Genome Res. 2012, 23, 665–678. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576. [Google Scholar] [CrossRef]
- Cowley, G.S.; Weir, B.A.; Vazquez, F.; Tamayo, P.; Scott, J.A.; Rusin, S.; Ali, L.D.; Gerath, W.; Pantel, S.E.; Lizotte, P.H.; et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci. Data 2014, 1, 140035. [Google Scholar] [CrossRef]
- McDonald, E.R.; De Weck, A.; Schlabach, M.R.; Billy, E.; Mavrakis, K.J.; Hoffman, G.; Belur, D.; Castelletti, D.; Frias, E.; Gampa, K.; et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 2017, 170, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Behan, F.M.; Iorio, F.; Picco, G.; Goncalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Carazo, F.; Campuzano, L.; Cendoya, X.; Planes, F.J.; Rubio, A. TranscriptAchilles: A genome-wide platform to predict isoform biomarkers of gene essentiality in cancer. GigaScience 2019, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.J.; Meyers, R.; Weir, B.A.; Vazquez, F.; Zhang, C.-Z.; Ben-David, U.; Cook, A.; Ha, G.; Harrington, W.F.; Doshi, M.B.; et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov. 2016, 6, 914–929. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.S.F.F.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.; Uhalte, E.C.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2016, 45, D945–D954. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2017, 46, D1074–D1082. [Google Scholar] [CrossRef]
- McFarland, J.M.; Ho, Z.V.; Kugener, G.; Dempster, J.M.; Montgomery, P.G.; Bryan, J.G.; Krill-Burger, J.M.; Green, T.M.; Vazquez, F.; Boehm, J.S.; et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun. 2018, 9, 4610. [Google Scholar] [CrossRef]
- Hart, T.; Brown, K.R.; Sircoulomb, F.; Rottapel, R.; Moffat, J. Measuring error rates in genomic perturbation screens: Gold standards for human functional genomics. Mol. Syst. Boil. 2014, 10, 733. [Google Scholar] [CrossRef]
- Ignatiadis, N.; Klaus, B.; Zaugg, J.B.; Huber, W. Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat. Methods 2016, 13, 577–580. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Lam, S.S.; Leung, A.Y. Overcoming Resistance to FLT3 Inhibitors in the Treatment of FLT3-Mutated AML. Int. J. Mol. Sci. 2020, 21, 1537. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; McKee, A.E.; Ning, Y.-M.; Hazarika, M.; Theoret, M.; Johnson, J.R.; Xu, Q.C.; Tang, S.; Sridhara, R.; Jiang, X.; et al. FDA Approval Summary: Vemurafenib for Treatment of Unresectable or Metastatic Melanoma with the BRAFV600E Mutation. Clin. Cancer Res. 2014, 20, 4994–5000. [Google Scholar] [CrossRef] [PubMed]
- Hay, T.; Matthews, J.R.; Pietzka, L.; Lau, A.; Cranston, A.; Clarke, A.; O’Connor, M.; Smith, G.; Douglas-Jones, A.; Martin, N.; et al. Poly(ADP-ribose) polymerase-1 inhibitor treatment regresses autochthonous brca2/p53-mutant mammary tumors in vivo and delays tumor relapse in combination with carboplatin. Cancer Res. 2009, 69, 3850–3855. [Google Scholar]
- Lord, C.J.; Ashworth, A. Targeted therapy for cancer using PARP inhibitors. Curr. Opin. Pharmacol. 2008, 8, 363–369. [Google Scholar] [CrossRef]
- Šmida, M.; De La Cruz, F.F.; Kerzendorfer, C.; Uras, I.Z.; Mair, B.; Mazouzi, A.; Suchankova, T.; Konopka, T.; Katz, A.M.; Paz, K.; et al. MEK inhibitors block growth of lung tumours with mutations in ataxia–telangiectasia mutated. Nat. Commun. 2016, 7, 13701. [Google Scholar] [CrossRef] [PubMed]
- Raymond, E.; Faivre, S.; Armand, J.P. Epidermal growth factor receptor tyrosine kinase as a target for anticancer therapy. Drugs 2000, 60, 15–23. [Google Scholar] [CrossRef]
- Weinstein, I.B.; Joe, A.; Felsher, D. Oncogene Addiction. Cancer Res. 2008, 68, 3077–3080. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Sharma, S.; Shah, B.; Portier, B.P.; Devaraj, S.G.T.; Liu, K.; Lyer, S.P.; Bearss, D.; Bhalla, K.N. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia 2016, 74, 27–34. [Google Scholar]
- Mohammad, H.P.; Smitheman, K.N.; Kamat, C.D.; Soong, D.; Federowicz, K.E.; Van Aller, G.S.; Schneck, J.L.; Carson, J.D.; Liu, Y.; Butticello, M.; et al. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell 2015, 28, 57–69. [Google Scholar] [CrossRef]
- Ito, T.; Kudoh, S.; Ichimura, T.; Fujino, K.; Hassan, W.; Udaka, N. Small cell lung cancer, an epithelial to mesenchymal transition (EMT)-like cancer: Significance of inactive Notch signaling and expression of achaete-scute complex homologue 1. Hum. Cell 2016, 30, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Abshire, C.F.; Carroll, J.L.; Dragoi, A.-M. FLASH protects ZEB1 from degradation and supports cancer cells’ epithelial-to-mesenchymal transition. Oncogenesis 2016, 5, e254. [Google Scholar] [CrossRef] [PubMed]
- Romero-Ferraro, O.A.; Torres-Diz, M.; Pros, E.; Moruno, A.G.; Salama, S.M.; Savola, S.; Montuenga, L.M.; Pio, R.; Yokota, J.; Sanchez-Cespedes, M. Abstract 2477: Max inactivation in small cell lung cancer disrupts the MYC-SWI/SNF programs and is synthetic lethal with BRG1. Mol. Cell. Bio. 2014, 74, 2477. [Google Scholar] [CrossRef]
- O’Connor, A.; Maffini, S.; Rainey, M.D.; Kaczmarczyk, A.; Gaboriau, D.C.A.; Musacchio, A.; Santocanale, C. Requirement for PLK1 kinase activity in the maintenance of a robust spindle assembly checkpoint. Biol. Open 2015, 5, 11–19. [Google Scholar] [CrossRef]
- Ritz, C.; Baty, F.; Streibig, J.C.; Gerhard, D. Dose-Response Analysis Using R. PLoS ONE 2015, 10, e0146021. [Google Scholar] [CrossRef] [PubMed]
- Cavalla, D.; Singal, C. Retrospective clinical analysis for drug rescue: For new indications or stratified patient groups. Drug Discov. Today 2012, 17, 104–109. [Google Scholar] [CrossRef]
- Smirnov, P.; Safikhani, Z.; El-Hachem, N.; Wang, D.; She, A.; Olsen, C.; Freeman, M.; Selby, H.; Gendoo, D.M.A.; Grossmann, P.; et al. PharmacoGx: An R package for analysis of large pharmacogenomic datasets. Bioinformatics 2015, 32, 1244–1246. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.; Lightfoot, H.; Forbes, S.; Sridhar, R.; Futreal, P.A.; Haber, D.; Stratton, M.; et al. Abstract 2206: Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Exp. Mol. Ther. 2013, 73, 2206. [Google Scholar] [CrossRef]
- Rudalska, R.; Dauch, D.; Longerich, T.; McJunkin, K.; Wuestefeld, T.; Kang, T.-W.; Hohmeyer, A.; Pesic, M.; Leibold, J.; Von Thun, A.; et al. In vivo RNAi screening identifies a mechanism of sorafenib resistance in liver cancer. Nat. Med. 2014, 20, 1138–1146. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A Potential Target for Cancer Therapy. Transl. Oncol. 2016, 10, 22–32. [Google Scholar] [CrossRef]
- Park, J.-E.; Hymel, D.; Burke, J.T.R.; Lee, K.S. Current progress and future perspectives in the development of anti-polo-like kinase 1 therapeutic agents. F1000 Research 2017, 6, 1024. [Google Scholar] [CrossRef] [PubMed]
- Creyghton, M.P.; Cheng, A.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.H.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lü, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Jia, D.; Augert, A.; Kim, N.-W.; Eastwood, E.; Wu, N.; Ibrahim, A.H.; Kim, K.-B.; Dunn, C.T.; Pillai, S.P.; Gazdar, A.F.; et al. Crebbp Loss Drives Small Cell Lung Cancer and Increases Sensitivity to HDAC Inhibition. Cancer Discov. 2018, 8, 1422–1437. [Google Scholar] [CrossRef] [PubMed]
- Major, M.L.; Lepe, R.; Costa, R.H. Forkhead box M1B transcriptional activity requires binding of Cdk-cyclin complexes for phosphorylation-dependent recruitment of p300/CBP coactivators. Mol. Cell. Biol. 2004, 24, 2649–2661. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Malureanu, L.; Huang, J.; Wang, W.; Li, H.; Van Deursen, J.M.; Tindall, D.J.; Chen, J. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nature 2008, 10, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, J.; Sebastian, M. Polo-like kinase 1 inhibition in NSCLC: Mechanism of action and emerging predictive biomarkers. Lung Cancer: Targets Ther. 2019, 10, 67–80. [Google Scholar] [CrossRef]
- Tatlow, P.; Piccolo, S.R. A cloud-based workflow to quantify transcript-expression levels in public cancer compendia. Sci. Rep. 2016, 6, 39259. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Harrow, J.; Frankish, A.; González, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef]
- Rouillard, A.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’Ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Troyanskaya, O.; Cantor, M.; Sherlock, G.; Brown, P.; Hastie, T.; Tibshirani, R.; Botstein, D.; Altman, R.B. Missing value estimation methods for DNA microarrays. Bioinformatics 2001, 17, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Efron, B.; Storey, J.D.; Tibshirani, R. Microarrays, empirical Bayes methods, and false discovery rates. Gen. Epi 2002, 1, 70–86. [Google Scholar] [CrossRef]
- Storey, J. A direct approach to false discovery rates. J. R. Stat. Soc. Ser. B Stat. Methodol. 2002, 64, 479–498. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Target | Mutation Biomarker | ∆Ess | % Mut Patient | p-Value | lfdr (adj. p-Value) |
|---|---|---|---|---|---|
| PLK1* | CREBBP | −3.9 | 10 | 1.45 × 10−3 | 0.15 |
| SMARCA4 | CREBBP | −7.33 | 10 | 3.20 × 10−3 | 0.19 |
| YY2 | TRRAP | 3.34 | 4.55 | 3.19 × 10−4 | 0.2 |
| SMARCB1 | CREBBP | −3.66 | 10 | 6.08 × 10−3 | 0.22 |
| XRCC6 | CREBBP | −3.95 | 10 | 7.05 × 10−3 | 0.22 |
| RANBP1 | TPR | −3.83 | 5.45 | 9.26 × 10−4 | 0.28 |
| SMARCA4 | NPM1 | 4.35 | NA | 4.73 × 10−2 | 0.42 |
| KDM1A | SMARCA4 | 3.73 | 4.55 | 2.26 × 10−2 | 0.43 |
| RAD21 | TRRAP | −3.42 | 4.55 | 1.61 × 10−3 | 0.46 |
| CDC45 | RB1 | −3.03 | 78.18 | 6.50 × 10−3 | 0.46 |
| UBE2I | RB1 | 3.47 | 78.18 | 8.67 × 10−3 | 0.47 |
| CDK2* | EPHA5 | −3.53 | 10.91 | 1.26 × 10−2 | 0.47 |
| ZNF548 | PRKG1 | −2.82 | 1.82 | 4.29 × 10−2 | 0.48 |
| RPS6 | TTBK1 | 3.46 | 4.55 | 3.82 × 10−2 | 0.49 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carazo, F.; Bértolo, C.; Castilla, C.; Cendoya, X.; Campuzano, L.; Serrano, D.; Gimeno, M.; Planes, F.J.; Pio, R.; Montuenga, L.M.; et al. DrugSniper, a Tool to Exploit Loss-Of-Function Screens, Identifies CREBBP as a Predictive Biomarker of VOLASERTIB in Small Cell Lung Carcinoma (SCLC). Cancers 2020, 12, 1824. https://doi.org/10.3390/cancers12071824
Carazo F, Bértolo C, Castilla C, Cendoya X, Campuzano L, Serrano D, Gimeno M, Planes FJ, Pio R, Montuenga LM, et al. DrugSniper, a Tool to Exploit Loss-Of-Function Screens, Identifies CREBBP as a Predictive Biomarker of VOLASERTIB in Small Cell Lung Carcinoma (SCLC). Cancers. 2020; 12(7):1824. https://doi.org/10.3390/cancers12071824
Chicago/Turabian StyleCarazo, Fernando, Cristina Bértolo, Carlos Castilla, Xabier Cendoya, Lucía Campuzano, Diego Serrano, Marian Gimeno, Francisco J. Planes, Ruben Pio, Luis M. Montuenga, and et al. 2020. "DrugSniper, a Tool to Exploit Loss-Of-Function Screens, Identifies CREBBP as a Predictive Biomarker of VOLASERTIB in Small Cell Lung Carcinoma (SCLC)" Cancers 12, no. 7: 1824. https://doi.org/10.3390/cancers12071824
APA StyleCarazo, F., Bértolo, C., Castilla, C., Cendoya, X., Campuzano, L., Serrano, D., Gimeno, M., Planes, F. J., Pio, R., Montuenga, L. M., & Rubio, A. (2020). DrugSniper, a Tool to Exploit Loss-Of-Function Screens, Identifies CREBBP as a Predictive Biomarker of VOLASERTIB in Small Cell Lung Carcinoma (SCLC). Cancers, 12(7), 1824. https://doi.org/10.3390/cancers12071824