Machine Learning-Based Ensemble Recursive Feature Selection of Circulating miRNAs for Cancer Tumor Classification

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Feature Selection
| Algorithm 1: Recursive ensemble feature selection. |
 |
2.2. Cancer Type Classification
2.3. Triple-Negative Breast Cancer Classification
3. Results
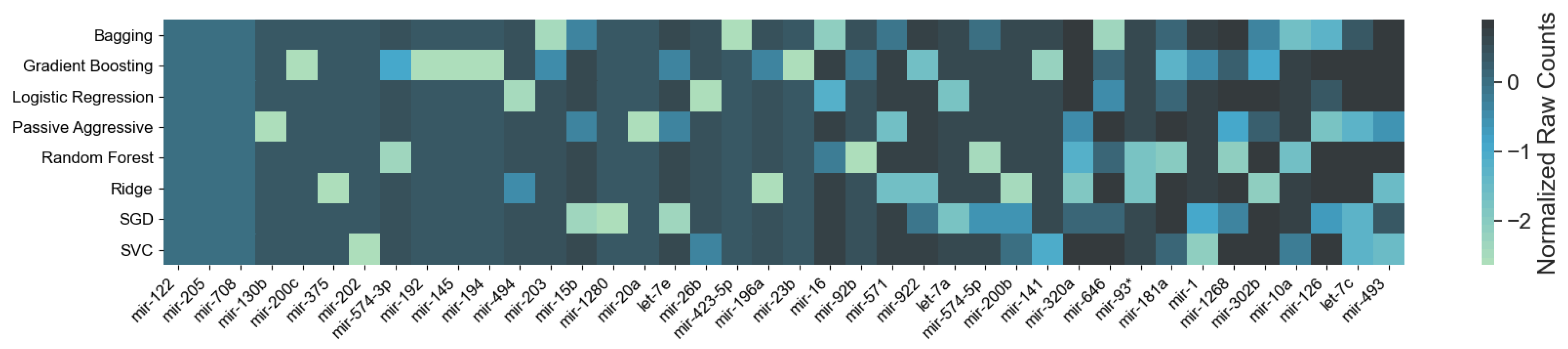
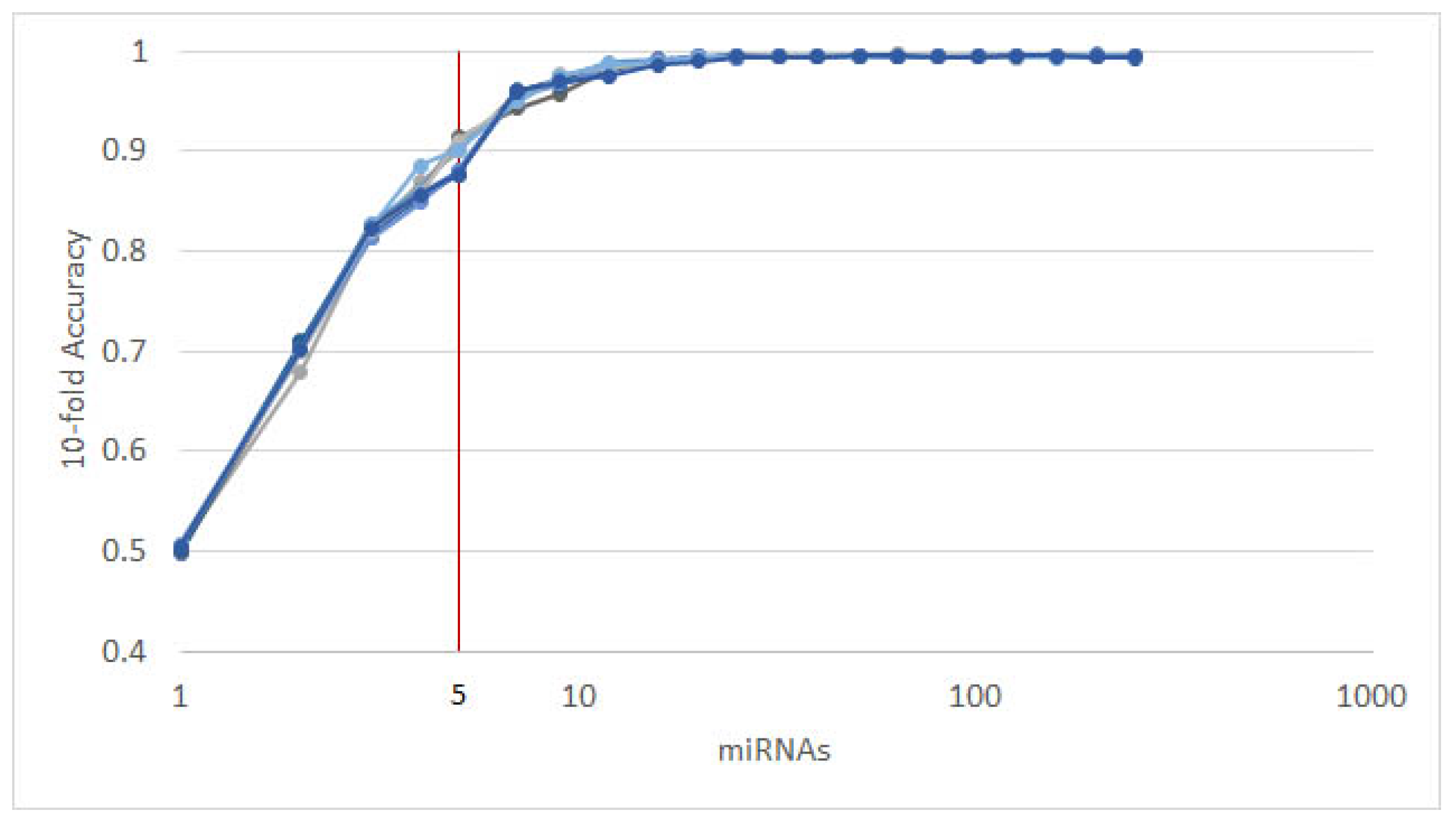
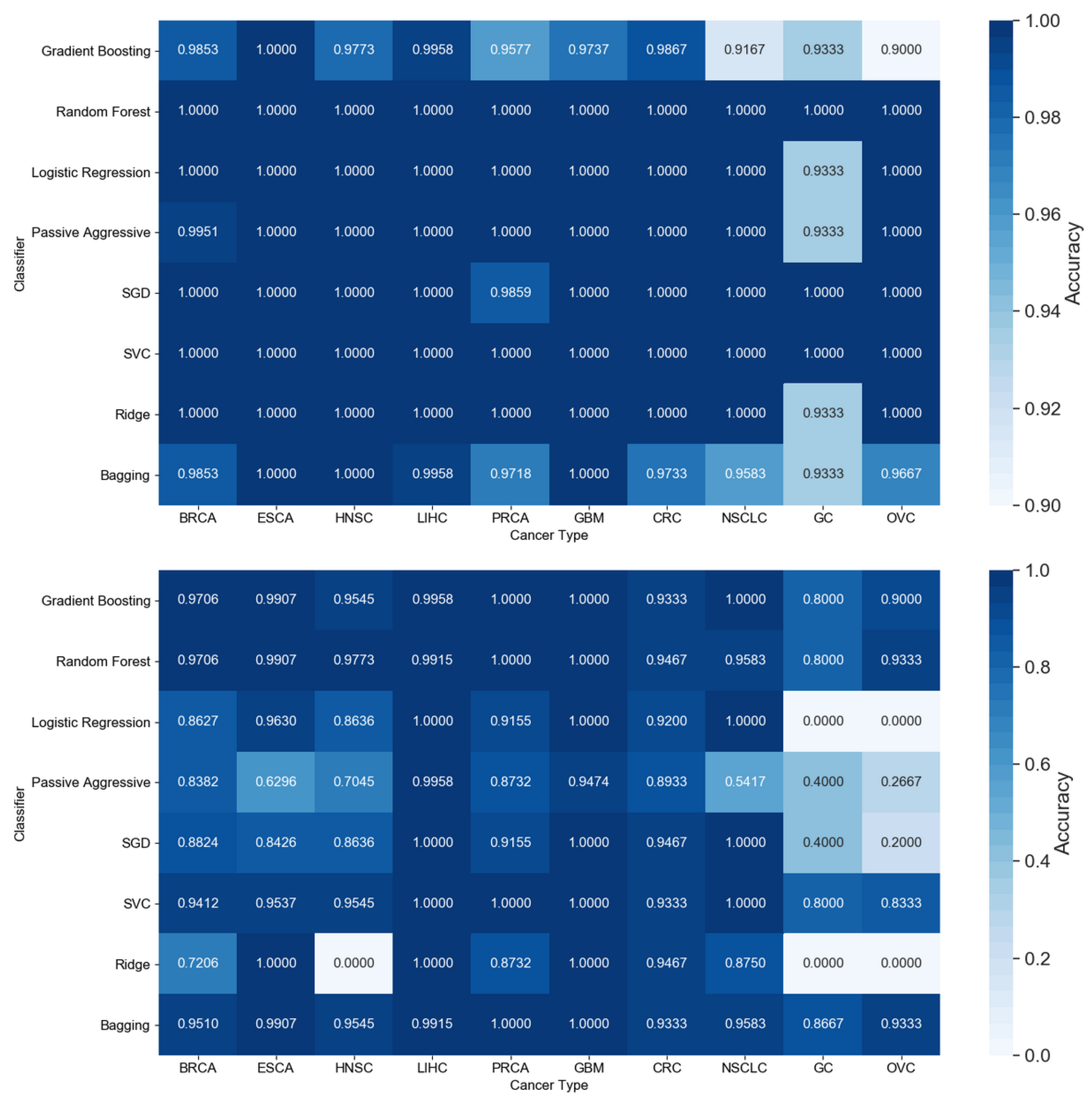
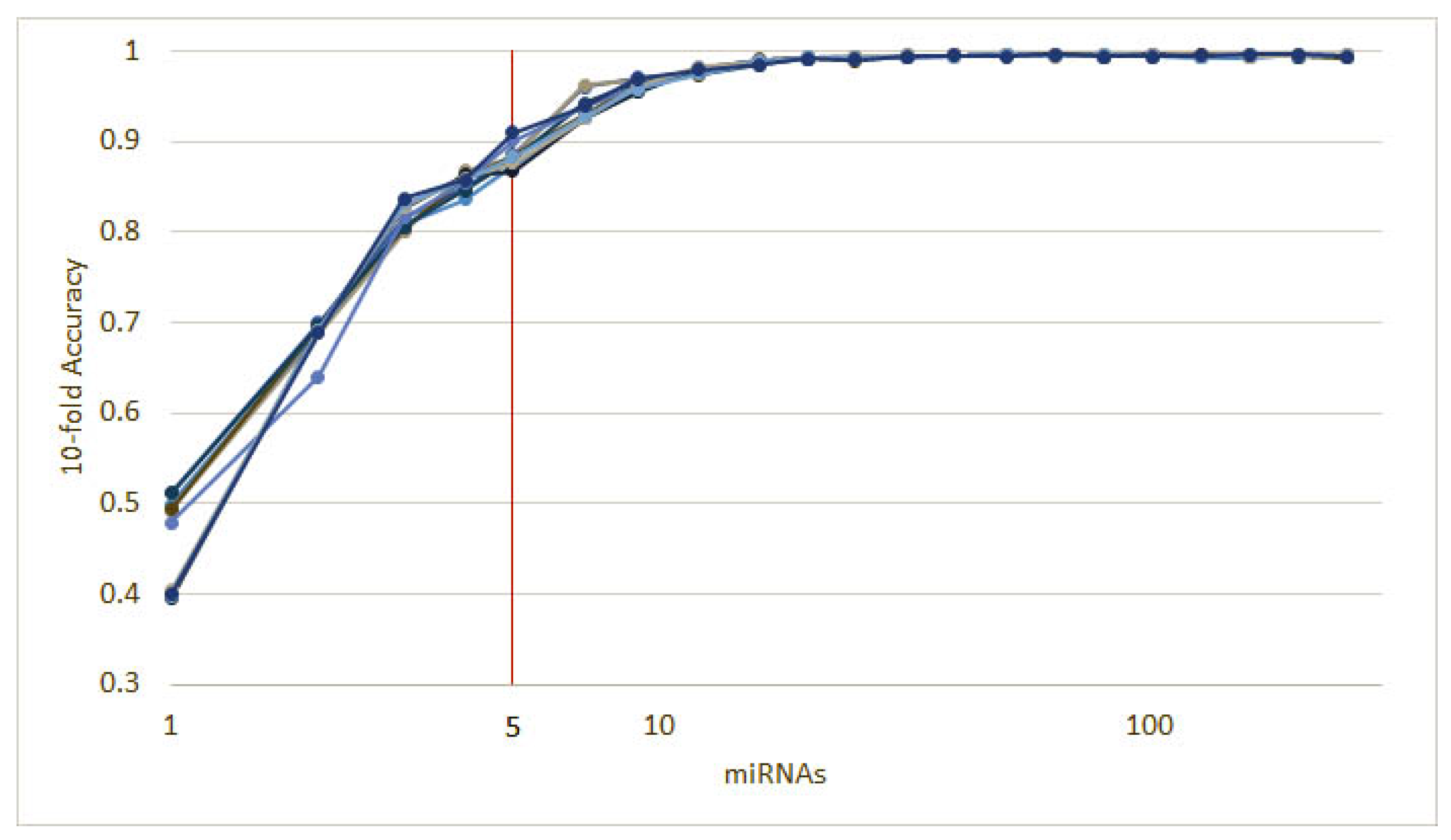
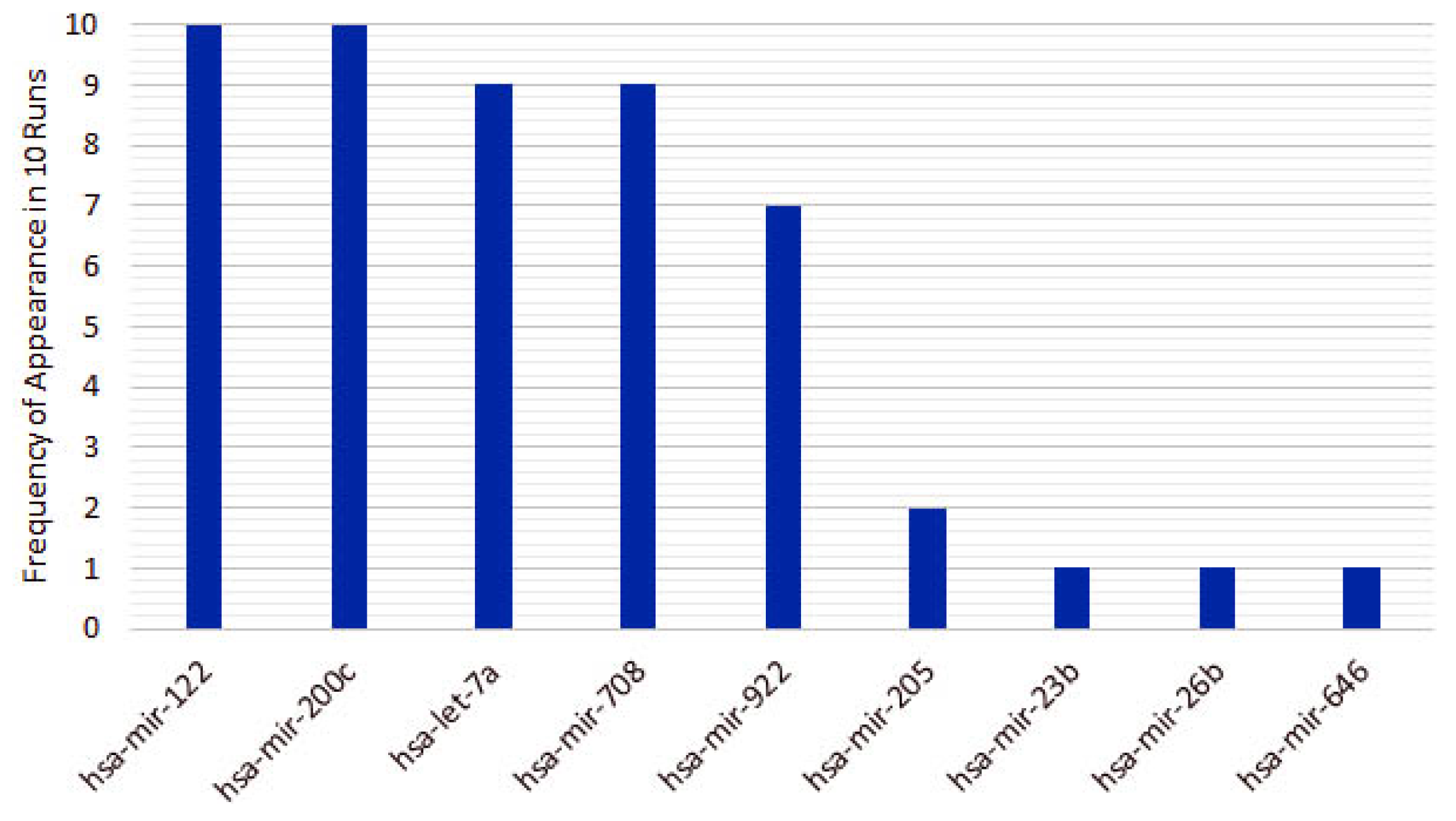
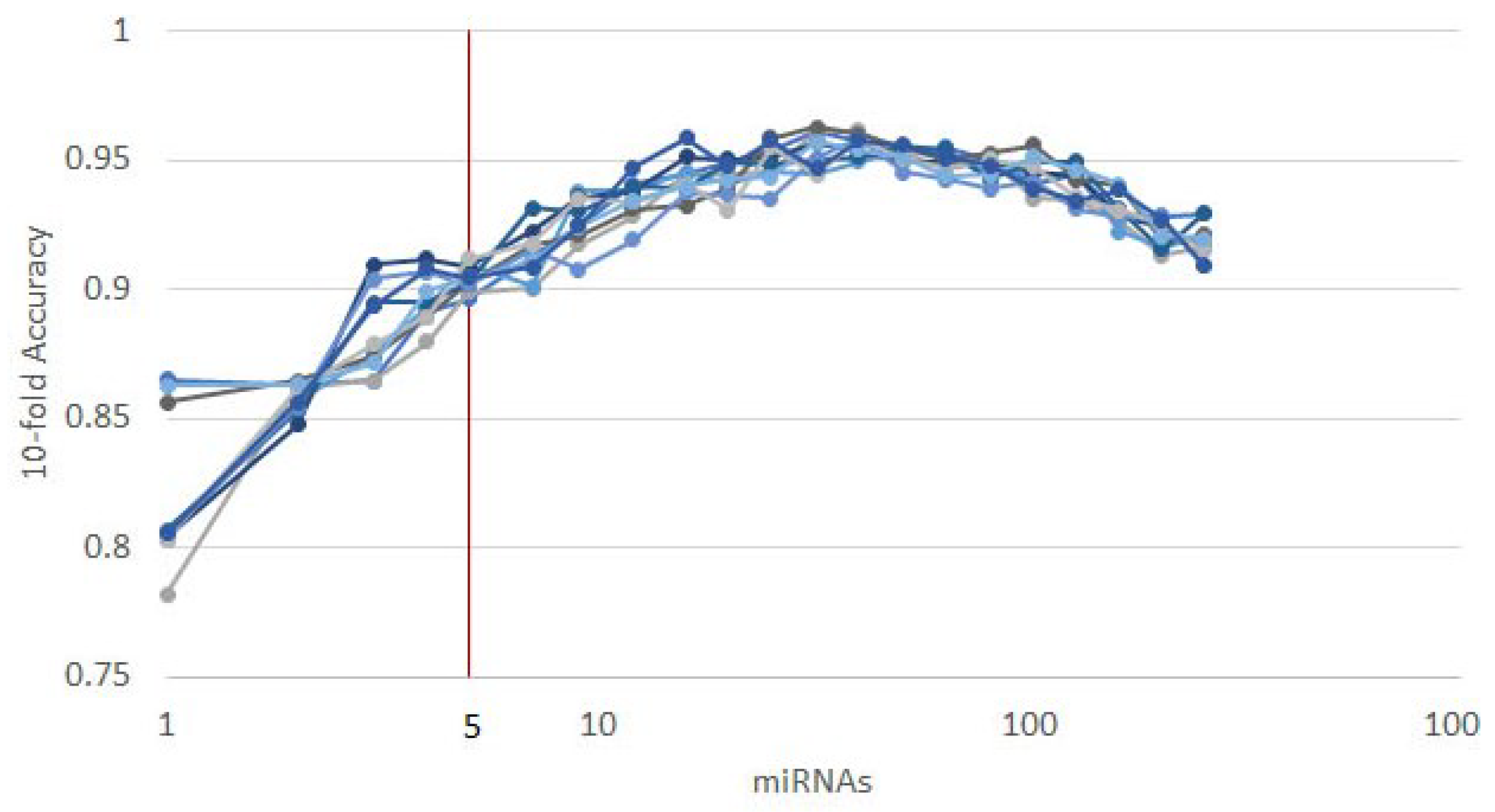
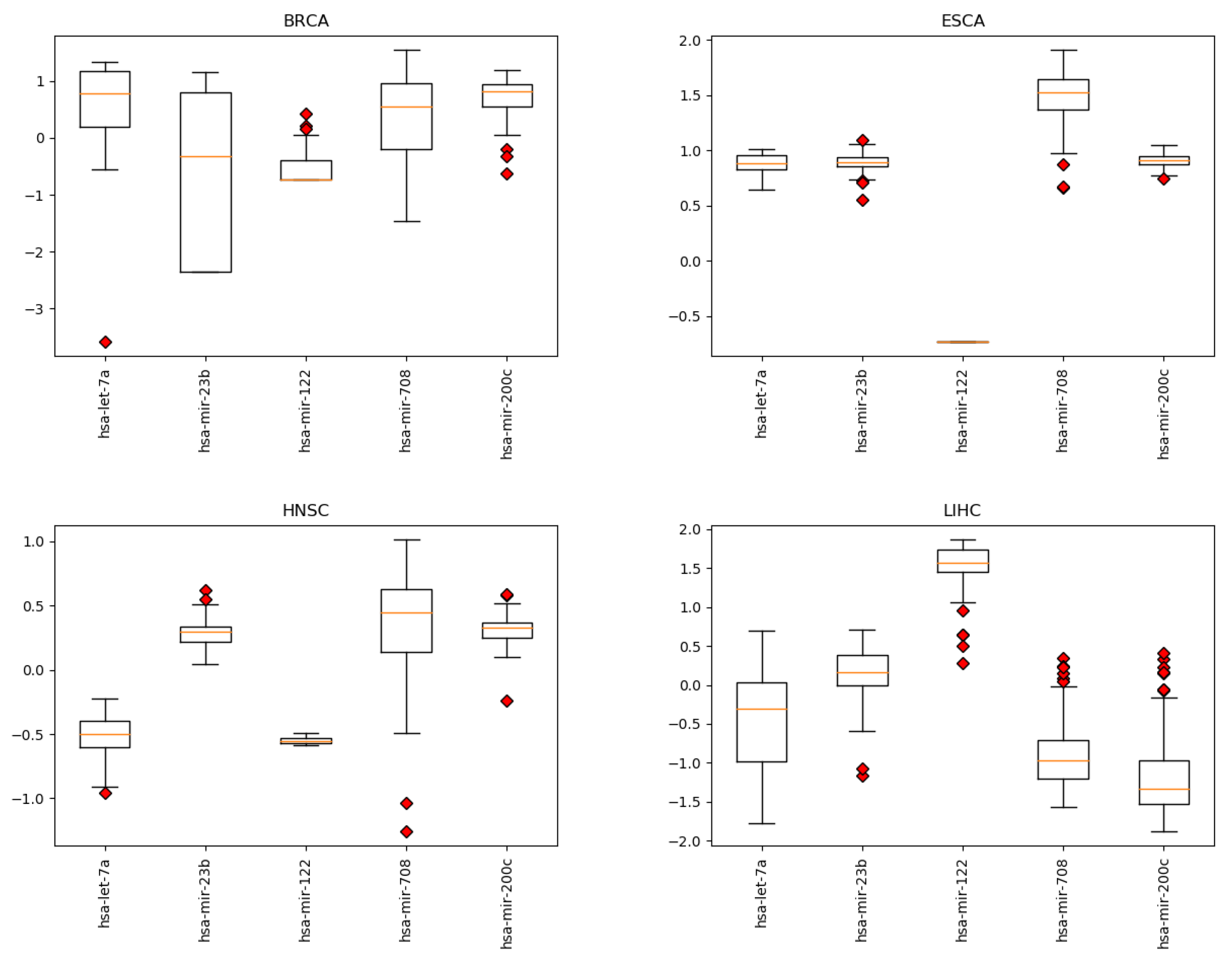
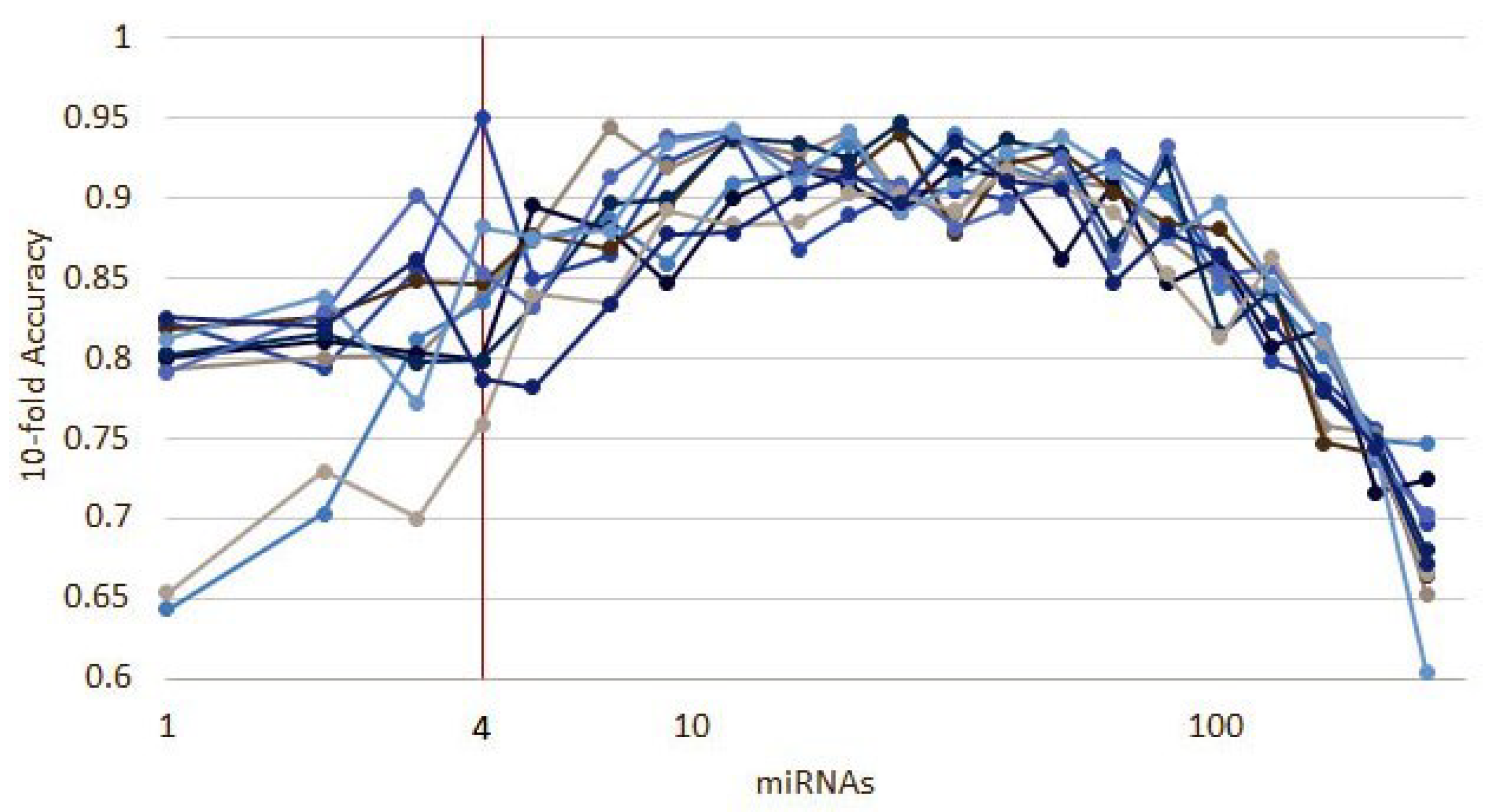
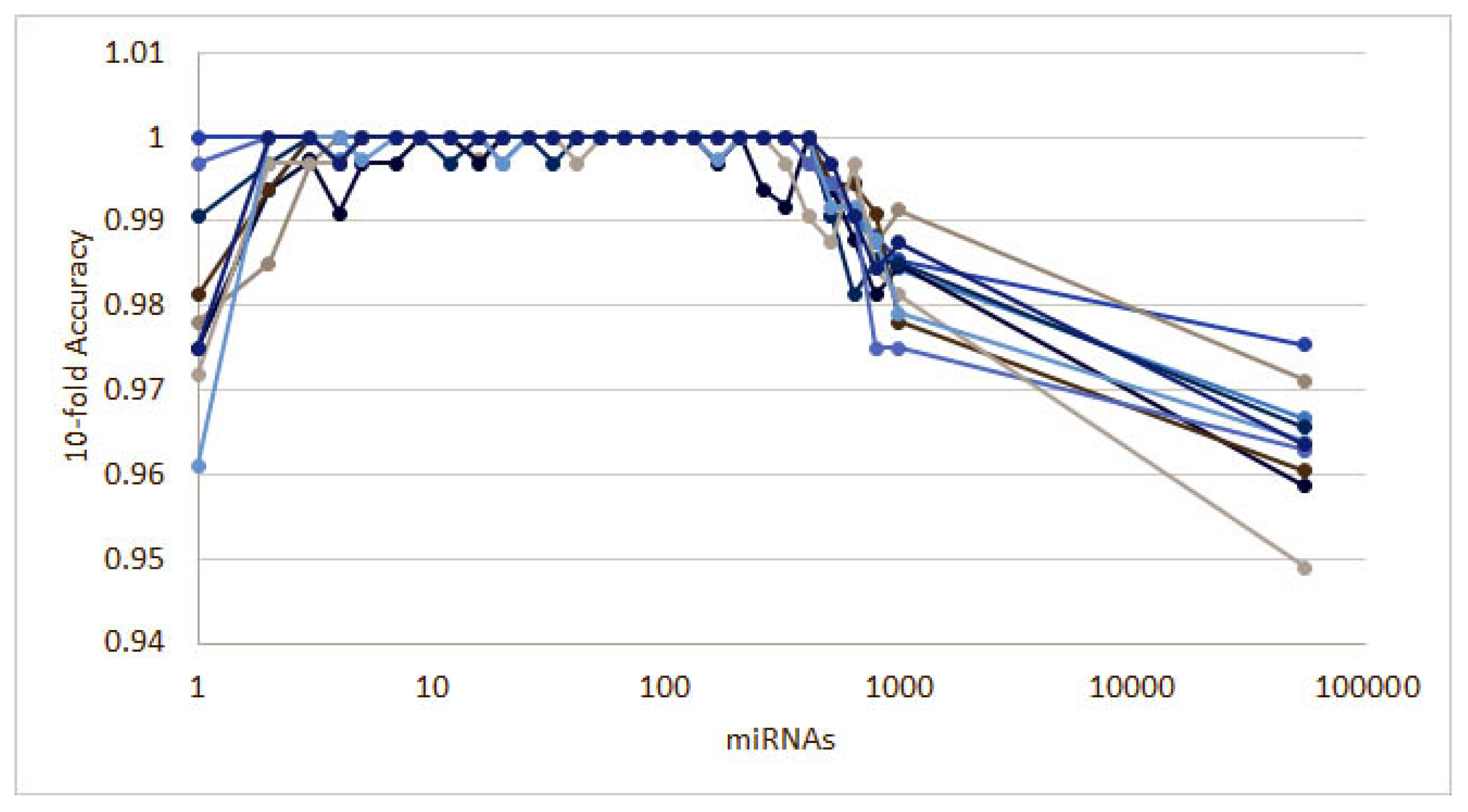
3.1. Cancer Type Classification
3.1.1. Numerical Validation
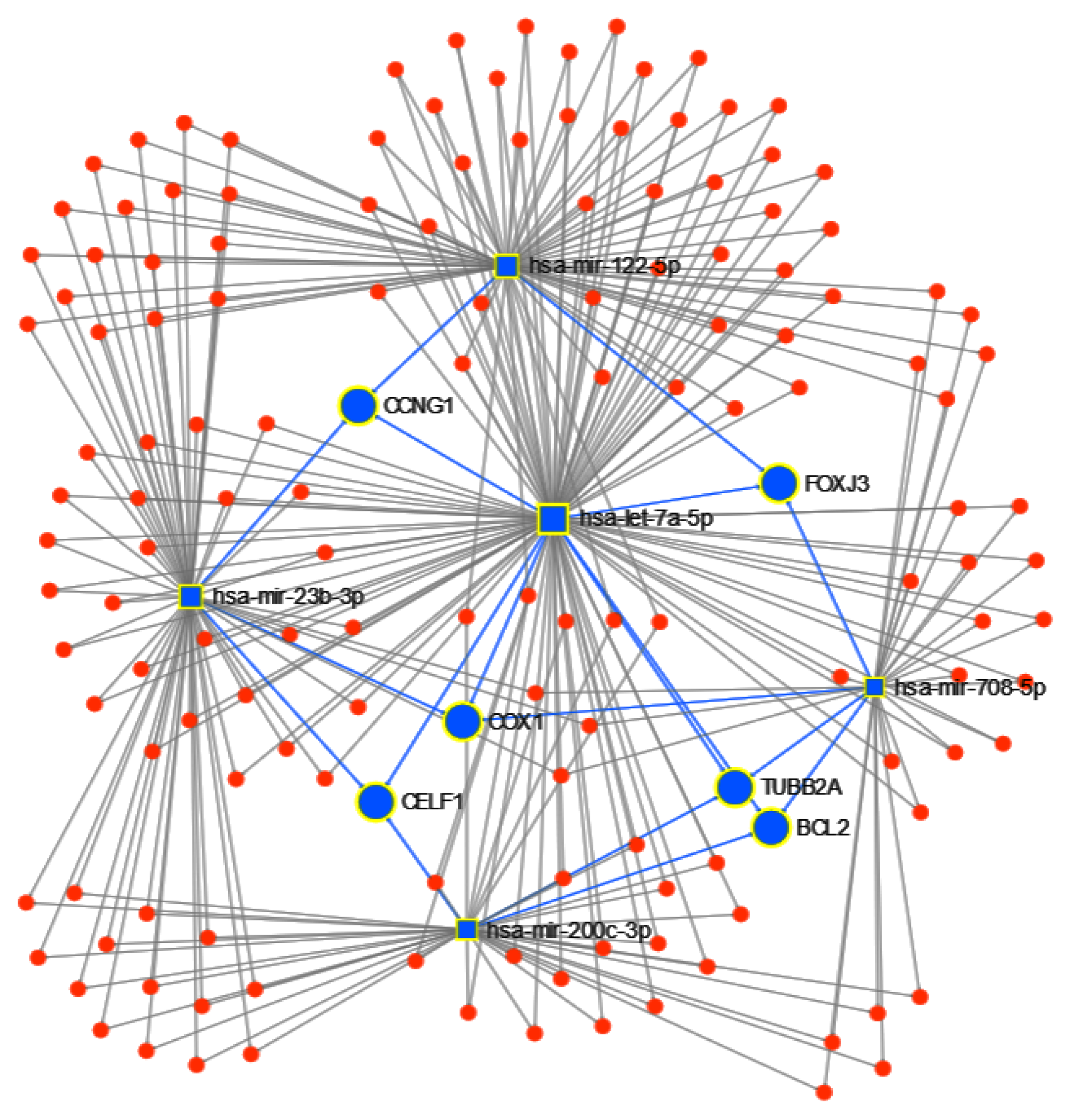
3.1.2. Pathway Analysis
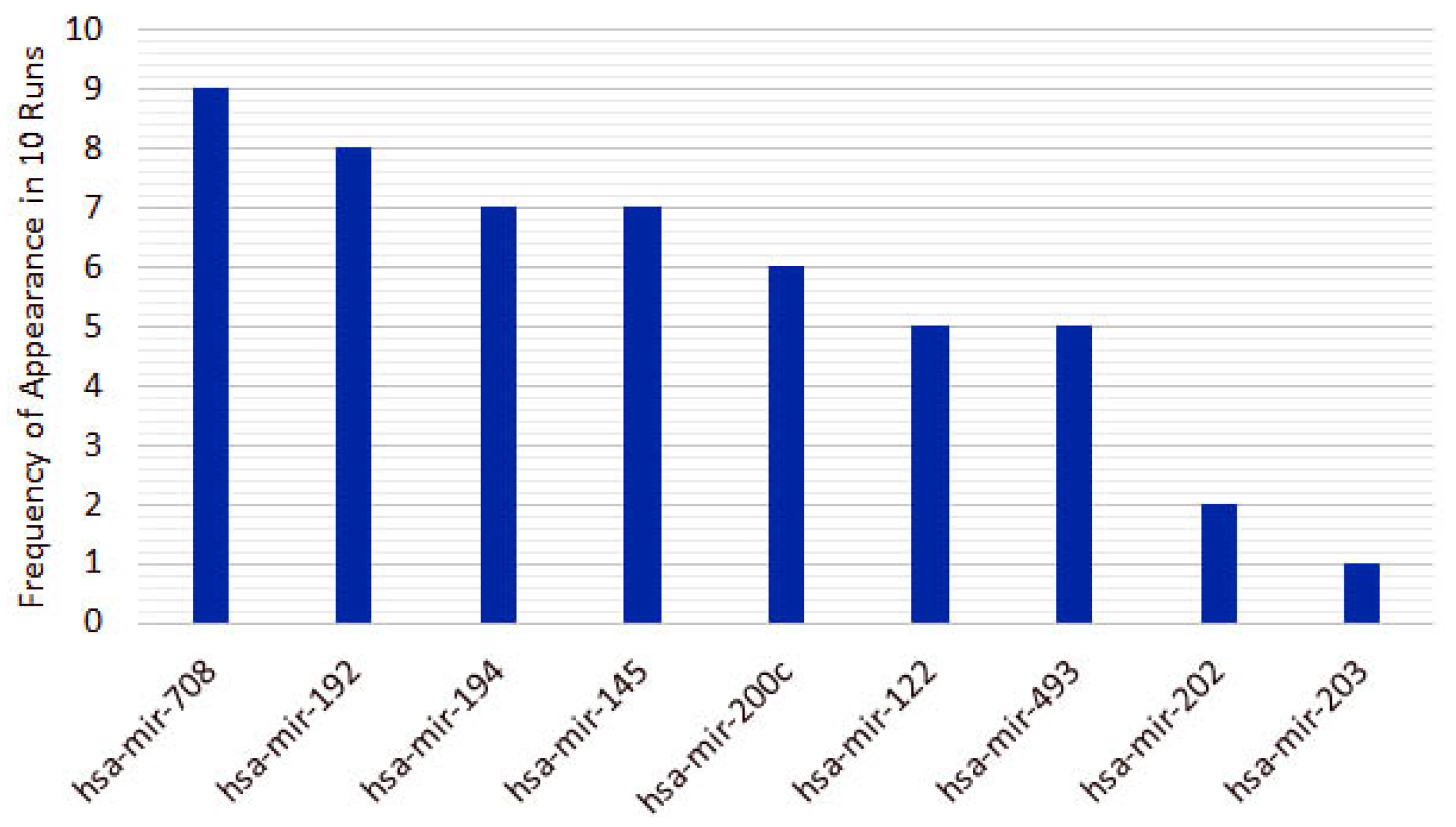
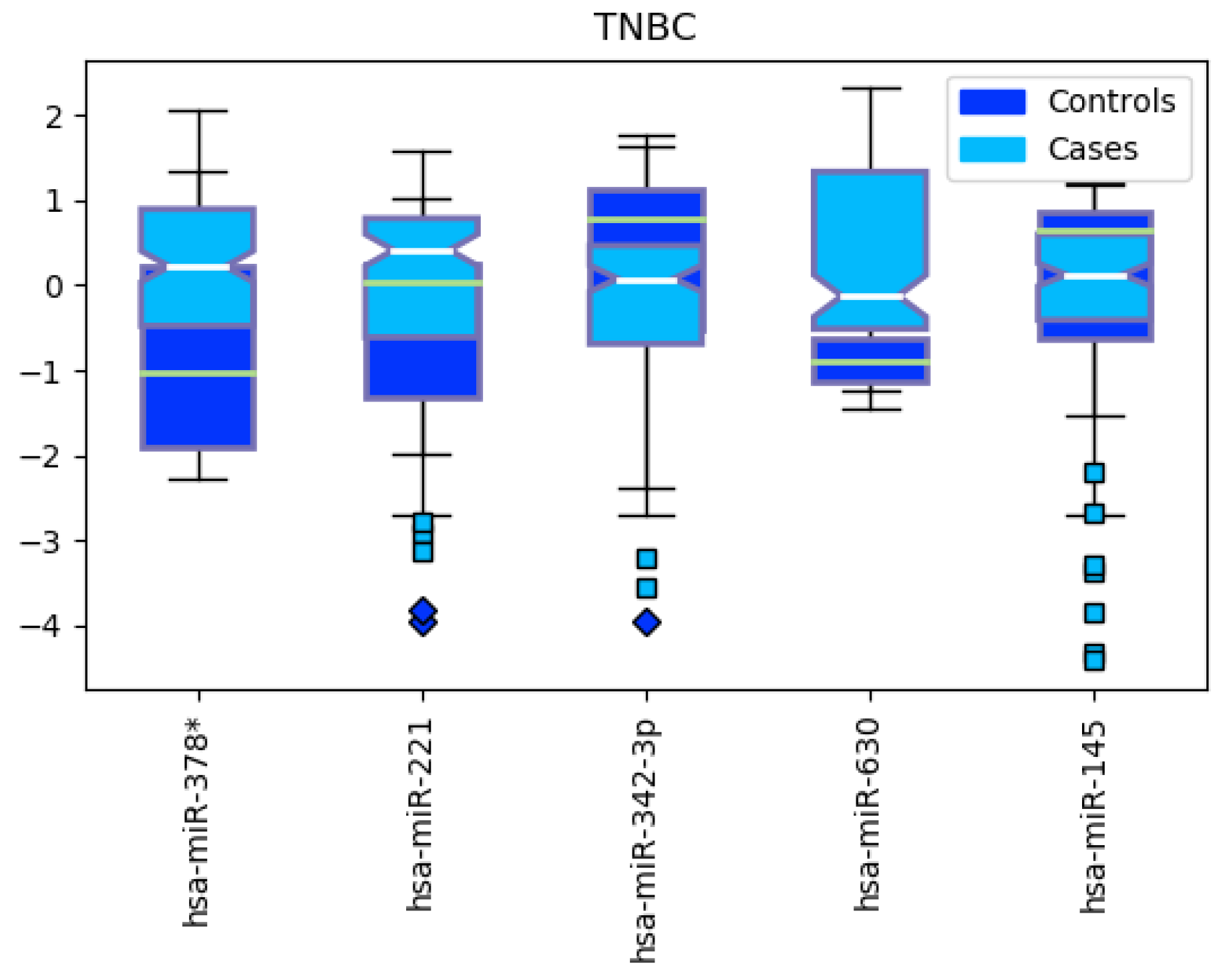
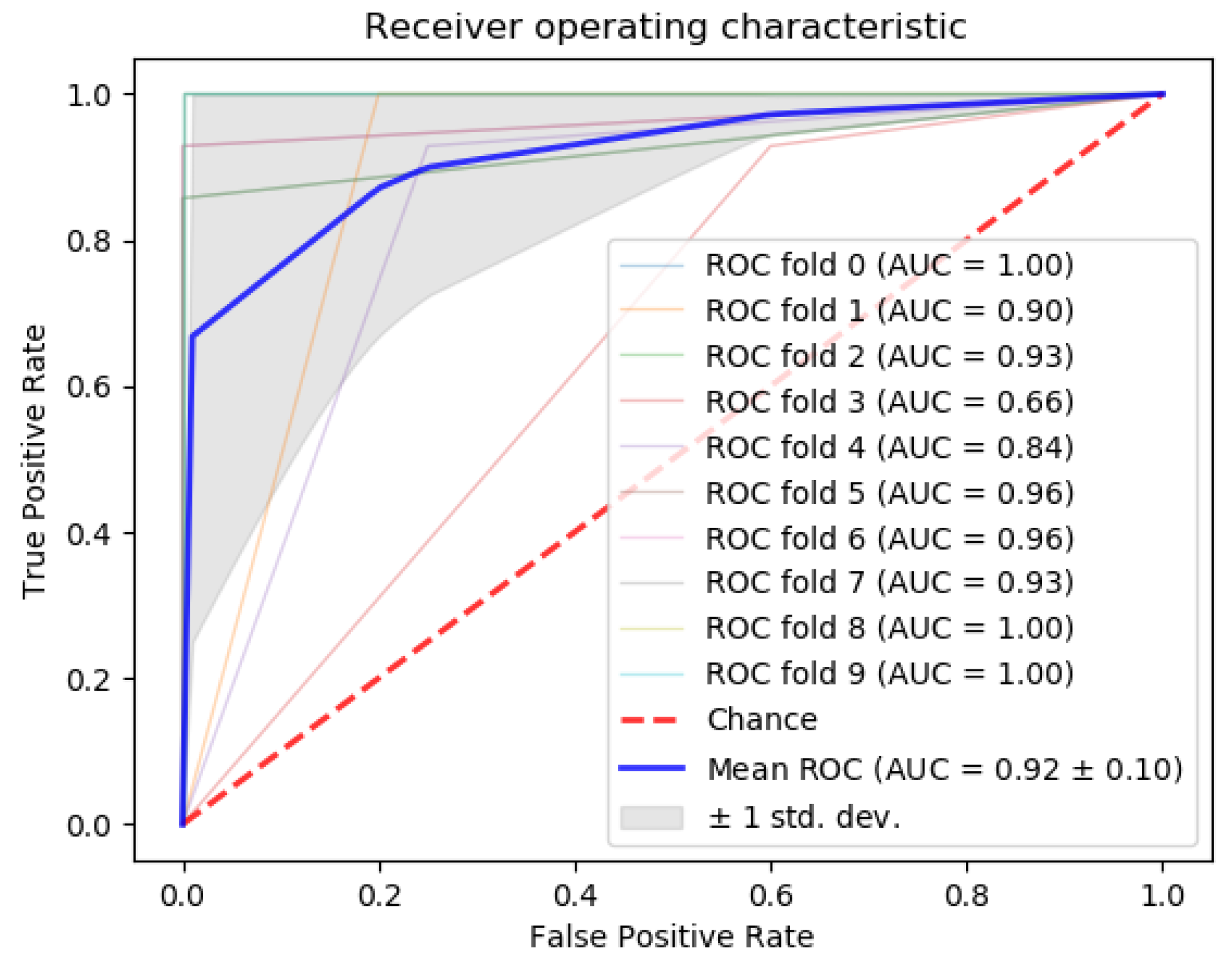
3.2. Triple-Negative Breast Cancer Classification
4. Discussion
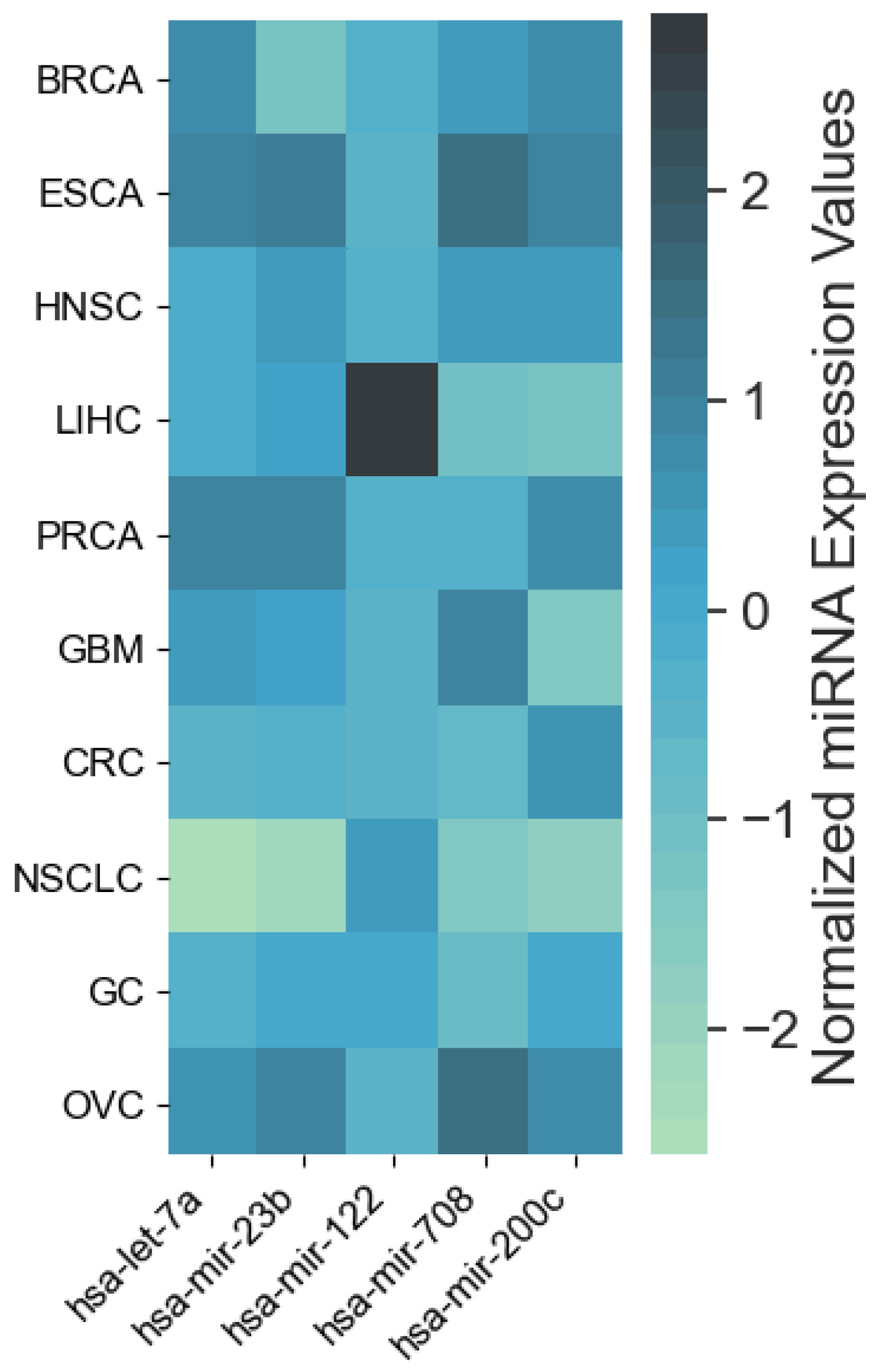
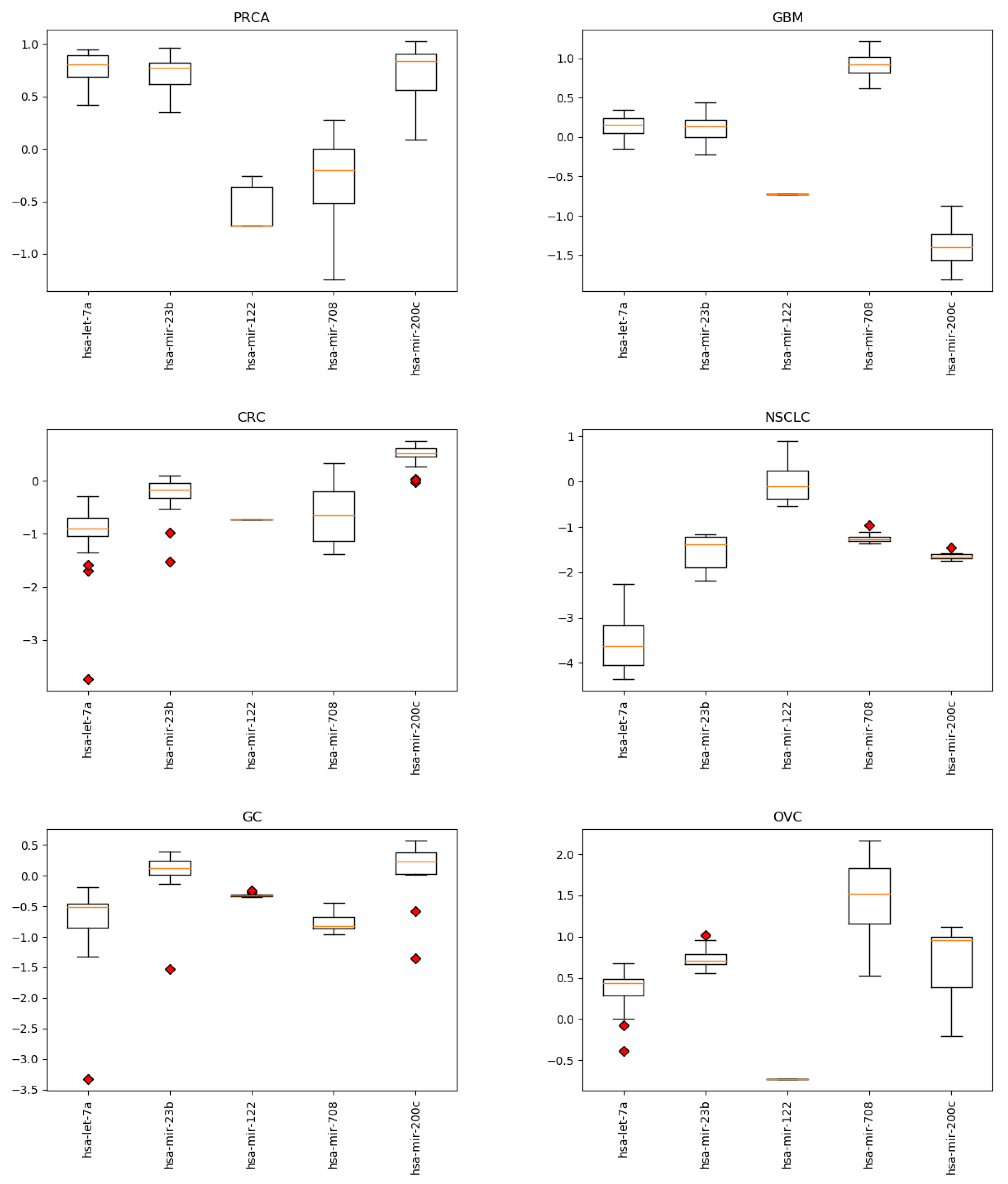
4.1. miRNAs from Cancer Type Classification
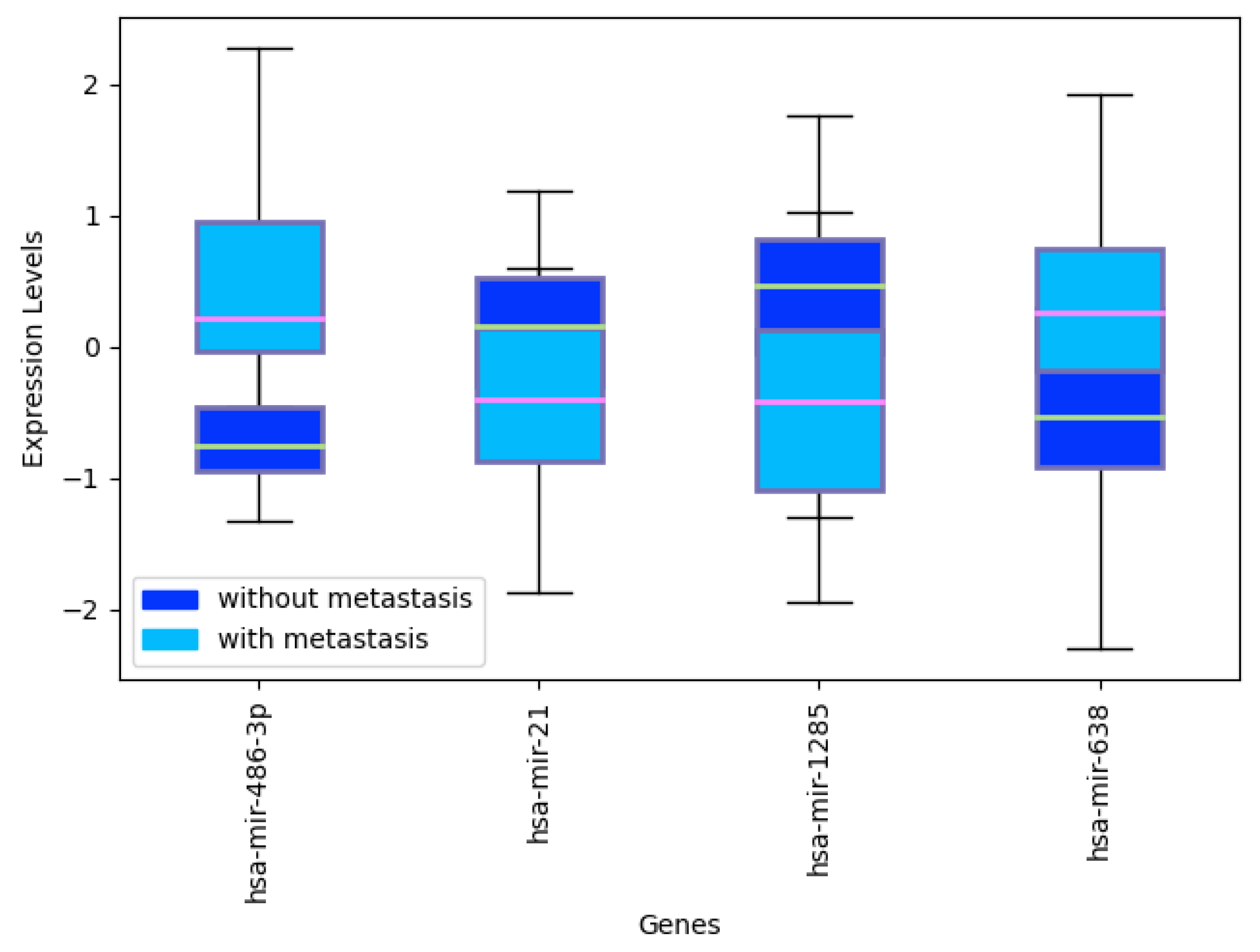
4.2. miRNAs from Triple-Negative Breast Cancer Classification
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Sample Availability
Appendix A. Circulating miRNAs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| let-7a | miR-140-3p | miR-19b | miR-335 | miR-513a-3p |
| let-7a* | miR-141 | miR-200a | miR-338-3p | miR-516b |
| let-7b | miR-142-3p | miR-200b | miR-338-5p | miR-518b |
| let-7c | miR-143 | miR-200c | miR-339-3p | miR-520a-3p |
| let-7d | miR-144 | miR-202 | miR-339-5p | miR-548b-5p |
| let-7e | miR-145 | miR-203 | miR-340* | miR-557 |
| let-7f | miR-146a | miR-205 | miR-342-3p | miR-564 |
| let-7g | miR-146b-3p | miR-206 | miR-345 | miR-566 |
| let-7i | miR-146b-5p | miR-20a | miR-346 | miR-571 |
| miR-1 | miR-148a | miR-20b | miR-34a | miR-574-3p |
| miR-100 | miR-148b | miR-21 | miR-34b | miR-574-5p |
| miR-101 | miR-150 | miR-210 | miR-361-3p | miR-587 |
| miR-106b | miR-150* | miR-212 | miR-365 | miR-589 |
| miR-107 | miR-151-5p | miR-214 | miR-371-5p | miR-595 |
| miR-10a | miR-152 | miR-215 | miR-372 | miR-601 |
| miR-10b | miR-155 | miR-218 | miR-373 | miR-616* |
| miR-1182 | miR-15a | miR-22 | miR-375 | miR-618 |
| miR-122 | miR-15b | miR-221 | miR-376a | miR-622 |
| miR-122* | miR-15b* | miR-222 | miR-376c | miR-625 |
| miR-1224-5p | miR-16 | miR-223 | miR-377 | miR-625* |
| miR-1229 | miR-16-2* | miR-23a | miR-378 | miR-628-3p |
| miR-1231 | miR-17 | miR-23b | miR-378* | miR-629 |
| miR-1245 | miR-181a | miR-24 | miR-379 | miR-630 |
| miR-1246 | miR-181a-2* | miR-25 | miR-382 | miR-638 |
| miR-1254 | miR-181b | miR-26a | miR-409-3p | miR-646 |
| miR-125b | miR-181d | miR-26b | miR-409-5p | miR-650 |
| miR-125b-2* | miR-182 | miR-27a | miR-410 | miR-652 |
| miR-126 | miR-1825 | miR-27b | miR-411 | miR-654-3p |
| miR-1260 | miR-183 | miR-296-5p | miR-421 | miR-656 |
| miR-1268 | miR-184 | miR-298 | miR-423-5p | miR-668 |
| miR-127-3p | miR-185 | miR-299-5p | miR-425 | miR-675 |
| miR-1275 | miR-186 | miR-29a | miR-425* | miR-7 |
| miR-128 | miR-187 | miR-29b | miR-429 | miR-708 |
| miR-1280 | miR-187* | miR-29c | miR-431 | miR-744 |
| miR-1284 | miR-18a | miR-301a | miR-431* | miR-744* |
| miR-1285 | miR-18b | miR-302b | miR-432 | miR-760 |
| miR-1288 | miR-18b* | miR-30a | miR-451 | miR-874 |
| miR-1290 | miR-190b | miR-30b | miR-452 | miR-885-5p |
| miR-1295 | miR-191 | miR-30c | miR-454 | miR-922 |
| miR-129-5p | miR-192 | miR-30c-1* | miR-454* | miR-92a |
| miR-1304 | miR-193a-3p | miR-30d | miR-483-3p | miR-92a-2* |
| miR-130a* | miR-193b | miR-30e | miR-483-5p | miR-92b |
| miR-130b | miR-194 | miR-31 | miR-484 | miR-93 |
| miR-1323 | miR-195 | miR-32 | miR-486-3p | miR-93* |
| miR-133a | miR-196a | miR-320a | miR-486-5p | miR-936 |
| miR-133b | miR-196b | miR-320c | miR-487b | miR-939 |
| miR-134 | miR-197 | miR-320d | miR-493 | miR-942 |
| miR-138 | miR-198 | miR-324-3p | miR-494 | miR-99a |
| miR-138-2* | miR-199a-3p | miR-326 | miR-497 | miR-99b |
| miR-139-3p | miR-199a-5p | miR-328 | miR-502-5p | |
| miR-139-5p | miR-19a | miR-331-3p | miR-504 |
Appendix B. miRNA Levels


Appendix C. miRNA Levels in CRC with and without Metastasis


Appendix D. Recursive Ensemble Feature Selection in mRNA Data


References
- Larrea, E.; Sole, C.; Manterola, L.; Goicoechea, I.; Armesto, M.; Arestin, M.; Caffarel, M.M.; Araujo, A.M.; Araiz, M.; Fernandez-Mercado, M.; et al. New concepts in cancer biomarkers: Circulating miRNAs in liquid biopsies. Int. J. Mol. Sci. 2016, 17, 627. [Google Scholar] [CrossRef]
- He, Y.; Lin, J.; Kong, D.; Huang, M.; Xu, C.; Kim, T.K.; Etheridge, A.; Luo, Y.; Ding, Y.; Wang, K. Current state of circulating microRNAs as cancer biomarkers. Clin. Chem. 2015, 61, 1138–1155. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Jeon, K.; Lee, J.T.; Kim, S.; Kim, V.N. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J. 2002, 21, 4663–4670. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.N. MicroRNA biogenesis: Coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol. 2005, 6, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Price, C.; Chen, J. MicroRNAs in cancer biology and therapy: Current status and perspectives. Genes Dis. 2014, 1, 53–63. [Google Scholar] [CrossRef]
- Han, J.; Lee, Y.; Yeom, K.H.; Kim, Y.K.; Jin, H.; Kim, V.N. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 18, 3016–3027. [Google Scholar] [CrossRef]
- Cheng, C.; Wang, Q.; You, W.; Chen, M.; Xia, J. MiRNAs as biomarkers of myocardial infarction: A meta-analysis. PLoS ONE 2014, 9, e88566. [Google Scholar] [CrossRef]
- Huang, J.T.; Wang, J.; Srivastava, V.; Sen, S.; Liu, S.M. MicroRNA machinery genes as novel biomarkers for cancer. Front. Oncol. 2014, 4, 113. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Jin, M.; Li, J.; Kong, X. Identifying circulating miRNA biomarkers for early diagnosis and monitoring of lung cancer. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 165847. [Google Scholar] [CrossRef]
- Zheng, D.; Ding, Y.; Ma, Q.; Zhao, L.; Guo, X.; Shen, Y.; He, Y.; Wei, W.; Liu, F. Identification of serum microRNAs as novel biomarkers in esophageal squamous cell carcinoma using feature selection algorithms. Front. Oncol. 2019, 8, 674. [Google Scholar] [CrossRef]
- Fehlmann, T.; Kahraman, M.; Ludwig, N.; Backes, C.; Galata, V.; Keller, V.; Geffers, L.; Mercaldo, N.; Hornung, D.; Weis, T.; et al. Evaluating the Use of Circulating MicroRNA Profiles for Lung Cancer Detection in Symptomatic Patients. JAMA Oncol. 2020, 6, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.; Feng, C.; Song, C.; Liu, W.; Shang, D.; Li, M.; Wang, Q.; Zhao, J.; Liu, Y.; Chen, J.; et al. Topologically inferring active miRNA-mediated subpathways toward precise cancer classification by directed random walk. Mol. Oncol. 2019, 13, 2211–2226. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Cheng, H.H.; Tewari, M. MicroRNA profiling: Approaches and considerations. Nat. Rev. Genet. 2012, 13, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Dutta, A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007, 21, 1025–1030. [Google Scholar] [CrossRef] [PubMed]
- Al-Sheikh, Y.A.; Ghneim, H.K.; Softa, K.I.; Al-Jobran, A.A.; Al-Obeed, O.; Mohamed, M.A.; Abdulla, M.; Aboul-Soud, M.A. Expression profiling of selected microRNA signatures in plasma and tissues of Saudi colorectal cancer patients by qPCR. Oncol. Lett. 2016, 11, 1406–1412. [Google Scholar] [CrossRef]
- Adam, L.; Wszolek, M.F.; Liu, C.G.; Jing, W.; Diao, L.; Zien, A.; Zhang, J.D.; Jackson, D.; Dinney, C.P. Plasma microRNA profiles for bladder cancer detection. In Urologic Oncology: Seminars and Original Investigations; Elsevier: Amsterdam, The Netherlands, 2013; Volume 31, pp. 1701–1708. [Google Scholar]
- Liao, Z.; Li, D.; Wang, X.; Li, L.; Zou, Q. Cancer diagnosis through IsomiR expression with machine learning method. Curr. Bioinform. 2018, 13, 57–63. [Google Scholar] [CrossRef]
- Rincon, A.L.; Tonda, A.; Elati, M.; Schwander, O.; Piwowarski, B.; Gallinari, P. Evolutionary Optimization of Convolutional Neural Networks for Cancer miRNA Biomarkers Classification. Appl. Soft Comput. 2018. [Google Scholar] [CrossRef]
- Yang, S.; Guo, L.; Shao, F.; Zhao, Y.; Chen, F. A systematic evaluation of feature selection and classification algorithms using simulated and real miRNA sequencing data. Comput. Math. Methods Med. 2015, 2015. [Google Scholar] [CrossRef]
- Saha, S.; Mitra, S.; Yadav, R.K. A stack-based ensemble framework for detecting cancer microRNA biomarkers. Genom. Proteom. Bioinform. 2017, 15, 381–388. [Google Scholar] [CrossRef]
- Lopez-Rincon, A.; Martinez-Archundia, M.; Martinez-Ruiz, G.U.; Tonda, A. Ensemble Feature Selection and Meta-Analysis of Cancer miRNA Biomarkers. bioRxiv 2018. [Google Scholar] [CrossRef]
- Lopez-Rincon, A.; Martinez-Archundia, M.; Martinez-Ruiz, G.U.; Schoenhuth, A.; Tonda, A. Automatic discovery of 100-miRNA signature for cancer classification using ensemble feature selection. BMC Bioinform. 2019, 20, 480. [Google Scholar] [CrossRef]
- Calore, F.; Lovat, F.; Garofalo, M. Non-coding RNAs and cancer. Int. J. Mol. Sci. 2013, 14, 17085–17110. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G. Circulating miRNAs: Roles in cancer diagnosis, prognosis and therapy. Adv. Drug Deliv. Rev. 2015, 81, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, K.Y.; Liu, S.M.; Sen, S. Tumor-associated circulating microRNAs as biomarkers of cancer. Molecules 2014, 19, 1912–1938. [Google Scholar] [CrossRef] [PubMed]
- Leshkowitz, D.; Horn-Saban, S.; Parmet, Y.; Feldmesser, E. Differences in microRNA detection levels are technology and sequence dependent. RNA 2013, 19, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Del Vescovo, V.; Meier, T.; Inga, A.; Denti, M.A.; Borlak, J. A cross-platform comparison of affymetrix and Agilent microarrays reveals discordant miRNA expression in lung tumors of c-Raf transgenic mice. PLoS ONE 2013, 8, e78870. [Google Scholar] [CrossRef]
- Bassani, N.; Ambrogi, F.; Biganzoli, E. Assessing agreement between miRNA microarray platforms. Microarrays 2014, 3, 302–321. [Google Scholar] [CrossRef]
- Abeel, T.; Helleputte, T.; Van de Peer, Y.; Dupont, P.; Saeys, Y. Robust biomarker identification for cancer diagnosis with ensemble feature selection methods. Bioinformatics 2009, 26, 392–398. [Google Scholar] [CrossRef]
- Saeys, Y.; Abeel, T.; Van de Peer, Y. Robust feature selection using ensemble feature selection techniques. In Proceedings of the Joint European Conference on Machine Learning and Knowledge Discovery in Databases, Antwerp, Belgium, 15–19 September 2008; pp. 313–325. [Google Scholar]
- Seijo-Pardo, B.; Porto-Diaz, I.; Bolon-Canedo, V.; Alonso-Betanzos, A. Ensemble feature selection: Homogeneous and heterogeneous approaches. Knowl.-Based Syst. 2017, 118, 124–139. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Peña-Chilet, M.; Martínez, M.T.; Pérez-Fidalgo, J.A.; Peiró-Chova, L.; Oltra, S.S.; Tormo, E.; Alonso-Yuste, E.; Martinez-Delgado, B.; Eroles, P.; Climent, J.; et al. MicroRNA profile in very young women with breast cancer. BMC Cancer 2014, 14, 529. [Google Scholar] [CrossRef] [PubMed]
- Romero-Cordoba, S.L.; Rodriguez-Cuevas, S.; Bautista-Pina, V.; Maffuz-Aziz, A.; D’Ippolito, E.; Cosentino, G.; Baroni, S.; Iorio, M.V.; Hidalgo-Miranda, A. Loss of function of miR-342-3p results in MCT1 over-expression and contributes to oncogenic metabolic reprogramming in triple negative breast cancer. Sci. Rep. 2018, 8, 12252. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.J.; Lee, H.S.; Burt, B.M.; Lee, G.K.; Yoon, K.A.; Park, Y.Y.; Sohn, B.H.; Kim, S.B.; Kim, M.S.; Lee, J.M.; et al. Integrated genomic analysis of recurrence-associated small non-coding RNAs in oesophageal cancer. Gut 2017, 66, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.C.; Chiu, Y.L.; Banerjee, S.; Park, K.; Mosquera, J.M.; Giannopoulou, E.; Alves, P.; Tewari, A.K.; Gerstein, M.B.; Beltran, H.; et al. Epigenetic repression of miR-31 disrupts androgen receptor homeostasis and contributes to prostate cancer progression. Cancer Res. 2013, 73, 1232–1244. [Google Scholar] [CrossRef]
- Casanova-Salas, I.; Rubio-Briones, J.; Calatrava, A.; Mancarella, C.; Masiá, E.; Casanova, J.; Fernández-Serra, A.; Rubio, L.; Ramírez-Backhaus, M.; Armiñán, A.; et al. Identification of miR-187 and miR-182 as biomarkers of early diagnosis and prognosis in patients with prostate cancer treated with radical prostatectomy. J. Urol. 2014, 192, 252–259. [Google Scholar] [CrossRef]
- Hermansen, S.K.; Sørensen, M.D.; Hansen, A.; Knudsen, S.; Alvarado, A.G.; Lathia, J.D.; Kristensen, B.W. A 4-miRNA signature to predict survival in glioblastomas. PLoS ONE 2017, 12, e0188090. [Google Scholar] [CrossRef]
- Jepsen, R.K.; Novotny, G.W.; Klarskov, L.L.; Bang-Berthelsen, C.H.; Haakansson, I.T.; Hansen, A.; Christensen, I.J.; Riis, L.B.; Høgdall, E. Early metastatic colorectal cancers show increased tissue expression of miR-17/92 cluster members in the invasive tumor front. Hum. Pathol. 2018, 80, 231–238. [Google Scholar] [CrossRef]
- Zhang, X.; Ni, Z.; Duan, Z.; Xin, Z.; Wang, H.; Tan, J.; Wang, G.; Li, F. Overexpression of E2F mRNAs associated with gastric cancer progression identified by the transcription factor and miRNA co-regulatory network analysis. PLoS ONE 2015, 10, e0116979. [Google Scholar] [CrossRef]
- Elgaaen, B.V.; Olstad, O.K.; Haug, K.B.F.; Brusletto, B.; Sandvik, L.; Staff, A.C.; Gautvik, K.M.; Davidson, B. Global miRNA expression analysis of serous and clear cell ovarian carcinomas identifies differentially expressed miRNAs including miR-200c-3p as a prognostic marker. BMC Cancer 2014, 14, 80. [Google Scholar]
- Trevino, V.; Falciani, F. GALGO: An R package for multivariate variable selection using genetic algorithms. Bioinformatics 2006, 22, 1154–1156. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Siklenka, K.; Arora, S.K.; Ribeiro, P.; Kimmins, S.; Xia, J. miRNet-dissecting miRNA-target interactions and functional associations through network-based visual analysis. Nucleic Acids Res. 2016, 44, W135–W141. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2016, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Falcon, S.; Gentleman, R. Using GOstats to test gene lists for GO term association. Bioinformatics 2006, 23, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929. [Google Scholar] [PubMed]
- Weiss, M. Your Guide to the Breast Cancer Pathology Report; Breastcancer. org: Ardmore, PA, USA, 2013. [Google Scholar]
- Jurman, G.; Riccadonna, S.; Furlanello, C. A comparison of MCC and CEN error measures in multi-class prediction. PLoS ONE 2012, 7, e41882. [Google Scholar] [CrossRef]
- Stegh, A.H. Targeting the p53 signaling pathway in cancer therapy–the promises, challenges and perils. Expert Opin. Ther. Targets 2012, 16, 67–83. [Google Scholar] [CrossRef]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef]
- Šimundić, A.M. Measures of diagnostic accuracy: Basic definitions. eJIFCC 2009, 19, 203. [Google Scholar]
- Mandrekar, J.N. Receiver operating characteristic curve in diagnostic test assessment. J. Thorac. Oncol. 2010, 5, 1315–1316. [Google Scholar] [CrossRef]
- Thakral, S.; Ghoshal, K. miR-122 is a unique molecule with great potential in diagnosis, prognosis of liver disease, and therapy both as miRNA mimic and antimir. Curr. Gene Ther. 2015, 15, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Bandiera, S.; Pfeffer, S.; Baumert, T.F.; Zeisel, M.B. miR-122—A key factor and therapeutic target in liver disease. J. Hepatol. 2015, 62, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Ergün, S.; Ulasli, M.; Igci, Y.Z.; Igci, M.; Kırkbes, S.; Borazan, E.; Balik, A.; Yumrutaş, Ö.; Camci, C.; Cakmak, E.A.; et al. The association of the expression of miR-122-5p and its target ADAM10 with human breast cancer. Mol. Biol. Rep. 2015, 42, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, X. miR-122-5p promotes aggression and epithelial-mesenchymal transition in triple-negative breast cancer by suppressing charged multivesicular body protein 3 through mitogen-activated protein kinase signaling. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef]
- Perez-Añorve, I.X.; Gonzalez-De la Rosa, C.H.; Soto-Reyes, E.; Beltran-Anaya, F.O.; Del Moral-Hernandez, O.; Salgado-Albarran, M.; Angeles-Zaragoza, O.; Gonzalez-Barrios, J.A.; Landero-Huerta, D.A.; Chavez-Saldaña, M.; et al. New insights into radioresistance in breast cancer identify a dual function of miR-122 as a tumor suppressor and oncomiR. Mol. Oncol. 2019, 13, 1249–1267. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, S.; Furuya, S.; Shiraishi, K.; Shimizu, H.; Akaike, H.; Hosomura, N.; Kawaguchi, Y.; Amemiya, H.; Kawaida, H.; Sudo, M.; et al. miR-122-5p as a novel biomarker for alpha-fetoprotein-producing gastric cancer. World J. Gastrointest. Oncol. 2018, 10, 344. [Google Scholar] [CrossRef]
- Lee, H.; Han, S.; Kwon, C.S.; Lee, D. Biogenesis and regulation of the let-7 miRNAs and their functional implications. Protein Cell 2016, 7, 100–113. [Google Scholar] [CrossRef]
- Liu, K.; Zhang, C.; Li, T.; Ding, Y.; Tu, T.; Zhou, F.; Qi, W.; Chen, H.; Sun, X. Let-7a inhibits growth and migration of breast cancer cells by targeting HMGA1. Int. J. Oncol. 2015, 46, 2526–2534. [Google Scholar] [CrossRef]
- Khalighfard, S.; Alizadeh, A.M.; Irani, S.; Omranipour, R. Plasma miR-21, miR-155, miR-10b, and Let-7a as the potential biomarkers for the monitoring of breast cancer patients. Sci. Rep. 2018, 8, 17981. [Google Scholar] [CrossRef]
- Zhao, W.; Hu, J.X.; Hao, R.M.; Zhang, Q.; Guo, J.Q.; Li, Y.J.; Xie, N.; Liu, L.Y.; Wang, P.Y.; Zhang, C.; et al. Induction of microRNA-let-7a inhibits lung adenocarcinoma cell growth by regulating cyclin D1. Oncol. Rep. 2018, 40, 1843–1854. [Google Scholar] [CrossRef]
- Yang, Q.; Jie, Z.; Cao, H.; Greenlee, A.R.; Yang, C.; Zou, F.; Jiang, Y. Low-level expression of let-7a in gastric cancer and its involvement in tumorigenesis by targeting RAB40C. Carcinogenesis 2011, 32, 713–722. [Google Scholar] [CrossRef]
- Grossi, I.; Salvi, A.; Baiocchi, G.; Portolani, N.; De Petro, G. Functional role of microRNA-23b-3p in cancer biology. MicroRNA 2018, 7, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Kou, C.H.; Zhou, T.; Han, X.L.; Zhuang, H.J.; Qian, H.X. Downregulation of mir-23b in plasma is associated with poor prognosis in patients with colorectal cancer. Oncol. Lett. 2016, 12, 4838–4844. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, K.; Han, K.; Tang, H.; Yin, X.; Zhang, J.; Zhang, X.; Zhang, L. Up-regulation of plasma miR-23b is associated with poor prognosis of gastric cancer. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2016, 22, 256. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, T.; Chen, G.; Yan, G.; Zhang, X.; Wan, Y.; Li, Q.; Zhu, B.; Zhuo, W. Identification of a serum microRNA expression signature for detection of lung cancer, involving miR-23b, miR-221, miR-148b and miR-423-3p. Lung Cancer 2017, 114, 6–11. [Google Scholar] [CrossRef]
- Chen, D.; Wu, X.; Xia, M.; Wu, F.; Ding, J.; Jiao, Y.; Zhan, Q.; An, F. Upregulated exosomic miR-23b-3p plays regulatory roles in the progression of pancreatic cancer. Oncol. Rep. 2017, 38, 2182–2188. [Google Scholar] [CrossRef]
- Monteleone, N.J.; Lutz, C.S. miR-708-5p: A microRNA with emerging roles in cancer. Oncotarget 2017, 8, 71292. [Google Scholar] [CrossRef]
- Jang, J.S.; Jeon, H.S.; Sun, Z.; Aubry, M.C.; Tang, H.; Park, C.H.; Rakhshan, F.; Schultz, D.A.; Kolbert, C.P.; Lupu, R.; et al. Increased miR-708 expression in NSCLC and its association with poor survival in lung adenocarcinoma from never smokers. Clin. Cancer Res. 2012, 18, 3658–3667. [Google Scholar] [CrossRef]
- Fedatto, P.F.; de Carvalho, T.I.; de Oliveira, J.C.; Antônio, D.S.M.; Pezuk, J.A.; da Cunha Tirapelli, D.P.; Féres, O.; da Rocha, J.J.R.; Scrideli, C.A.; Tone, L.G.; et al. MiR-708-5p as a Predictive Marker of Colorectal Cancer Prognosis. J. Anal. Oncol. 2016, 5, 14–23. [Google Scholar]
- Song, T.; Zhang, X.; Zhang, L.; Dong, J.; Cai, W.; Gao, J.; Hong, B. miR-708 promotes the development of bladder carcinoma via direct repression of Caspase-2. J. Cancer Res. Clin. Oncol. 2013, 139, 1189–1198. [Google Scholar] [CrossRef]
- Saini, S.; Yamamura, S.; Majid, S.; Shahryari, V.; Hirata, H.; Tanaka, Y.; Dahiya, R. MicroRNA-708 induces apoptosis and suppresses tumorigenicity in renal cancer cells. Cancer Res. 2011, 71, 6208–6219. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, D.R.; Howe, E.N.; Spoelstra, N.S.; Richer, J.K. Loss of miR-200c: A marker of aggressiveness and chemoresistance in female reproductive cancers. J. Oncol. 2010, 2010. [Google Scholar] [CrossRef]
- Liu, X.G.; Zhu, W.Y.; Huang, Y.Y.; Ma, L.N.; Zhou, S.Q.; Wang, Y.K.; Zeng, F.; Zhou, J.H.; Zhang, Y.K. High expression of serum miR-21 and tumor miR-200c associated with poor prognosis in patients with lung cancer. Med. Oncol. 2012, 29, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, M.; Raza, U.; Saatci, Ö.; Eyüpoğlu, E.; Yurdusev, E.; Şahin, Ö. miR-200c: A versatile watchdog in cancer progression, EMT, and drug resistance. J. Mol. Med. 2016, 94, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Zhao, Y.; Feng, Z. MicroRNA control of p53. J. Cell. Biochem. 2017, 118, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Takwi, A.; Li, Y. The p53 pathway encounters the microRNA world. Curr. Genom. 2009, 10, 194–197. [Google Scholar] [CrossRef]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microRNA processing by p53. Nature 2009, 460, 529–533. [Google Scholar] [CrossRef]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 2010. [Google Scholar] [CrossRef]
- Chen, M.; Xie, S. Therapeutic targeting of cellular stress responses in cancer. Thorac. Cancer 2018, 9, 1575–1582. [Google Scholar] [CrossRef]
- Babar, I.A.; Slack, F.J.; Weidhaas, J.B. miRNA modulation of the cellular stress response. Future Oncol. 2008. [Google Scholar] [CrossRef]
- Eichner, L.J.; Perry, M.C.; Dufour, C.R.; Bertos, N.; Park, M.; St-Pierre, J.; Giguère, V. miR-378 mediates metabolic shift in breast cancer cells via the PGC-1β/ERRγ transcriptional pathway. Cell Metab. 2010, 12, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.Y.; Deng, Z.Q.; Liu, F.Q.; Qian, J.; Lin, J.; Tang, Q.; Wen, X.M.; Zhou, J.D.; Zhang, Y.Y.; Zhu, X.W. Association between mir-24 and mir-378 in formalin-fixed paraffin-embedded tissues of breast cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 4261. [Google Scholar] [PubMed]
- He, Y.J.; Wu, J.Z.; Ji, M.H.; Ma, T.; Qiao, E.Q.; Ma, R.; Tang, J.H. miR-342 is associated with estrogen receptor-α expression and response to tamoxifen in breast cancer. Exp. Ther. Med. 2013, 5, 813–818. [Google Scholar] [CrossRef]
- Wei, Y.; Lai, X.; Yu, S.; Chen, S.; Ma, Y.; Zhang, Y.; Li, H.; Zhu, X.; Yao, L.; Zhang, J. Exosomal miR-221/222 enhances tamoxifen resistance in recipient ER-positive breast cancer cells. Breast Cancer Res. Treat. 2014, 147, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.J.; Lin, J.; Yang, H.; Kong, W.; He, L.; Ma, X.; Coppola, D.; Cheng, J.Q. MicroRNA-221/222 negatively regulates estrogen receptorα and is associated with tamoxifen resistance in breast cancer. J. Biol. Chem. 2008, 283, 31079–31086. [Google Scholar] [CrossRef] [PubMed]
- Cittelly, D.M.; Das, P.M.; Spoelstra, N.S.; Edgerton, S.M.; Richer, J.K.; Thor, A.D.; Jones, F.E. Downregulation of miR-342 is associated with tamoxifen resistant breast tumors. Mol. Cancer 2010, 9, 317. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.X.; Wang, C.L.; Yu, A.L.; Wang, Q.Y.; Zhan, M.N.; Tang, J.; Gong, X.F.; Yin, Q.Q.; He, M.; He, J.R.; et al. MiR-630 suppresses breast cancer progression by targeting metadherin. Oncotarget 2016, 7, 1288. [Google Scholar] [CrossRef]
- Wang, S.; Bian, C.; Yang, Z.; Bo, Y.; Li, J.; Zeng, L.; Zhou, H.; Zhao, R.C. miR-145 inhibits breast cancer cell growth through RTKN. Int. J. Oncol. 2009, 34, 1461–1466. [Google Scholar]
- Yan, X.; Chen, X.; Liang, H.; Deng, T.; Chen, W.; Zhang, S.; Liu, M.; Gao, X.; Liu, Y.; Zhao, C.; et al. miR-143 and miR-145 synergistically regulate ERBB3 to suppress cell proliferation and invasion in breast cancer. Mol. Cancer 2014, 13, 220. [Google Scholar] [CrossRef]
- Santolla, M.F.; Lappano, R.; Cirillo, F.; Rigiracciolo, D.C.; Sebastiani, A.; Abonante, S.; Tassone, P.; Tagliaferri, P.; Di Martino, M.T.; Maggiolini, M.; et al. miR-221 stimulates breast cancer cells and cancer- associated fibroblasts (CAFs) through selective interference with the A20/c-Rel/CTGF signaling. J. Exp. Clin. Cancer Res. 2018, 37, 94. [Google Scholar] [CrossRef]
- Chen, W.X.; Hu, Q.; Qiu, M.T.; Zhong, S.L.; Xu, J.J.; Tang, J.H.; Zhao, J.H. miR-221/222: Promising biomarkers for breast cancer. Tumor Biol. 2013, 34, 1361–1370. [Google Scholar] [CrossRef]
- Stinson, S.; Lackner, M.R.; Adai, A.T.; Yu, N.; Kim, H.J.; O’Brien, C.; Spoerke, J.; Jhunjhunwala, S.; Boyd, Z.; Januario, T.; et al. TRPS1 targeting by miR-221/222 promotes the epithelial-to-mesenchymal transition in breast cancer. Sci. Signal. 2011, 4, ra41. [Google Scholar]
- Liu, Y.X.; Zhao, L.P.; Zhang, Y.L.; Dong, Y.Y.; Ren, H.Y.; Diao, K.X.; Mi, X.Y. MiR-630 inhibits cells migration and invasion by targeting SOX4 in triple-negative breast cancer. Int. J. Clin. Exp. Pathol. 2016, 9, 9097–9105. [Google Scholar]
- Spizzo, R.; Nicoloso, M.; Lupini, L.; Lu, Y.; Fogarty, J.; Rossi, S.; Zagatti, B.; Fabbri, M.; Veronese, A.; Liu, X.; et al. miR-145 participates with TP53 in a death-promoting regulatory loop and targets estrogen receptor-α in human breast cancer cells. Cell Death Differ. 2010, 17, 246–254. [Google Scholar] [CrossRef]
- Zare, M.; Bastami, M.; Solali, S.; Alivand, M.R. Aberrant miRNA promoter methylation and EMT-involving miRNAs in breast cancer metastasis: Diagnosis and therapeutic implications. J. Cell. Physiol. 2018, 233, 3729–3744. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2007, 36, D154–D158. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.L.; Wang, Z.Q.; Zeng, Z.L.; Wu, W.J.; Zhang, D.S.; Luo, H.Y.; Wang, F.; Qiu, M.Z.; Wang, D.S.; Ren, C.; et al. Identification of microRNA-214 as a negative regulator of colorectal cancer liver metastasis by way of regulation of fibroblast growth factor receptor 1 expression. Hepatology 2014, 60, 598–609. [Google Scholar] [CrossRef]
- Dodd, L.E.; Sengupta, S.; Chen, I.H.; Den Boon, J.A.; Cheng, Y.J.; Westra, W.; Newton, M.A.; Mittl, B.F.; McShane, L.; Chen, C.J.; et al. Genes involved in DNA repair and nitrosamine metabolism and those located on chromosome 14q32 are dysregulated in nasopharyngeal carcinoma. Cancer Epidemiol. Prev. Biomark. 2006, 15, 2216–2225. [Google Scholar] [CrossRef] [PubMed]
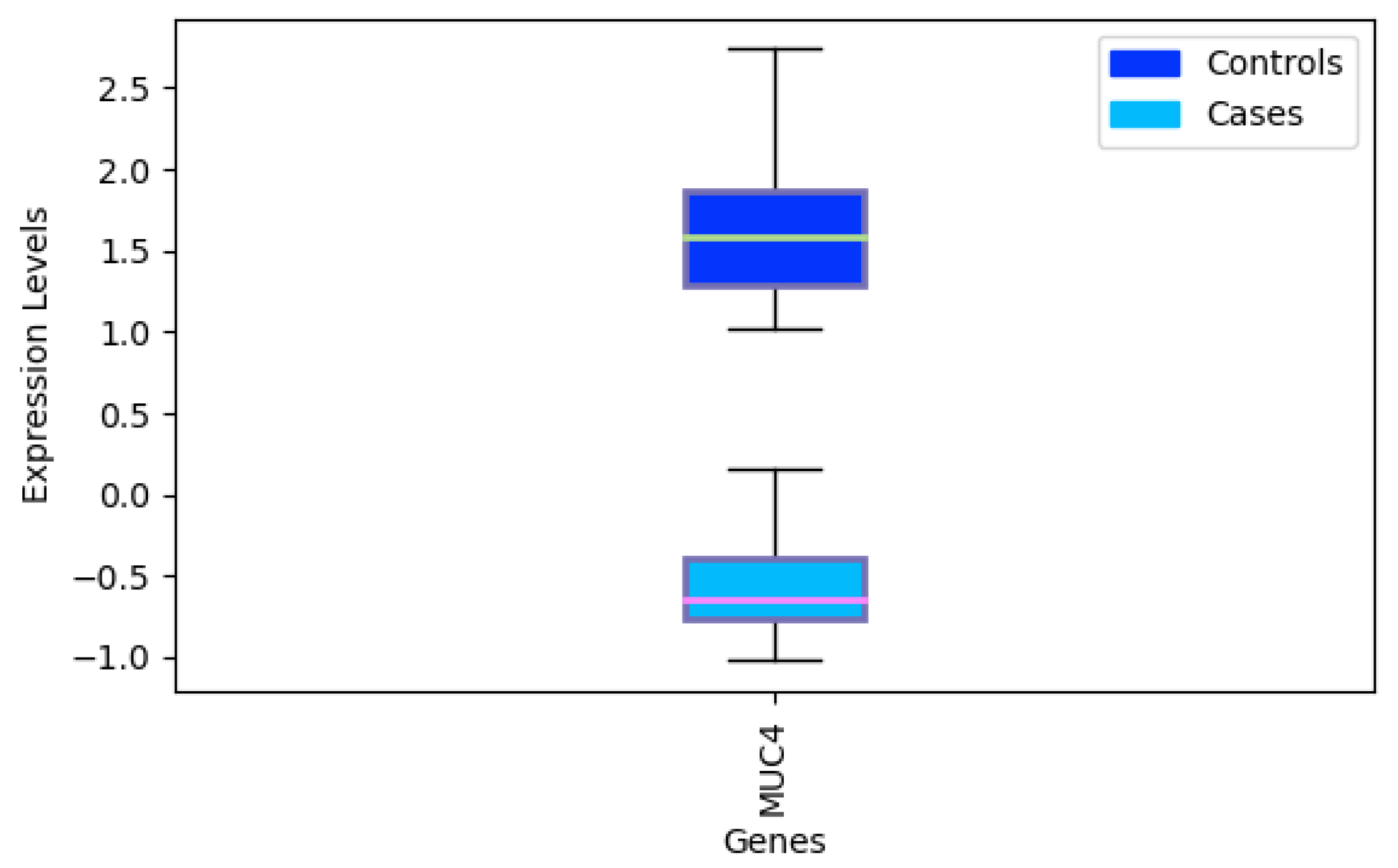
- Chakraborty, S.; Jain, M.; Sasson, A.R.; Batra, S.K. MUC4 as a diagnostic marker in cancer. Expert Opin. Med. Diagn. 2008, 2, 891–910. [Google Scholar] [CrossRef]
- Wei, Y.C.; Yang, S.F.; Chang, S.L.; Chen, T.J.; Lee, S.W.; Chen, H.S.; Lin, L.C.; Li, C.F. Periostin overexpression is associated with worse prognosis in nasopharyngeal carcinoma from endemic area: A cohort study. OncoTargets Ther. 2018, 11, 3205. [Google Scholar] [CrossRef] [PubMed]













| Dataset | Samples | Type | Reference | Class | Platform |
|---|---|---|---|---|---|
| GSE48088 | 33 | BRCA | [34] | 0 | GPL14613 |
| GSE86277 | 72 | BRCA | [35] | 0 | GPL14613 |
| GSE86278 | 49 | BRCA | [35] | 0 | GPL14613 |
| GSE86281 | 50 | BRCA | [35] | 0 | GPL16384 |
| GSE55856 | 108 | ESCA | [36] | 1 | GPL14613 |
| GSE34496 | 44 | HSNC | - | 2 | GPL8786 |
| GSE67138 | 57 | LIHC | - | 3 | GPL8786 |
| GSE67139 | 115 | LIHC | - | 3 | GPL8786 |
| GSE116182 | 64 | LIHC | - | 3 | GPL14613 |
| GSE36802 | 21 | PRCA | [37] | 4 | GPL8786 |
| GSE45604 | 50 | PRCA | [38] | 4 | GPL14613 |
| GSE104554 | 38 | GBM | [39] | 5 | GPL14613 |
| GSE110402 | 75 | CRC | [40] | 6 | GPL14613 |
| GSE46729 | 24 | NSCLC | - | 7 | GPL8786 |
| GSE63121 | 15 | GC | [41] | 8 | GPL8786 |
| GSE47841 | 30 | OVC | [42] | 9 | GPL14613 |
| Heterogeneous | Homogeneous | GALGO | ||||||
|---|---|---|---|---|---|---|---|---|
| Ens. 5 Feats. | Ens. 5 Feats. | 5 Feats. | 253 Feats. | |||||
| Gradient Boosting | 0.9751 | 0.0134 | 0.9797 | 0.0154 | 0.8374 | 0.0453 | 0.975 | 0.0128 |
| Random Forest | 0.9761 | 0.0192 | 0.9854 | 0.0155 | 0.8656 | 0.0383 | 1 | 0 |
| Logistic Regression | 0.8877 | 0.0239 | 0.8777 | 0.0281 | 0.4954 | 0.0416 | 1 | 0 |
| Passive Aggressive | 0.8239 | 0.0544 | 0.8144 | 0.0707 | 0.4545 | 0.0590 | 1 | 0 |
| SGD | 0.8937 | 0.0305 | 0.8632 | 0.0362 | 0.5204 | 0.0832 | 0.9941 | 0.0078 |
| SVC | 0.9620 | 0.0197 | 0.9499 | 0.0186 | 0.5308 | 0.0454 | 1 | 0 |
| Ridge | 0.8083 | 0.0272 | 0.6900 | 0.0173 | 0.5010 | 0.0451 | 0.9977 | 0.0045 |
| Bagging | 0.9702 | 0.0193 | 0.9643 | 0.0165 | 0.8418 | 0.0425 | 0.9894 | 0.0121 |
| Global | 0.9121 | 0.0260 | 0.8906 | 0.0273 | 0.6309 | 0.0500 | 0.9945 | 0.0047 |
| Heterogeneous | Homogeneous | Univariate | GALGO | Random 1 | Random 2 | Random 3 | Shuffle | |
|---|---|---|---|---|---|---|---|---|
| Gradient Boosting (n_estimators = 300) | 0.9412 | 0.9294 | 0.9412 | 0.9176 | 0.8824 | 0.8353 | 0.8000 | 0.2471 |
| Random Forest (n_estimators = 300) | 0.9412 | 0.9529 | 0.9412 | 0.9059 | 0.8941 | 0.8235 | 0.8235 | 0.2471 |
| Logistic Regression | 0.9059 | 0.8824 | 0.8588 | 0.8706 | 0.6353 | 0.5412 | 0.5882 | 0.2824 |
| Passive Aggressive | 0.8706 | 0.7765 | 0.7176 | 0.8471 | 0.4235 | 0.4118 | 0.5294 | 0.1765 |
| SGD | 0.8824 | 0.8588 | 0.7765 | 0.7882 | 0.5294 | 0.4235 | 0.3765 | 0.2353 |
| SVC(linear) | 0.9765 | 0.9176 | 0.8941 | 0.8588 | 0.6235 | 0.6235 | 0.5412 | 0.2824 |
| Ridge | 0.8118 | 0.7059 | 0.7412 | 0.7059 | 0.5882 | 0.4588 | 0.4000 | 0.2706 |
| Bagging (n_estimators = 300) | 0.9412 | 0.9294 | 0.9176 | 0.8824 | 0.8706 | 0.8471 | 0.8235 | 0.2118 |
| Average | 0.9089 | 0.8691 | 0.8485 | 0.8471 | 0.6809 | 0.6206 | 0.6103 | 0.2442 |
| Heterogeneous | Homogeneous | Univariate | GALGO | Random 1 | Random 2 | Random 3 | Shuffle | |
|---|---|---|---|---|---|---|---|---|
| Gradient Boosting (n_estimators = 300) | 0.9346 | 0.9216 | 0.9346 | 0.9085 | 0.8693 | 0.8170 | 0.7778 | 0.1634 |
| Random Forest (n_estimators = 300) | 0.9346 | 0.9477 | 0.9346 | 0.8954 | 0.8824 | 0.8039 | 0.8039 | 0.1634 |
| Logistic Regression | 0.8954 | 0.8693 | 0.8431 | 0.8562 | 0.5948 | 0.4902 | 0.5425 | 0.2026 |
| Passive Aggressive | 0.8562 | 0.7516 | 0.6863 | 0.8301 | 0.3595 | 0.3464 | 0.4771 | 0.0850 |
| SGD | 0.8693 | 0.8431 | 0.7516 | 0.7647 | 0.4771 | 0.3595 | 0.3072 | 0.1503 |
| SVC(linear) | 0.9739 | 0.9085 | 0.8824 | 0.8431 | 0.5817 | 0.5817 | 0.4902 | 0.2026 |
| Ridge | 0.7908 | 0.6732 | 0.7124 | 0.6732 | 0.5425 | 0.3987 | 0.3333 | 0.1895 |
| Bagging (n_estimators = 300) | 0.9346 | 0.9216 | 0.9085 | 0.8693 | 0.8562 | 0.8301 | 0.8039 | 0.1242 |
| Average | 0.8987 | 0.8546 | 0.8317 | 0.8301 | 0.6454 | 0.5784 | 0.5670 | 0.1601 |
| Accuracy | MCC | |||
|---|---|---|---|---|
| Heterogeneous | 0.8840 | 0.0120 | 0.8691 | 0.0156 |
| Homogeneous | 0.8518 | 0.0183 | 0.8353 | 0.0204 |
| GALGO | 0.8227 | 0.0255 | 0.8132 | 0.0338 |
| Pathway | Total | Expected | Hits | Pval |
|---|---|---|---|---|
| p53 signaling pathway | 68 | 1 | 10 | 3.70 × 10 |
| Pathways in cancer | 310 | 4.57 | 19 | 3.70 × 10 |
| Prostate cancer | 87 | 1.28 | 11 | 3.70 × 10 |
| Glioma | 65 | 0.958 | 8 | 0.000207 |
| Melanoma | 68 | 1 | 7 | 0.00196 |
| Bladder cancer | 29 | 0.428 | 5 | 0.00196 |
| Colorectal cancer | 49 | 0.722 | 6 | 0.00217 |
| Chronic myeloid leukemia | 73 | 1.08 | 7 | 0.00227 |
| Focal adhesion | 200 | 2.95 | 11 | 0.00327 |
| Small cell lung cancer | 80 | 1.18 | 7 | 0.00327 |
| Pathway | Total | Expected | Hits | Pval |
|---|---|---|---|---|
| Cellular response to stress | 1620 | 15.4 | 44 | 3.03 × 10 |
| Positive regulation of cell proliferation | 786 | 7.43 | 27 | 1.66 × 10 |
| Response to hypoxia | 245 | 2.31 | 15 | 2.53 × 10 |
| Regulation of cell cycle | 886 | 8.37 | 28 | 2.53 × 10 |
| Regulation of cell proliferation | 1430 | 13.5 | 36 | 4.30 × 10 |
| Response to abiotic stimulus | 876 | 8.28 | 27 | 5.44 × 10 |
| Negative regulation of cell cycle | 520 | 4.91 | 20 | 1.04 × 10 |
| Regulation of molecular function | 2250 | 21.2 | 46 | 1.08 × 10 |
| Regulation of cyclin-dependent protein kinase activity | 89 | 0.841 | 9 | 1.40 × 10 |
| Negative regulation of apoptotic process | 679 | 6.42 | 22 | 2.56 × 10 |
| 5 Feats. | 253 Feats. | Romero et al. | (31 Feats.) | |||
|---|---|---|---|---|---|---|
| Classifier | ||||||
| GradientBoosting | 0.9345 | 0.0523 | 0.9134 | 0.0487 | 0.9239 | 0.0485 |
| RandomForest | 0.9354 | 0.0617 | 0.9459 | 0.0416 | 0.9184 | 0.0432 |
| LogisticRegression | 0.9243 | 0.0487 | 0.9406 | 0.0612 | 0.8958 | 0.0643 |
| PassiveAggressive | 0.9076 | 0.0550 | 0.9076 | 0.0778 | 0.8797 | 0.0637 |
| SGDClassifier | 0.9085 | 0.0628 | 0.8918 | 0.0770 | 0.8692 | 0.0700 |
| SVC(linear) | 0.9243 | 0.0487 | 0.9242 | 0.0655 | 0.8572 | 0.0400 |
| Ridge | 0.9079 | 0.0754 | 0.9085 | 0.0533 | 0.8856 | 0.0611 |
| Bagging | 0.9295 | 0.0411 | 0.9076 | 0.0544 | 0.9341 | 0.0412 |
| Global | 0.9215 | 0.0557 | 0.9175 | 0.0599 | 0.8955 | 0.0540 |
| Pathway | Total | Expected | Hits | Pval |
|---|---|---|---|---|
| p53 signaling pathway | 68 | 0.509 | 6 | 0.000518 |
| Pancreatic cancer | 69 | 0.516 | 6 | 0.000518 |
| Glioma | 65 | 0.486 | 6 | 0.000518 |
| Melanoma | 68 | 0.509 | 6 | 0.000518 |
| Chronic myeloid leukemia | 73 | 0.546 | 6 | 0.000576 |
| Bladder cancer | 29 | 0.217 | 4 | 0.00197 |
| Cell cycle | 124 | 0.927 | 6 | 0.00821 |
| Pathways in cancer | 310 | 2.32 | 9 | 0.009 |
| Non-small cell lung cancer | 52 | 0.389 | 4 | 0.0133 |
| Adherens junction | 70 | 0.524 | 4 | 0.0368 |
| Pathway | Total | Expected | Hits | Pval |
|---|---|---|---|---|
| negative regulation of cell proliferation | 585 | 2.7 | 12 | 0.00631 |
| regulation of cell proliferation | 1430 | 6.6 | 19 | 0.00631 |
| cell proliferation | 1900 | 8.79 | 22 | 0.00674 |
| G1 phase of mitotic cell cycle | 47 | 0.217 | 4 | 0.00882 |
| enzyme linked receptor protein signaling pathway | 1180 | 5.43 | 16 | 0.00882 |
| myeloid cell differentiation | 296 | 1.37 | 8 | 0.00882 |
| G1 phase | 49 | 0.226 | 4 | 0.00882 |
| response to endogenous stimulus | 1360 | 6.3 | 17 | 0.0114 |
| positive regulation of cell proliferation | 786 | 3.63 | 12 | 0.0166 |
| response to organic substance | 2500 | 11.5 | 24 | 0.0166 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopez-Rincon, A.; Mendoza-Maldonado, L.; Martinez-Archundia, M.; Schönhuth, A.; Kraneveld, A.D.; Garssen, J.; Tonda, A. Machine Learning-Based Ensemble Recursive Feature Selection of Circulating miRNAs for Cancer Tumor Classification. Cancers 2020, 12, 1785. https://doi.org/10.3390/cancers12071785
Lopez-Rincon A, Mendoza-Maldonado L, Martinez-Archundia M, Schönhuth A, Kraneveld AD, Garssen J, Tonda A. Machine Learning-Based Ensemble Recursive Feature Selection of Circulating miRNAs for Cancer Tumor Classification. Cancers. 2020; 12(7):1785. https://doi.org/10.3390/cancers12071785
Chicago/Turabian StyleLopez-Rincon, Alejandro, Lucero Mendoza-Maldonado, Marlet Martinez-Archundia, Alexander Schönhuth, Aletta D. Kraneveld, Johan Garssen, and Alberto Tonda. 2020. "Machine Learning-Based Ensemble Recursive Feature Selection of Circulating miRNAs for Cancer Tumor Classification" Cancers 12, no. 7: 1785. https://doi.org/10.3390/cancers12071785
APA StyleLopez-Rincon, A., Mendoza-Maldonado, L., Martinez-Archundia, M., Schönhuth, A., Kraneveld, A. D., Garssen, J., & Tonda, A. (2020). Machine Learning-Based Ensemble Recursive Feature Selection of Circulating miRNAs for Cancer Tumor Classification. Cancers, 12(7), 1785. https://doi.org/10.3390/cancers12071785