Bone, a Secondary Growth Site of Breast and Prostate Carcinomas: Role of Osteocytes



Abstract
1. Introduction
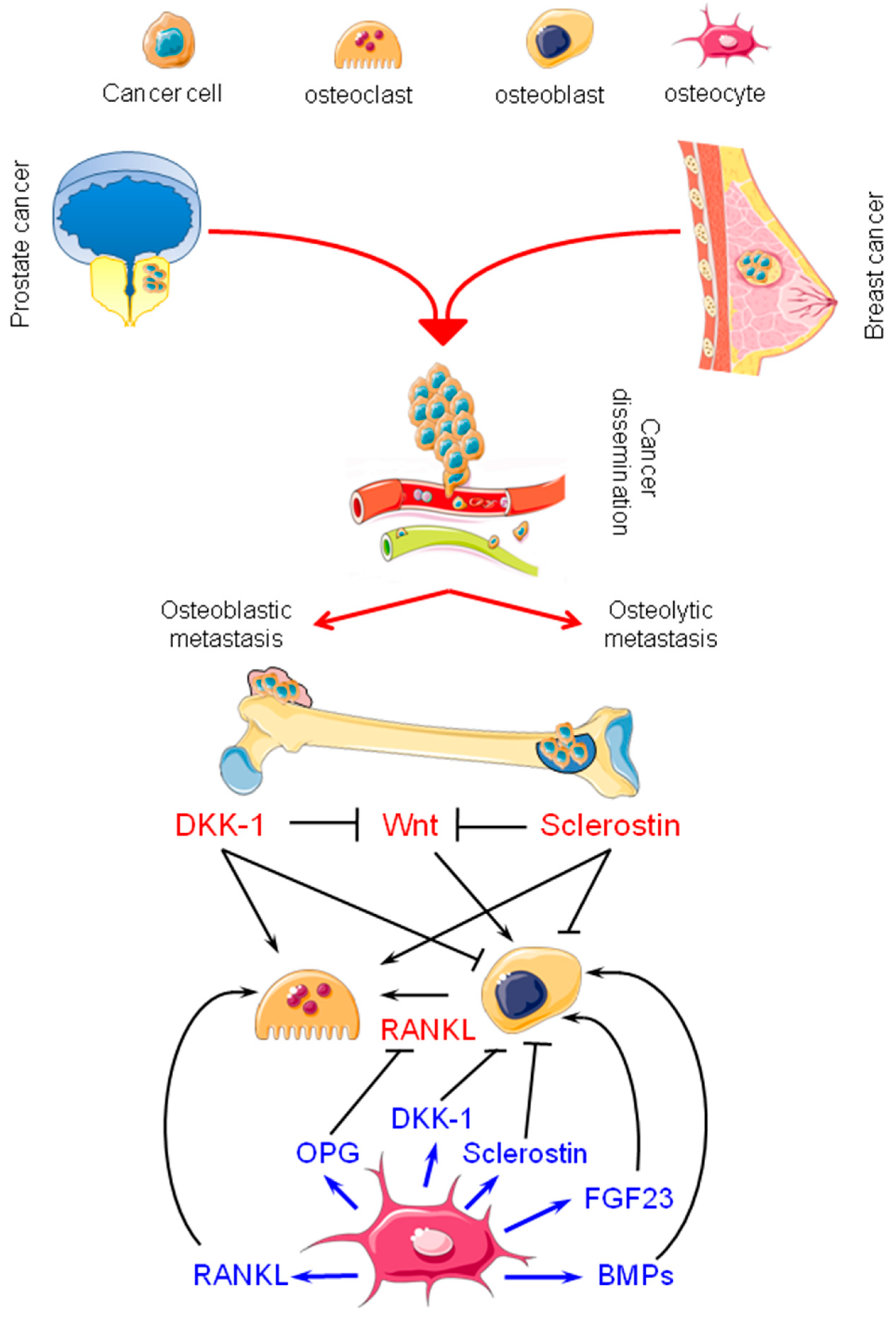
2. Bone Metastasis from Breast and Prostate Carcinomas: Osteolytic and Osteoblastic Lesions
3. Osteocytes: Their Physiology and Possible Contributions to Bone Metastasis
3.1. Osteocytes in Bone Physiology
3.2. Osteocytes in Bone Metastasis from Breast and Prostate Carcinomas: Active Players
3.3. Sclerostin, Dickkopf-1 (DKK-1), and Fibroblast Growth Factor 23 (FGF23) in Breast and Prostate Cancer: New Therapeutic Opportunities?
3.4. Cx43 Hemichannels: A New Way to Explore
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mattiuzzi, C.; Lippi, G. Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, L.A.; Fournier, P.G.J.; Chirgwin, J.M.; Guise, T.A. Molecular Biology of Bone Metastasis. Mol. Cancer Ther. 2007, 6, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, N.; Ferretti, M.; Bolzoni, M.; Storti, P.; Lazzaretti, M.; Dalla Palma, B.; Bonomini, S.; Martella, E.; Agnelli, L.; Neri, A.; et al. Increased Osteocyte Death in Multiple Myeloma Patients: Role in Myeloma-Induced Osteoclast Formation. Leukemia 2012, 26, 1391–1401. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Gomarasca, M.; Maroni, P.; Banfi, G.; Lombardi, G. microRNAs in the Antitumor Immune Response and in Bone Metastasis of Breast Cancer: From Biological Mechanisms to Therapeutics. Int. J. Mol. Sci. 2020, 21, 2805. [Google Scholar] [CrossRef]
- Haider, M.A.; Smit, D.J.; Taipaleenmäki, H. The Endosteal Niche in Breast Cancer Bone Metastasis. Front. Oncol. 2020, 10, 335. [Google Scholar] [CrossRef]
- Casimiro, S.; Ferreira, A.R.; Mansinho, A.; Alho, I.; Costa, L. Molecular Mechanisms of Bone Metastasis: Which Targets Came from the Bench to the Bedside? Int. J. Mol. Sci. 2016, 17, 1415. [Google Scholar] [CrossRef]
- Roodman, G.D.; Silbermann, R. Mechanisms of Osteolytic and Osteoblastic Skeletal Lesions. BoneKEy Rep. 2015, 4, 753. [Google Scholar] [CrossRef]
- Clezardin, P.; Teti, A. Bone Metastasis: Pathogenesis and Therapeutic Implications. Clin. Exp. Metastasis 2007, 24, 599–608. [Google Scholar] [CrossRef]
- Wong, S.K.; Mohamad, N.V.; Giaze, T.R.; Chin, K.Y.; Mohamed, N.; Ima-Nirwana, S. Prostate Cancer and Bone Metastases: The Underlying Mechanisms. Int. J. Mol. Sci. 2019, 20, 2587. [Google Scholar] [CrossRef] [PubMed]
- Clines, G.A.; Guise, T.A. Molecular mechanisms and treatment of bone metastasis. Expert Rev. Mol. Med. 2008, 10, e7. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.-T.; Taipaleenmäki, H. Targeting the Metastatic Bone Microenvironment by MicroRNAs. Front. Endocrinol. 2018, 9, 202. [Google Scholar] [CrossRef]
- Guise, T.A.; Yin, J.J.; Taylor, S.D.; Kumagai, Y.; Dallas, M.; Boyce, B.F.; Yoneda, T.; Mundy, G.R. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J. Clin. Investig. 1996, 98, 1544–1549. [Google Scholar] [CrossRef]
- Suominen, M.I.; Wilson, T.; Käkönen, S.-M.; Scholz, A. The Mode-of-Action of Targeted Alpha Therapy Radium-223 as an Enabler for Novel Combinations to Treat Patients with Bone Metastasis. Int. J. Mol. Sci. 2019, 20, 3899. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Docherty, F.E.; Brown, H.K.; Reeves, K.J.; Fowles, A.C.; Ottewell, P.D.; Dear, T.N.; Holen, I.; Croucher, P.I.; Eaton, C.L. Prostate Cancer Cells Preferentially Home to Osteoblast-rich Areas in the Early Stages of Bone Metastasis: Evidence From In Vivo Models: Spatial Distribution of Prostate Cancer Cells in Mouse Bone. J. Bone Min. Res. 2014, 29, 2688–2696. [Google Scholar] [CrossRef]
- Logothetis, C.; Morris, M.J.; Den, R.; Coleman, R.E. Current perspectives on bone metastases in castrate-resistant prostate cancer. Cancer Metastasis Rev. 2018, 37, 189–196. [Google Scholar] [CrossRef]
- Quiroz-Munoz, M.; Izadmehr, S.; Arumugam, D.; Wong, B.; Kirschenbaum, A.; Levine, A.C. Mechanisms of Osteoblastic Bone Metastasis in Prostate Cancer: Role of Prostatic Acid Phosphatase. J. Endocr. Soc. 2019, 3, 655–664. [Google Scholar] [CrossRef]
- Sekita, A.; Matsugaki, A.; Nakano, T. Disruption of collagen/apatite alignment impairs bone mechanical function in osteoblastic metastasis induced by prostate cancer. Bone 2017, 97, 83–93. [Google Scholar] [CrossRef]
- Matsugaki, A.; Aramoto, G.; Ninomiya, T.; Sawada, H.; Hata, S.; Nakano, T. Abnormal arrangement of a collagen/apatite extracellular matrix orthogonal to osteoblast alignment is constructed by a nanoscale periodic surface structure. Biomaterials 2015, 37, 134–143. [Google Scholar] [CrossRef]
- Clines, G.A.; Mohammad, K.S.; Bao, Y.; Stephens, O.W.; Suva, L.J.; Shaughnessy, J.D.; Fox, J.W., Jr.; Chirgwin, J.M.; Guise, T.A. Dickkopf homolog 1 mediates endothelin-1-stimulated new bone formation. Mol. Endocrinol. 2007, 21, 486–498. [Google Scholar] [CrossRef]
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Gonçalves, F. Bone metastases: An overview. Oncol. Rev. 2017, 11. [Google Scholar] [CrossRef]
- Chen, X.; Wang, L.; Zhao, K.; Wang, H. Osteocytogenesis: Roles of Physicochemical Factors, Collagen Cleavage, and Exogenous Molecules. Tissue Eng. Part. B Rev. 2018, 24, 215–225. [Google Scholar] [CrossRef]
- Bonewald, L.F. The amazing osteocyte. J. Bone Min. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Bellido, T. Osteocytes and Skeletal Pathophysiology. Curr. Mol. Bio. Rep. 2015, 1, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Capulli, M.; Paone, R.; Rucci, N. Osteoblast and osteocyte: Games without frontiers. Arch. Biochem. Biophys. 2014, 561, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.C.; McGregor, N.E.; Poulton, I.J.; Pompolo, S.; Allan, E.H.; Quinn, J.M.; Gillespie, M.T.; Martin, T.J.; Sims, N.A. Cardiotrophin-1 Is an Osteoclast-Derived Stimulus of Bone Formation Required for Normal Bone Remodeling. J. Bone Min. Res. 2008, 23, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Knothe Tate, M.L.; Niederer, P.; Knothe, U. In Vivo tracer transport through the lacunocanalicular system of rat bone in an environment devoid of mechanical loading. Bone 1998, 22, 107–117. [Google Scholar] [CrossRef]
- Uda, Y.; Azab, E.; Sun, N.; Shi, C.; Pajevic, P.D. Osteocyte Mechanobiology. Currosteoporos Rep. 2017, 15, 318–325. [Google Scholar] [CrossRef]
- Balemans, W. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum. Mol. Genet. 2001, 10, 537–543. [Google Scholar] [CrossRef]
- Brunkow, M.E.; Gardner, J.C.; Van Ness, J.; Paeper, B.W.; Kovacevich, B.R.; Proll, S.; Skonier, J.E.; Zhao, L.; Sabo, P.J.; Fu, Y.-H.; et al. Bone Dysplasia Sclerosteosis Results from Loss of the SOST Gene Product, a Novel Cystine Knot–Containing Protein. Am. J. Hum. Genet. 2001, 68, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ominsky, M.S.; Niu, Q.-T.; Sun, N.; Daugherty, B.; D’Agostin, D.; Kurahara, C.; Gao, Y.; Cao, J.; Gong, J.; et al. Targeted Deletion of the Sclerostin Gene in Mice Results in Increased Bone Formation and Bone Strength. J. Bone Min. Res. 2008, 23, 860–869. [Google Scholar] [CrossRef]
- Compton, J.T.; Lee, F.Y. A Review of Osteocyte Function and the Emerging Importance of Sclerostin. J. Bone Jt. Surg. Am. 2014, 96, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Genetos, D.C.; Yellowley, C.E.; Loots, G.G. Prostaglandin E2 signals through PTGER2 to regulate sclerostin expression. PLoS ONE 2011, 6, e17772. [Google Scholar] [CrossRef]
- Prasadam, I.; Zhou, Y.; Du, Z.; Chen, J.; Crawford, R.; Xiao, Y. Osteocyte-induced angiogenesis via VEGF–MAPK-dependent pathways in endothelial cells. Mol. Cell. Biochem. 2014, 386, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef]
- Santos, A.; Bakker, A.D.; Willems, H.M.E.; Bravenboer, N.; Bronckers, A.L.J.J.; Klein-Nulend, J. Mechanical Loading Stimulates BMP7, But Not BMP2, Production by Osteocytes. Calcif. Tissue Int. 2011, 89, 318–326. [Google Scholar] [CrossRef][Green Version]
- Krause, C.; Korchynskyi, O.; de Rooij, K.; Weidauer, S.E.; de Gorter, D.J.J.; van Bezooijen, R.L.; Hatsell, S.; Economides, A.N.; Mueller, T.D.; Löwik, C.W.G.M.; et al. Distinct Modes of Inhibition by Sclerostin on Bone Morphogenetic Protein and Wnt Signaling Pathways. J. Biol. Chem. 2010, 285, 41614–41626. [Google Scholar] [CrossRef]
- Oranger, A.; Brunetti, G.; Colaianni, G.; Tamma, R.; Carbone, C.; Lippo, L.; Mori, G.; Pignataro, P.; Cirulli, N.; Zerlotin, R.; et al. Sclerostin stimulates angiogenesis in human endothelial cells. Bone 2017, 101, 26–36. [Google Scholar] [CrossRef]
- Myers, T.J.; Longobardi, L.; Willcockson, H.; Temple, J.D.; Tagliafierro, L.; Ye, P.; Li, T.; Esposito, A.; Moats-Staats, B.M.; Spagnoli, A. BMP2 regulation of CXCL12 cellular, temporal, and spatial expression is essential during fracture repair. J. Bone Min. Res. 2015, 30, 2014–2027. [Google Scholar] [CrossRef]
- Cui, Y.-X.; Evans, B.A.J.; Jiang, W.G. New Roles of Osteocytes in Proliferation, Migration and Invasion of Breast and Prostate Cancer Cells. Anticancer Res. 2016, 36, 1193–1201. [Google Scholar] [PubMed]
- Chen, A.; Wang, L.; Liu, S.; Wang, Y.; Liu, Y.; Wang, M.; Nakshatri, H.; Li, B.Y.; Yokota, H. Attraction and compaction of migratory breast cancer cells by bone matrix proteins through tumor-osteocyte interactions. Sci. Rep. 2018, 8, 5420. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Fan, Y.; Chen, A.; Jalali, A.; Minami, K.; Ogawa, K.; Nakshatri, H.; Li, B.Y.; Yokota, H. Osteocyte-driven downregulation of Snail restrains effects of Drd2 inhibitors on mammary tumor cells. Cancer Res. 2018, 78, 3865–3876. [Google Scholar] [CrossRef] [PubMed]
- Matteucci, E.; Maroni, P.; Luzzati, A.; Perrucchini, G.; Bendinelli, P.; Desiderio, M.A. Bone metastatic process of breast cancer involves methylation state affecting E-cadherin expression through TAZ and WWOX nuclear effectors. Eur. J. Cancer 2013, 49, 231–244. [Google Scholar] [CrossRef]
- Mei, X.; Middleton, K.; Shim, D.; Wan, Q.; Xu, L.; Ma, Y.V.; Devadas, D.; Walji, N.; Wang, L.; Young, E.W.K.; et al. Microfluidic platform for studying osteocyte mechanoregulation of breast cancer bone metastasis. Integr. Biol. 2019, 11, 119–129. [Google Scholar] [CrossRef]
- Ma, Y.V.; Xu, L.; Mei, X.; Middleton, K.; You, L. Mechanically stimulated osteocytes reduce the bone-metastatic potential of breast cancer cells in vitro by signaling through endothelial cells. J. Cell. Biochem. 2019, 120, 7590–7601. [Google Scholar] [CrossRef]
- Ma, Y.V.; Lam, C.; Dalmia, S.; Gao, P.; Young, J.; Middleton, K.; Liu, C.; Xu, H.; You, L. Mechanical regulation of breast cancer migration and apoptosis via direct and indirect osteocyte signaling. J. Cell. Biochem. 2018, 119, 5665–5675. [Google Scholar] [CrossRef]
- Fan, Y.; Jalali, A.; Chen, A.; Zhao, X.; Liu, S.; Teli, M.; Guo, Y.; Li, F.; Li, J.; Siegel, A.; et al. Skeletal loading regulates breast cancer-associated osteolysis in a loading intensity-dependent fashion. Bone Res. 2020, 8, 9. [Google Scholar] [CrossRef]
- Wang, W.; Sarazin, B.A.; Kornilowicz, G.; Lynch, M.E. Mechanically-Loaded Breast Cancer Cells Modify Osteocyte Mechanosensitivity by Secreting Factors That Increase Osteocyte Dendrite Formation and Downstream Resorption. Front. Endocrinol. 2018, 9, 352. [Google Scholar] [CrossRef]
- Wang, W.; Yang, X.; Dai, J.; Lu, Y.; Zhang, J.; Keller, E.T. Prostate cancer promotes a vicious cycle of bone metastasis progression through inducing osteocytes to secrete GDF15 that stimulates prostate cancer growth and invasion. Oncogene 2019, 38, 4540–4559. [Google Scholar] [CrossRef]
- Sottnik, J.L.; Dai, J.; Zhang, H.; Campbell, B.; Keller, E.T. Tumor-Induced Pressure in the Bone Microenvironment Causes Osteocytes to Promote the Growth of Prostate Cancer Bone Metastases. Cancer Res. 2015, 75, 2151–2158. [Google Scholar] [CrossRef]
- Plotkin, L.I. Apoptotic Osteocytes and the Control of Targeted Bone Resorption. Curr. Osteoporos. Rep. 2014, 12, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Noble, B. Microdamage and apoptosis. Eur. J. Morphol. 2005, 42, 91–98. [Google Scholar] [CrossRef]
- Toscani, D.; Bolzoni, M.; Accardi, F.; Aversa, F.; Giuliani, N. The osteoblastic niche in the contest of multiple myeloma. Ann. NY Acad. Sci. 2015, 1335, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Mathis, K.M.; Sturgeon, K.M.; Winkels, R.M.; Wiskemann, J.; de Souza, M.J.; Schmitz, K.H. Bone resorption and bone metastasis risk. Med. Hypotheses 2018, 118, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Shandala, T.; Ng, S.Y.; Hopwood, B.; Yip, Y.C.; Foster, B.K.; Xian, C.J. The role of osteocyte apoptosis in cancer chemotherapy-induced bone loss. J. Cell. Physiol. 2012, 227, 2889–2897. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Ma, Q.; Ding, N.; Luo, F.; Bai, Y.; Kang, F.; Gong, X.; Dong, R.; Dai, J.; Dai, Q.; et al. IL-11 is essential in promoting osteolysis in breast cancer bone metastasis via RANKL-independent activation of osteoclastogenesis. Cell Death Dis. 2019, 10, 353. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Hiasa, M.; Chirgwin, J.M.; Carlesso, N.; Yoneda, T.; Mohammad, K.S.; Plotkin, L.I.; Roodman, G.D.; et al. Bidirectional Notch signaling and osteocyte-derived factors in the bone marrow microenvironment promote tumor cell proliferation and bone destruction in multiple myeloma. Cancer Res. 2016, 76, 1089–1100. [Google Scholar] [CrossRef]
- Atkinson, E.G.; Delgado-Calle, J. The Emerging Role of Osteocytes in Cancer in Bone. JBMR Plus 2019, 3, e10186. [Google Scholar] [CrossRef]
- Galliera, E.; Massaccesi, L.; de Benedettis, E.; Longhi, E.; de Toma, D.; Corsi Romanelli, M.M.; Banfi, G. Longitudinal evaluation of Wnt inhibitors and comparison with others serum osteoimmunological biomarkers in osteolytic bone metastasis. J. Leukoc. Biol. 2020. [Google Scholar] [CrossRef]
- Quarles, L.D. Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism. Nat. Rev. Endocrinol. 2012, 8, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Atkins, G.J.; Rowe, P.S.; Lim, H.P.; Welldon, K.J.; Ormsby, R.; Wijenayaka, A.R.; Zelenchuk, L.; Evdokiou, A.; Findlay, D.M. Sclerostin is a locally acting regulator of late osteoblast/preosteocyte differentiation and regulates mineralization through a MEPE-ASARM-dependent mechanism. J. Bone Min. Res. 2011, 26, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Wijenayaka, A.R.; Kogawa, M.; Lim, H.P.; Bonewald, L.F.; Findlay, D.M.; Atkins, G.J. Sclerostin Stimulates Osteocyte Support of Osteoclast Activity by a RANKL-Dependent Pathway. PLoS ONE 2011, 6, e25900. [Google Scholar] [CrossRef] [PubMed]
- Kogawa, M.; Khalid, K.A.; Wijenayaka, A.R.; Ormsby, R.T.; Evdokiou, A.; Anderson, P.H.; Findlay, D.M.; Atkins, G.J. Recombinant sclerostin antagonizes effects of ex vivo mechanical loading in trabecular bone and increases osteocyte lacunar size. Am. J. Physiol. Cell Physiol. 2018, 314, C53–C61. [Google Scholar] [CrossRef]
- Bendinelli, P.; Maroni, P.; Matteucci, E.; Luzzati, A.; Perrucchini, G.; Desiderio, M.A. Microenvironmental stimuli affect Endothelin-1 signaling responsible for invasiveness and osteomimicry of bone metastasis from breast cancer. BBA Mol. Cell Res. 2014, 1843, 815–826. [Google Scholar] [CrossRef]
- Gkotzamanidou, M.; Dimopoulos, M.A.; Kastritis, E.; Christoulas, D.; Moulopoulos, L.A.; Terpos, E. Sclerostin: A possible target for the management of cancer-induced bone disease. Expert Opin. Ther. Targets 2012, 16, 761–769. [Google Scholar] [CrossRef]
- Zhu, M.; Liu, C.; Li, S.; Zhang, S.; Yao, Q.; Song, Q. Sclerostin induced tumor growth, bone metastasis and osteolysis in breast cancer. Sci. Rep. 2017, 7, 11399. [Google Scholar] [CrossRef]
- Hesse, E.; Schröder, S.; Brandt, D.; Pamperin, J.; Saito, H.; Taipaleenmäki, H. Sclerostin inhibition alleviates breast cancer–induced bone metastases and muscle weakness. JCI Insight 2019, 4, e125543. [Google Scholar] [CrossRef]
- Taipaleenmäki, H.; Farina, N.H.; van Wijnen, A.J.; Stein, J.L.; Hesse, E.; Stein, G.S.; Lian, J.B. Antagonizing miR-218-5p attenuates Wnt signaling and reduces metastatic bone disease of triple negative breast cancer cells. Oncotarget 2016, 7. [Google Scholar] [CrossRef]
- Hassan, M.Q.; Maeda, Y.; Taipaleenmaki, H.; Zhang, W.; Jafferji, M.; Gordon, J.A.R.; Li, Z.; Croce, C.M.; van Wijnen, A.J.; Stein, J.L.; et al. miR-218 Directs a Wnt Signaling Circuit to Promote Differentiation of Osteoblasts and Osteomimicry of Metastatic Cancer Cells. J. Biol Chem. 2012, 287, 42084–42092. [Google Scholar] [CrossRef]
- Padhi, D.; Jang, G.; Stouch, B.; Fang, L.; Posvar, E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J. Bone Min. Res. 2011, 26, 19–26. [Google Scholar] [CrossRef]
- Padhi, D.; Allison, M.; Kivitz, A.J.; Gutierrez, M.J.; Stouch, B.; Wang, C.; Jang, G. Multiple doses of sclerostin antibody romosozumab in healthy men and postmenopausal women with low bone mass: A randomized, double-blind, placebo-controlled study. J. Clin. Pharm. 2014, 54, 168–178. [Google Scholar] [CrossRef]
- Appelman-Dijkstra, N.M.; Papapoulos, S.E. Sclerostin Inhibition in the Management of Osteoporosis. Calcif. Tissue Int. 2016, 98, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Yamabuki, T.; Takano, A.; Koinuma, J.; Aragaki, M.; Masuda, K.; Ishikawa, N.; Kohno, N.; Ito, H.; Miyamoto, M.; et al. Wnt Inhibitor Dickkopf-1 as a Target for Passive Cancer Immunotherapy. Cancer Res. 2010, 70, 5326–5336. [Google Scholar] [CrossRef] [PubMed]
- Kagey, M.H.; He, X. Rationale for targeting the Wnt signalling modulator Dickkopf-1 for oncology. Br. J. Pharm. 2017, 174, 4637–4650. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Seo, E.-M.; Sharma, A.R.; Ganbold, B.; Park, J.; Sharma, G.; Kang, Y.-H.; Song, D.-K.; Lee, S.-S.; Nam, J.-S. Regulation of Wnt signaling activity for growth suppression induced by quercetin in 4T1 murine mammary cancer cells. Int. J. Oncol. 2013, 43, 1319–1325. [Google Scholar] [CrossRef]
- Wang, B.-E.; Wang, X.-D.; Ernst, J.A.; Polakis, P.; Gao, W.Q. Regulation of Epithelial Branching Morphogenesis and Cancer Cell Growth of the Prostate by Wnt Signaling. PLoS ONE 2008, 3, e2186. [Google Scholar] [CrossRef]
- Aufderklamm, S.; Hennenlotter, J.; Leidenberger, P.; Rausch, S.; Hohneder, A.; Kühs, U.; Maas, M.; Schwentner, C.; Bedke, J.; Stenzl, A.; et al. Systemic Alterations of Wnt Inhibitors in Patients with Prostate Cancer and Bone Metastases. Dis. Markers 2018, 2018, 1–5. [Google Scholar] [CrossRef]
- Hall, C.L.; Daignault, S.D.; Shah, R.B.; Pienta, K.J.; Keller, E.T. Dickkopf-1 expression increases early in prostate cancer development and decreases during progression from primary tumor to metastasis. Prostate 2008, 68, 1396–1404. [Google Scholar] [CrossRef]
- Hall, C.L.; Zhang, H.; Baile, S.; Ljungman, M.; Kuhstoss, S.; Keller, E.T. p21CIP-1/WAF-1 Induction Is Required to Inhibit Prostate Cancer Growth Elicited by Deficient Expression of the Wnt Inhibitor Dickkopf-1. Cancer Res. 2010, 70, 9916–9926. [Google Scholar] [CrossRef]
- Yavropoulou, M.P.; van Lierop, A.H.; Hamdy, N.A.; Rizzoli, R.; Papapoulos, S.E. Serum sclerostin levels in Paget’s disease and prostate cancer with bone metastases with a wide range of bone turnover. Bone 2012, 51, 153–157. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choudhary, S.; Ramasundaram, P.; Dziopa, E.; Mannion, C.; Kissin, Y.; Tricoli, L.; Albanese, C.; Lee, W.; Zilberberg, J. Human ex vivo 3D bone model recapitulates osteocyte response to metastatic prostate cancer. Sci. Rep. 2018, 8, 17975. [Google Scholar] [CrossRef] [PubMed]
- Sottnik, J.L.; Hall, C.L.; Zhang, J.; Keller, E.T. Wnt and Wnt inhibitors in bone metastasis. BoneKEy Rep. 2012, 1, 101. [Google Scholar] [CrossRef] [PubMed]
- Mansinho, A.; Ferreira, A.R.; Casimiro, S.; Alho, I.; Vendrell, I.; Costa, A.L.; Sousa, R.; Abreu, C.; Pulido, C.; Macedo, D.; et al. Levels of Circulating Fibroblast Growth Factor 23 (FGF23) and Prognosis in Cancer Patients with Bone Metastases. Int. J. Mol. Sci. 2019, 20, 695. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Wang, J.; Zhang, Y.; Creighton, C.J.; Ittmann, M. FGF23 promotes prostate cancer progression. Oncotarget 2015, 6, 17291–17301. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, K.H.; Lee, J.; Oh, J.J.; Cheong, H.S.; Wong, E.L.; Yang, B.S.; Byun, S.S.; Myung, S.C. Single nucleotide polymorphisms in fibroblast growth factor 23 gene, FGF23, are associated with prostate cancer risk. BJU Int. 2014, 114, 303–310. [Google Scholar] [CrossRef]
- Bultynck, G. The anti-metastatic micro-environment of the bone: Importance of osteocyte Cx43 hemichannels. Biochim. Biophys. Acta 2016, 1866, 121–127. [Google Scholar] [CrossRef]
- Zhou, J.Z.; Riquelme, M.A.; Gu, S.; Kar, R.; Gao, X.; Sun, L.; Jiang, J.X. Osteocytic connexin hemichannels suppress breast cancer growth and bone metastasis. Oncogene 2016, 35, 5597–5607. [Google Scholar] [CrossRef]
- Bivi, N.; Condon, K.W.; Allen, M.R.; Farlow, N.; Passeri, G.; Brun, L.R.; Rhee, Y.; Bellido, T.; Plotkin, L.I. Cell autonomous requirement of connexin 43 for osteocyte survival: Consequences for endocortical resorption and periosteal bone formation. J. Bone Min. Res. 2012, 27, 374–389. [Google Scholar] [CrossRef]
- Bonewald, L.F.; Johnson, M.L. Osteocytes, mechanosensing and Wnt signaling. Bone 2008, 42, 606–615. [Google Scholar] [CrossRef]


{kind=link}
| Study | Effects on Breast Cancer | Effects on Prostate Cancer |
|---|---|---|
| Osteocyte-derived conditioned medium |  proliferation, migration, and invasion [41] proliferation, migration, and invasion [41] | proliferation, migration, and invasion [41] |
| Tumor 3D spheroids grown in presence of osteocytes spheroids | Tumor shrank [42] MET via down-regulation of Snail [43] | |
| 3D microfluidic tissue |  Cancer cells extravasation [45] Cancer cells extravasation [45] | |
| MMP9 expression and invasion of cancer cells [46] Interaction breast cancer cells/osteoclasts/endothelial cells [47] | ||
| Mechanical loading of osteocytes (MLO) |
Low level MLO: OPN, tumor migration; Medium level MLO: OPN, tumor migration [48] | |
| In vitro and in vivo experiments | Growth and invasion via GDF15 [50] | |
| Osteocytes under hydrostatic pressure | viability, invasion, and migration via RANTES and MMPs [51] | viability, invasion, and migration via RANTES and MMPs [51] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maroni, P.; Bendinelli, P. Bone, a Secondary Growth Site of Breast and Prostate Carcinomas: Role of Osteocytes. Cancers 2020, 12, 1812. https://doi.org/10.3390/cancers12071812
Maroni P, Bendinelli P. Bone, a Secondary Growth Site of Breast and Prostate Carcinomas: Role of Osteocytes. Cancers. 2020; 12(7):1812. https://doi.org/10.3390/cancers12071812
Chicago/Turabian StyleMaroni, Paola, and Paola Bendinelli. 2020. "Bone, a Secondary Growth Site of Breast and Prostate Carcinomas: Role of Osteocytes" Cancers 12, no. 7: 1812. https://doi.org/10.3390/cancers12071812
APA StyleMaroni, P., & Bendinelli, P. (2020). Bone, a Secondary Growth Site of Breast and Prostate Carcinomas: Role of Osteocytes. Cancers, 12(7), 1812. https://doi.org/10.3390/cancers12071812